
Antibody–Drug Conjugates as an Emerging Therapy in Oncodermatology

 , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. ADCs in Oncology
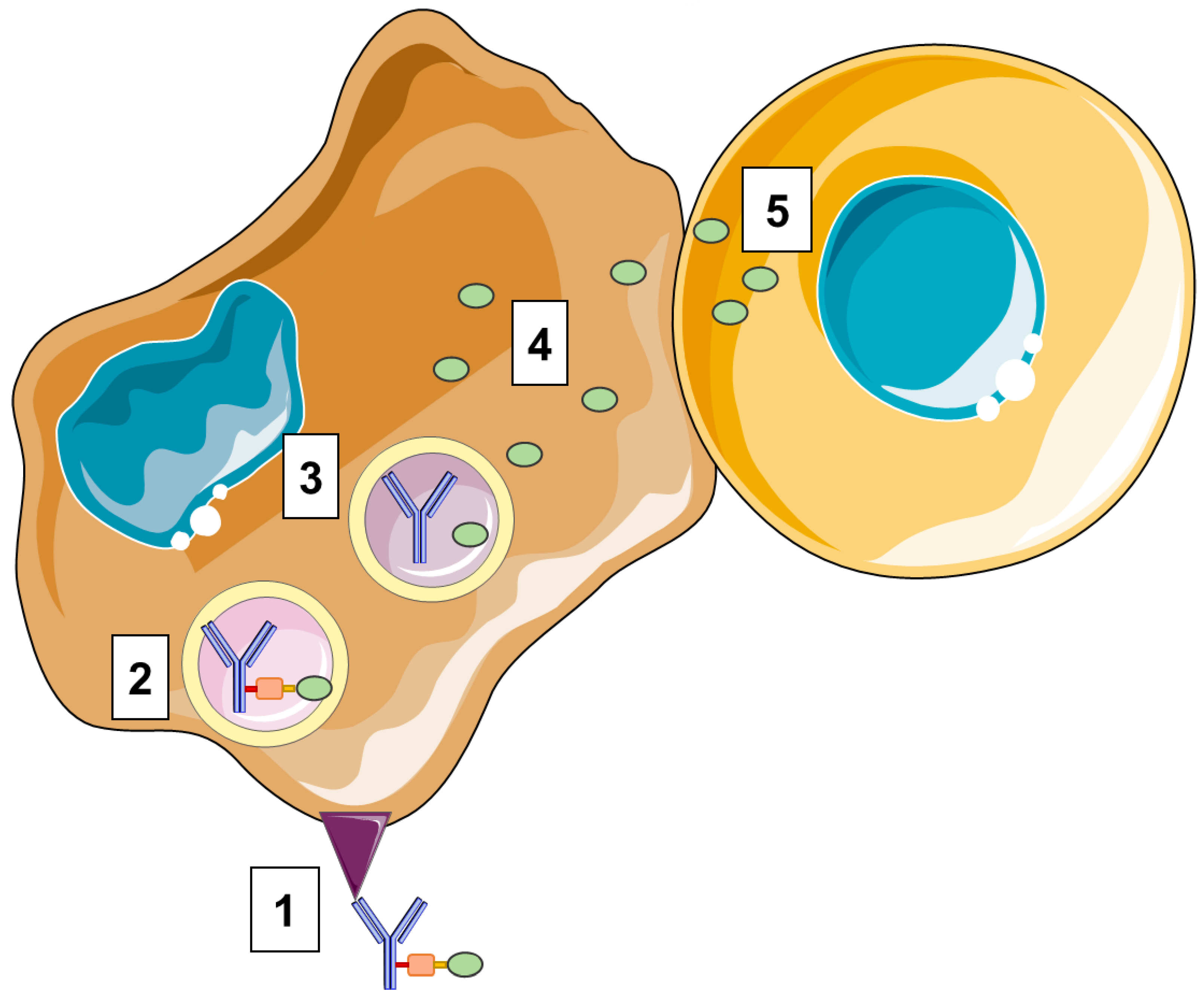
2.1. Structure of ADCs
2.2. Targets
2.3. Antibodies
2.4. Payloads
2.5. Linker
2.6. Linker–Antibody Conjugation
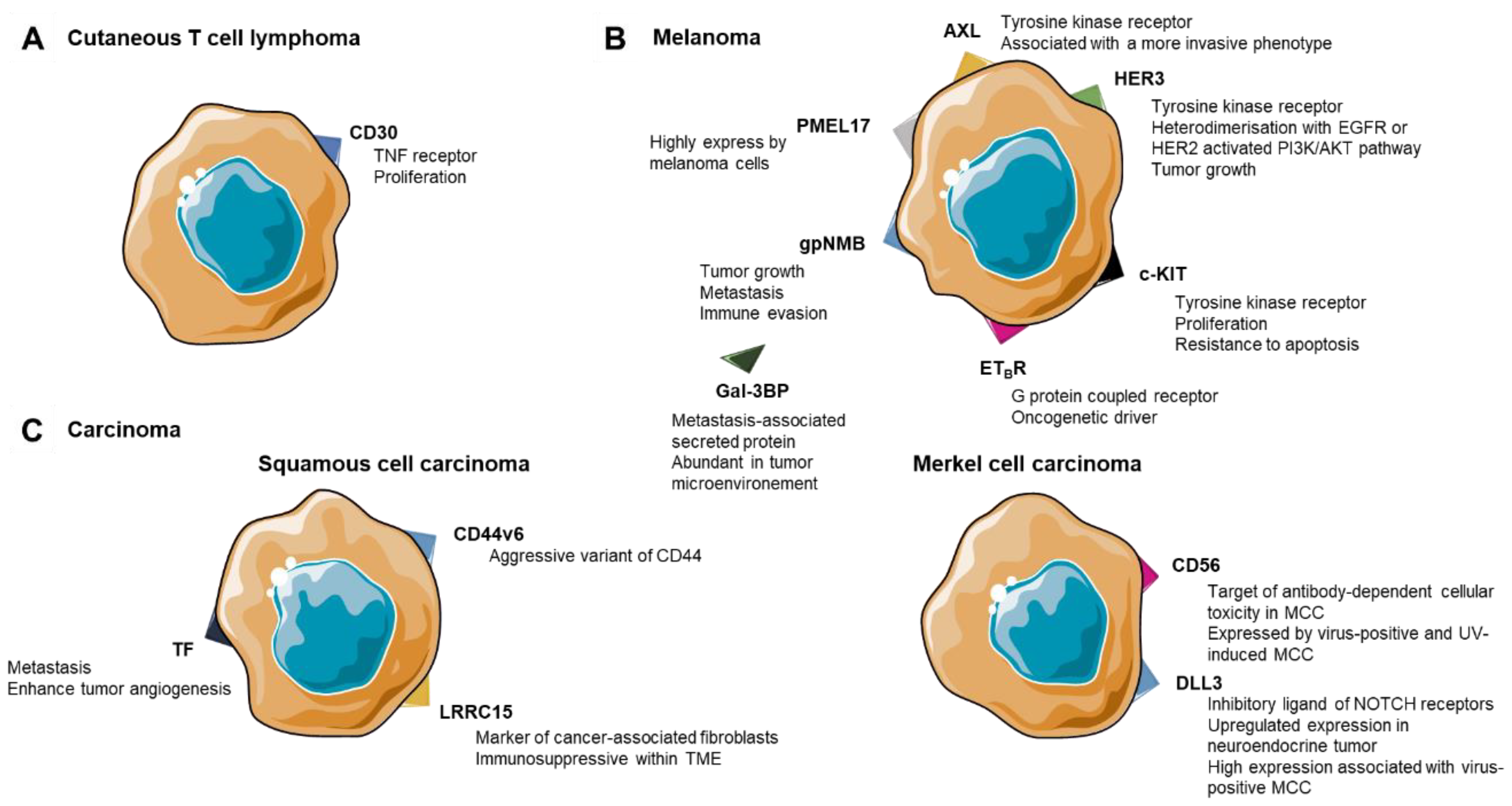
3. ADCs in Cutaneous T-Cell Lymphoma
4. ADCs in Melanoma
4.1. Membrane Protein as Targets
4.2. Tyrosine Kinase Receptor
4.3. Soluble Target for ADCs
5. ADCs in Skin Carcinomas
5.1. Squamous Cell Carcinoma
5.2. Merkel Cell Carcinoma
6. Strengths and Weaknesses of ADCs: Challenges and Perspectives
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADC | antibody–drug conjugate |
| CTCL | cutaneous T-cell lymphoma |
| DAR | drug to antibody ratio |
| ETBR | endothelin B receptor |
| FcRn | neonatal Fc receptor |
| FDA | food and drug administration |
| Gal-3BP | galectin-3-binding protein |
| gpNMB | glycoprotein-NMB |
| ICI | immune checkpoint inhibitors |
| LRRC15 | leucine-rich repeat containing 15 mAb: monoclonal antibody |
| MCC | Merkel cell carcinoma |
| MF | mycosis fungoides |
| MMAE | monomethyl auristatin E |
| MMAF | monomethyl auristatin F |
| PBD | pyrrolobenzodiazepines |
| PDX | patient-derived xenograft |
| RTK | receptor tyrosine kinase |
| SCC | squamous cell carcinoma |
| SS | Sézary syndrome |
| TF | tissue factor |
| TME | tumor microenvironment |
References
- Schwartz, R.S. Paul Ehrlich’s Magic Bullets. N. Engl. J. Med. 2004, 350, 1079–1080. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s Magic Bullet Concept: 100 Years of Progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Kakadia, S.; Yarlagadda, N.; Awad, R.; Kundranda, M.; Niu, J.; Naraev, B.; Mina, L.; Dragovich, T.; Gimbel, M.; Mahmoud, F. Mechanisms of Resistance to BRAF and MEK Inhibitors and Clinical Update of US Food and Drug Administration-Approved Targeted Therapy in Advanced Melanoma. OncoTargets Ther. 2018, 11, 7095–7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody-Drug Conjugates for Cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef] [PubMed]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody–Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and Challenges for the next Generation of Antibody-Drug Conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Boni, V.; Sharma, M.R.; Patnaik, A. The Resurgence of Antibody Drug Conjugates in Cancer Therapeutics: Novel Targets and Payloads. Am. Soc. Clin. Oncol. Educ. Book Am. Soc. Clin. Oncol. Annu. Meet. 2020, 40, e58–e74. [Google Scholar] [CrossRef]
- Amani, N.; Dorkoosh, F.A.; Mobedi, H. ADCs, as Novel Revolutionary Weapons for Providing a Step Forward in Targeted Therapy of Malignancies. Curr. Drug Deliv. 2020, 17, 23–51. [Google Scholar] [CrossRef]
- Teicher, B.A. Antibody-Drug Conjugate Targets. Curr. Cancer Drug Targets 2009, 9, 982–1004. [Google Scholar] [CrossRef]
- Lai, J.; Wang, Y.; Wu, S.-S.; Ding, D.; Sun, Z.-Y.; Zhang, Y.; Zhou, J.; Zhou, Z.; Xu, Y.-C.; Pan, L.-Q.; et al. Elimination of Melanoma by Sortase A-Generated TCR-like Antibody-Drug Conjugates (TL-ADCs) Targeting Intracellular Melanoma Antigen MART-1. Biomaterials 2018, 178, 158–169. [Google Scholar] [CrossRef]
- Staudacher, A.H.; Brown, M.P. Antibody Drug Conjugates and Bystander Killing: Is Antigen-Dependent Internalisation Required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef]
- Damelin, M.; Zhong, W.; Myers, J.; Sapra, P. Evolving Strategies for Target Selection for Antibody-Drug Conjugates. Pharm. Res. 2015, 32, 3494–3507. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.F.; Thurston, D.E.; Crescioli, S.; et al. Antibody Structure and Engineering Considerations for the Design and Function of Antibody Drug Conjugates (ADCs). OncoImmunology 2018, 7, e1395127. [Google Scholar] [CrossRef] [PubMed]
- Vaklavas, C.; Forero-Torres, A. Safety and Efficacy of Brentuximab Vedotin in Patients with Hodgkin Lymphoma or Systemic Anaplastic Large Cell Lymphoma. Ther. Adv. Hematol. 2012, 3, 209–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef]
- Fayad, L.; Offner, F.; Smith, M.R.; Verhoef, G.; Johnson, P.; Kaufman, J.L.; Rohatiner, A.; Advani, A.; Foran, J.; Hess, G.; et al. Safety and Clinical Activity of a Combination Therapy Comprising Two Antibody-Based Targeting Agents for the Treatment of Non-Hodgkin Lymphoma: Results of a Phase I/II Study Evaluating the Immunoconjugate Inotuzumab Ozogamicin With Rituximab. J. Clin. Oncol. 2013, 31, 573–583. [Google Scholar] [CrossRef]
- Sievers, E.L.; Senter, P.D. Antibody-Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef]
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody–Drug Conjugates for Cancer Therapy. Lancet Oncol. 2016, 17, e254–e262. [Google Scholar] [CrossRef]
- Perrino, E.; Steiner, M.; Krall, N.; Bernardes, G.J.L.; Pretto, F.; Casi, G.; Neri, D. Curative Properties of Noninternalizing Antibody–Drug Conjugates Based on Maytansinoids. Cancer Res. 2014, 74, 2569–2578. [Google Scholar] [CrossRef] [Green Version]
- Gébleux, R.; Stringhini, M.; Casanova, R.; Soltermann, A.; Neri, D. Non-Internalizing Antibody–Drug Conjugates Display Potent Anti-Cancer Activity upon Proteolytic Release of Monomethyl Auristatin E in the Subendothelial Extracellular Matrix. Int. J. Cancer 2017, 140, 1670–1679. [Google Scholar] [CrossRef] [Green Version]
- O’Malley, D.M.; Matulonis, U.A.; Birrer, M.J.; Castro, C.M.; Gilbert, L.; Vergote, I.; Martin, L.P.; Mantia-Smaldone, G.M.; Martin, A.G.; Bratos, R.; et al. Phase Ib Study of Mirvetuximab Soravtansine, a Folate Receptor Alpha (FRα)-Targeting Antibody-Drug Conjugate (ADC), in Combination with Bevacizumab in Patients with Platinum-Resistant Ovarian Cancer. Gynecol. Oncol. 2020, 157, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Castaneda, D.C.; Brachet, G.; Goupille, C.; Ouldamer, L.; Gouilleux-Gruart, V. The Neonatal Fc Receptor in Cancer FcRn in Cancer. Cancer Med. 2020, 9, 4736–4742. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Wang, W.; Fauty, S.; Fang, Y.; Hamuro, L.; Hussain, A.; Prueksaritanont, T. The Effect of the Neonatal Fc Receptor on Human IgG Biodistribution in Mice. mAbs 2014, 6, 502–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma Receptors as Regulators of Immune Responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Aguiar, S.; Dias, J.; Manuel, A.M.; Russo, R.; Gois, P.M.P.; da Silva, F.A.; Goncalves, J. Chimeric Small Antibody Fragments as Strategy to Deliver Therapeutic Payloads. Adv. Protein Chem. Struct. Biol. 2018, 112, 143–182. [Google Scholar] [CrossRef]
- Carter, P.J. Potent Antibody Therapeutics by Design. Nat. Rev. Immunol. 2006, 6, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Gauzy-Lazo, L.; Sassoon, I.; Brun, M.-P. Advances in Antibody-Drug Conjugate Design: Current Clinical Landscape and Future Innovations. SLAS Discov. Adv. Sci. Drug Discov. 2020, 25, 843–868. [Google Scholar] [CrossRef]
- Lucas, A.T.; Robinson, R.; Schorzman, A.N.; Piscitelli, J.A.; Razo, J.F.; Zamboni, W.C. Pharmacologic Considerations in the Disposition of Antibodies and Antibody-Drug Conjugates in Preclinical Models and in Patients. Antibodies Basel Switz. 2019, 8, 3. [Google Scholar] [CrossRef] [Green Version]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [Green Version]
- Buecheler, J.W.; Winzer, M.; Weber, C.; Gieseler, H. Alteration of Physicochemical Properties for Antibody-Drug Conjugates and Their Impact on Stability. J. Pharm. Sci. 2020, 109, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buettner, M.J.; Shah, S.R.; Saeui, C.T.; Ariss, R.; Yarema, K.J. Improving Immunotherapy Through Glycodesign. Front. Immunol. 2018, 9, 2485. [Google Scholar] [CrossRef] [PubMed]
- Schrama, D.; Reisfeld, R.A.; Becker, J.C. Antibody Targeted Drugs as Cancer Therapeutics. Nat. Rev. Drug Discov. 2006, 5, 147–159. [Google Scholar] [CrossRef] [PubMed]
- King, D.M.; Albertini, M.R.; Schalch, H.; Hank, J.A.; Gan, J.; Surfus, J.; Mahvi, D.; Schiller, J.H.; Warner, T.; Kim, K.; et al. Phase I Clinical Trial of the Immunocytokine EMD 273063 in Melanoma Patients. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2004, 22, 4463–4473. [Google Scholar] [CrossRef]
- Teicher, B.A.; Chari, R.V.J. Antibody Conjugate Therapeutics: Challenges and Potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.M.; Senter, P.D. Arming Antibodies: Prospects and Challenges for Immunoconjugates. Nat. Biotechnol. 2005, 23, 1137–1146. [Google Scholar] [CrossRef]
- Milenic, D.E.; Brady, E.D.; Brechbiel, M.W. Antibody-Targeted Radiation Cancer Therapy. Nat. Rev. Drug Discov. 2004, 3, 488–499. [Google Scholar] [CrossRef] [Green Version]
- Leung, D.; Wurst, J.M.; Liu, T.; Martinez, R.M.; Datta-Mannan, A.; Feng, Y. Antibody Conjugates-Recent Advances and Future Innovations. Antibodies 2020, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Yaghoubi, S.; Karimi, M.H.; Lotfinia, M.; Gharibi, T.; Mahi-Birjand, M.; Kavi, E.; Hosseini, F.; Sepehr, K.S.; Khatami, M.; Bagheri, N.; et al. Potential Drugs Used in the Antibody–Drug Conjugate (ADC) Architecture for Cancer Therapy. J. Cell. Physiol. 2020, 235, 31–64. [Google Scholar] [CrossRef]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef]
- Abdollahpour-Alitappeh, M.; Lotfinia, M.; Gharibi, T.; Mardaneh, J.; Farhadihosseinabadi, B.; Larki, P.; Faghfourian, B.; Sepehr, K.S.; Abbaszadeh-Goudarzi, K.; Abbaszadeh-Goudarzi, G.; et al. Antibody–Drug Conjugates (ADCs) for Cancer Therapy: Strategies, Challenges, and Successes. J. Cell. Physiol. 2019, 234, 5628–5642. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.M.; Nesterova, A.; Alley, S.C.; Torgov, M.Y.; Carter, P.J. Potent Cytotoxicity of an Auristatin-Containing Antibody-Drug Conjugate Targeting Melanoma Cells Expressing Melanotransferrin/P97. Mol. Cancer Ther. 2006, 5, 1474–1482. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.-T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel Anti-B-Cell Maturation Antigen Antibody-Drug Conjugate (GSK2857916) Selectively Induces Killing of Multiple Myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Sborov, D.; Suvannasankha, A.; et al. Longer Term Outcomes with Single-Agent Belantamab Mafodotin in Patients with Relapsed or Refractory Multiple Myeloma: 13-Month Follow-up from the Pivotal DREAMM-2 Study. Cancer 2021, 127, 4198–4212. [Google Scholar] [CrossRef]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-Selective Modification Strategies in Antibody–Drug Conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef]
- Burke, P.J.; Hamilton, J.Z.; Jeffrey, S.C.; Hunter, J.H.; Doronina, S.O.; Okeley, N.M.; Miyamoto, J.B.; Anderson, M.E.; Stone, I.J.; Ulrich, M.L.; et al. Optimization of a PEGylated Glucuronide-Monomethylauristatin E Linker for Antibody-Drug Conjugates. Mol. Cancer Ther. 2017, 16, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viricel, W.; Fournet, G.; Beaumel, S.; Perrial, E.; Papot, S.; Dumontet, C.; Joseph, B. Monodisperse Polysarcosine-Based Highly-Loaded Antibody-Drug Conjugates. Chem. Sci. 2019, 10, 4048–4053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-Specific Conjugation of a Cytotoxic Drug to an Antibody Improves the Therapeutic Index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable Linkers in Antibody–Drug Conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Juen, L.; Baltus, C.B.; Gély, C.; Feuillâtre, O.; Desgranges, A.; Viaud-Massuard, M.-C.; Martin, C. Innovative Bioconjugation Technology for Antibody-Drug Conjugates: Proof of Concept in a CD30-Positive Lymphoma Mouse Model. Bioconjugate Chem. 2021, 32, 595–606. [Google Scholar] [CrossRef]
- Van Geel, R.; Wijdeven, M.A.; Heesbeen, R.; Verkade, J.M.M.; Wasiel, A.A.; van Berkel, S.S.; van Delft, F.L. Chemoenzymatic Conjugation of Toxic Payloads to the Globally Conserved N-Glycan of Native MAbs Provides Homogeneous and Highly Efficacious Antibody–Drug Conjugates. Bioconjug. Chem. 2015, 26, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Willemze, R.; Cerroni, L.; Kempf, W.; Berti, E.; Facchetti, F.; Swerdlow, S.H.; Jaffe, E.S. The 2018 Update of the WHO-EORTC Classification for Primary Cutaneous Lymphomas. Blood 2019, 133, 1703–1714. [Google Scholar] [CrossRef] [PubMed]
- Scarisbrick, J.J.; Prince, H.M.; Vermeer, M.H.; Quaglino, P.; Horwitz, S.; Porcu, P.; Stadler, R.; Wood, G.S.; Beylot-Barry, M.; Pham-Ledard, A.; et al. Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sézary Syndrome: Effect of Specific Prognostic Markers on Survival and Development of a Prognostic Model. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3766–3773. [Google Scholar] [CrossRef]
- Welborn, M.; Duvic, M. Antibody-Based Therapies for Cutaneous T-Cell Lymphoma. Am. J. Clin. Dermatol. 2019, 20, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.M.R.; Mehta, A. Diagnostic, Prognostic and Therapeutic Role of CD30 in Lymphoma. Expert Rev. Hematol. 2017, 10, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Mehra, T.; Ikenberg, K.; Moos, R.M.; Benz, R.; Nair, G.; Schanz, U.; Haralambieva, E.; Hoetzenecker, W.; Dummer, R.; French, L.E.; et al. Brentuximab as a Treatment for CD30+ Mycosis Fungoides and Sézary Syndrome. JAMA Dermatol. 2015, 151, 73–77. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Tavallaee, M.; Sundram, U.; Salva, K.A.; Wood, G.S.; Li, S.; Rozati, S.; Nagpal, S.; Krathen, M.; Reddy, S.; et al. Phase II Investigator-Initiated Study of Brentuximab Vedotin in Mycosis Fungoides and Sézary Syndrome with Variable CD30 Expression Level: A Multi-Institution Collaborative Project. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3750–3758. [Google Scholar] [CrossRef]
- Chen, R.; Hou, J.; Newman, E.; Kim, Y.; Donohue, C.; Liu, X.; Thomas, S.H.; Forman, S.J.; Kane, S.E. CD30 Downregulation, MMAE Resistance, and MDR1 Upregulation Are All Associated with Resistance to Brentuximab Vedotin. Mol. Cancer Ther. 2015, 14, 1376–1384. [Google Scholar] [CrossRef] [Green Version]
- Merli, M.; Ferrario, A.; Maffioli, M.; Olivares, C.; Stasia, A.; Arcaini, L.; Passamonti, F. New Uses for Brentuximab Vedotin and Novel Antibody Drug Conjugates in Lymphoma. Expert Rev. Hematol. 2016, 9, 767–780. [Google Scholar] [CrossRef]
- Amatore, F.; Ortonne, N.; Lopez, M.; Barré, M.; Orlanducci, F.; Castellano, R.; Ingen-Housz-Oro, S.; De Croos, A.; Gorvel, L.; Goubard, A.; et al. ICOS Is Widely Expressed in Cutaneous T-Cell Lymphoma and Its Targeting Promotes Potent Killing of Malignant Cells. Blood 2021, 138, 790. [Google Scholar] [CrossRef]
- Choudhary, R.K.; Jones, R.J.; Kuiatse, I.; Wang, H.; Vega, F.; Bouska, A.C.; Lone, W.G.; Iqbal, J.; Orlowski, R.Z. A Novel Antibody Drug Conjugate (ADC) Targeting Cell Surface Heat Shock Protein 70 (CsHSP70) Is Active Against Pre-Clinical Models of Peripheral T-Cell Lymphoma (PTCL). Blood 2021, 138, 870. [Google Scholar] [CrossRef]
- Seth, R.; Messersmith, H.; Kaur, V.; Kirkwood, J.M.; Kudchadkar, R.; McQuade, J.L.; Provenzano, A.; Swami, U.; Weber, J.; Alluri, K.C.; et al. Systemic Therapy for Melanoma: ASCO Guideline. J. Clin. Oncol. 2020, 38, 3947–3970. [Google Scholar] [CrossRef]
- Albittar, A.A.; Alhalabi, O.; Oliva, I.C.G. Immunotherapy for Melanoma. Adv. Exp. Med. Biol. 2020, 1244, 51–68. [Google Scholar] [CrossRef] [PubMed]
- Gide, T.N.; Wilmott, J.S.; Scolyer, R.A.; Long, G.V. Primary and Acquired Resistance to Immune Checkpoint Inhibitors in Metastatic Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1260–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuske, M.; Westphal, D.; Wehner, R.; Schmitz, M.; Beissert, S.; Praetorius, C.; Meier, F. Immunomodulatory Effects of BRAF and MEK Inhibitors: Implications for Melanoma Therapy. Pharmacol. Res. 2018, 136, 151–159. [Google Scholar] [CrossRef]
- Rose, A.A.N.; Biondini, M.; Curiel, R.; Siegel, P.M. Targeting GPNMB with Glembatumumab Vedotin: Current Developments and Future Opportunities for the Treatment of Cancer. Pharmacol. Ther. 2017, 179, 127–141. [Google Scholar] [CrossRef]
- Williams, M.D.; Esmaeli, B.; Soheili, A.; Simantov, R.; Gombos, D.S.; Bedikian, A.Y.; Hwu, P. GPNMB Expression in Uveal Melanoma: A Potential for Targeted Therapy. Melanoma Res. 2010, 20, 184–190. [Google Scholar] [CrossRef]
- Taya, M.; Hammes, S.R. Glycoprotein Non-Metastatic Melanoma Protein B (GPNMB) and Cancer: A Novel Potential Therapeutic Target. Steroids 2018, 133, 102–107. [Google Scholar] [CrossRef]
- Tse, K.F.; Jeffers, M.; Pollack, V.A.; McCabe, D.A.; Shadish, M.L.; Khramtsov, N.V.; Hackett, C.S.; Shenoy, S.G.; Kuang, B.; Boldog, F.L.; et al. CR011, a Fully Human Monoclonal Antibody-Auristatin E Conjugate, for the Treatment of Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Ott, P.A.; Hamid, O.; Pavlick, A.C.; Kluger, H.; Kim, K.B.; Boasberg, P.D.; Simantov, R.; Crowley, E.; Green, J.A.; Hawthorne, T.; et al. Phase I/II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Patients with Advanced Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 3659–3666. [Google Scholar] [CrossRef] [Green Version]
- Rose, A.A.N.; Annis, M.G.; Frederick, D.T.; Biondini, M.; Dong, Z.; Kwong, L.; Chin, L.; Keler, T.; Hawthorne, T.; Watson, I.R.; et al. MAPK Pathway Inhibitors Sensitize BRAF-Mutant Melanoma to an Antibody-Drug Conjugate Targeting GPNMB. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 6088–6098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocana, A.; Vera-Badillo, F.; Seruga, B.; Templeton, A.; Pandiella, A.; Amir, E. HER3 Overexpression and Survival in Solid Tumors: A Meta-Analysis. J. Natl. Cancer Inst. 2013, 105, 266–273. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Liu, S.; Lyu, H.; Riker, A.I.; Zhang, Y.; Liu, B. Development of Effective Therapeutics Targeting HER3 for Cancer Treatment. Biol. Proced. Online 2019, 21, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abel, E.V.; Basile, K.J.; Kugel, C.H.; Witkiewicz, A.K.; Le, K.; Amaravadi, R.K.; Karakousis, G.C.; Xu, X.; Xu, W.; Schuchter, L.M.; et al. Melanoma Adapts to RAF/MEK Inhibitors through FOXD3-Mediated Upregulation of ERBB3. J. Clin. Investig. 2013, 123, 2155–2168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Chang, Y.; Rios, A.; An, Z. HER3/ErbB3, an Emerging Cancer Therapeutic Target. Acta Biochim. Biophys. Sin. 2016, 48, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Capone, E.; Lamolinara, A.; D’Agostino, D.; Rossi, C.; De Laurenzi, V.; Iezzi, M.; Iacobelli, S.; Sala, G. EV20-Mediated Delivery of Cytotoxic Auristatin MMAF Exhibits Potent Therapeutic Efficacy in Cutaneous Melanoma. J. Control. Release Off. J. Control. Release Soc. 2018, 277, 48–56. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Koyama, K.; Kamai, Y.; Hirotani, K.; Ogitani, Y.; Zembutsu, A.; Abe, M.; Kaneda, Y.; Maeda, N.; Shiose, Y.; et al. A Novel HER3-Targeting Antibody–Drug Conjugate, U3-1402, Exhibits Potent Therapeutic Efficacy through the Delivery of Cytotoxic Payload by Efficient Internalization. Clin. Cancer Res. 2019, 25, 7151–7161. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chalouni, C.; Tan, C.; Clark, R.; Venook, R.; Ohri, R.; Raab, H.; Firestein, R.; Mallet, W.; Polakis, P. The Melanosomal Protein PMEL17 as a Target for Antibody Drug Conjugate Therapy in Melanoma. J. Biol. Chem. 2012, 287, 24082–24091. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.; Bagnato, A.; Battistini, B.; Nisen, P. The Endothelin Axis: Emerging Role in Cancer. Nat. Rev. Cancer 2003, 3, 110–116. [Google Scholar] [CrossRef]
- Saldana-Caboverde, A.; Kos, L. Roles of Endothelin Signaling in Melanocyte Development and Melanoma. Pigment. Cell Melanoma Res. 2010, 23, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Bagnato, A.; Rosanò, L.; Spinella, F.; Di Castro, V.; Tecce, R.; Natali, P.G. Endothelin B Receptor Blockade Inhibits Dynamics of Cell Interactions and Communications in Melanoma Cell Progression. Cancer Res. 2004, 64, 1436–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asundi, J.; Reed, C.; Arca, J.; McCutcheon, K.; Ferrando, R.; Clark, S.; Luis, E.; Tien, J.; Firestein, R.; Polakis, P. An Antibody-Drug Conjugate Targeting the Endothelin B Receptor for the Treatment of Melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 965–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, S.; McNeil, C.M.; LoRusso, P.; Patel, M.R.; Kabbarah, O.; Li, C.; Sanabria, S.; Flanagan, W.M.; Yeh, R.-F.; Brunstein, F.; et al. Phase I Study of the Anti-Endothelin B Receptor Antibody-Drug Conjugate DEDN6526A in Patients with Metastatic or Unresectable Cutaneous, Mucosal, or Uveal Melanoma. Investig. New Drugs 2019, 38, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Asundi, J.; Lacap, J.A.; Clark, S.; Nannini, M.; Roth, L.; Polakis, P. MAPK Pathway Inhibition Enhances the Efficacy of an Anti-Endothelin B Receptor Drug Conjugate by Inducing Target Expression in Melanoma. Mol. Cancer Ther. 2014, 13, 1599–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An Epithelial-Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Boshuizen, J.; Koopman, L.A.; Krijgsman, O.; Shahrabi, A.; van den Heuvel, E.G.-; Ligtenberg, M.A.; Vredevoogd, D.W.; Kemper, K.; Kuilman, T.; Song, J.-Y.; et al. Cooperative Targeting of Melanoma Heterogeneity with an AXL Antibody-Drug Conjugate and BRAF/MEK Inhibitors. Nat. Med. 2018, 24, 203–212. [Google Scholar] [CrossRef]
- Cichoń, M.A.; Szentpetery, Z.; Caley, M.P.; Papadakis, E.S.; Mackenzie, I.C.; Brennan, C.H.; O’Toole, E.A. The Receptor Tyrosine Kinase Axl Regulates Cell-Cell Adhesion and Stemness in Cutaneous Squamous Cell Carcinoma. Oncogene 2014, 33, 4185–4192. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, M.; Lasota, J. KIT (CD117): A Review on Expression in Normal and Neoplastic Tissues, and Mutations and Their Clinicopathologic Correlation. Appl. Immunohistochem. Mol. Morphol. AIMM 2005, 13, 205–220. [Google Scholar] [CrossRef]
- Gramza, A.W.; Corless, C.L.; Heinrich, M.C. Resistance to Tyrosine Kinase Inhibitors in Gastrointestinal Stromal Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 7510–7518. [Google Scholar] [CrossRef] [Green Version]
- Abrams, T.; Connor, A.; Fanton, C.; Cohen, S.B.; Huber, T.; Miller, K.; Hong, E.E.; Niu, X.; Kline, J.; Ison-Dugenny, M.; et al. Preclinical Antitumor Activity of a Novel Anti-c-KIT Antibody-Drug Conjugate against Mutant and Wild-Type c-KIT-Positive Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4297–4308. [Google Scholar] [CrossRef] [Green Version]
- Giansanti, F.; Capone, E.; Ponziani, S.; Piccolo, E.; Gentile, R.; Lamolinara, A.; Di Campli, A.; Sallese, M.; Iacobelli, V.; Cimini, A.; et al. Secreted Gal-3BP Is a Novel Promising Target for Non-Internalizing Antibody–Drug Conjugates. J. Control. Release 2019, 294, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Cesinaro, A.M.; Trentini, G.P.; Natoli, C.; Grassadonia, A.; Tinari, N.; Iacobelli, S. Expression of the 90K Tumor-Associated Protein in Benign and Malignant Melanocytic Lesions. J. Investig. Dermatol. 2002, 119, 187–190. [Google Scholar] [CrossRef] [Green Version]
- Motaparthi, K.; Kapil, J.P.; Velazquez, E.F. Cutaneous Squamous Cell Carcinoma: Review of the Eighth Edition of the American Joint Committee on Cancer Staging Guidelines, Prognostic Factors, and Histopathologic Variants. Adv. Anat. Pathol. 2017, 24, 171–194. [Google Scholar] [CrossRef] [PubMed]
- Markham, A.; Duggan, S. Cemiplimab: First Global Approval. Drugs 2018, 78, 1841–1846. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.R.; Petersen, E.; Patel, R.; Migden, M.R. Cemiplimab-Rwlc as First and Only Treatment for Advanced Cutaneous Squamous Cell Carcinoma. Expert Rev. Clin. Pharmacol. 2019, 12, 947–951. [Google Scholar] [CrossRef]
- Stratigos, A.J.; Garbe, C.; Dessinioti, C.; Lebbe, C.; Bataille, V.; Bastholt, L.; Dreno, B.; Fargnoli, M.C.; Forsea, A.M.; Frenard, C.; et al. European Interdisciplinary Guideline on Invasive Squamous Cell Carcinoma of the Skin: Part 2. Treatment. Eur. J. Cancer 2020, 128, 83–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breij, E.C.W.; de Goeij, B.E.C.G.; Verploegen, S.; Schuurhuis, D.H.; Amirkhosravi, A.; Francis, J.; Miller, V.B.; Houtkamp, M.; Bleeker, W.K.; Satijn, D.; et al. An Antibody-Drug Conjugate That Targets Tissue Factor Exhibits Potent Therapeutic Activity against a Broad Range of Solid Tumors. Cancer Res. 2014, 74, 1214–1226. [Google Scholar] [CrossRef] [Green Version]
- Kasthuri, R.S.; Taubman, M.B.; Mackman, N. Role of Tissue Factor in Cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 4834–4838. [Google Scholar] [CrossRef] [Green Version]
- Van den Berg, Y.W.; Osanto, S.; Reitsma, P.H.; Versteeg, H.H. The Relationship between Tissue Factor and Cancer Progression: Insights from Bench and Bedside. Blood 2012, 119, 924–932. [Google Scholar] [CrossRef] [Green Version]
- De Bono, J.S.; Concin, N.; Hong, D.S.; Thistlethwaite, F.C.; Machiels, J.-P.; Arkenau, H.-T.; Plummer, R.; Jones, R.H.; Nielsen, D.; Windfeld, K.; et al. Tisotumab Vedotin in Patients with Advanced or Metastatic Solid Tumours (InnovaTV 201): A First-in-Human, Multicentre, Phase 1-2 Trial. Lancet Oncol. 2019, 20, 383–393. [Google Scholar] [CrossRef]
- Purcell, J.W.; Tanlimco, S.G.; Hickson, J.; Fox, M.; Sho, M.; Durkin, L.; Uziel, T.; Powers, R.; Foster, K.; McGonigal, T.; et al. LRRC15 Is a Novel Mesenchymal Protein and Stromal Target for Antibody-Drug Conjugates. Cancer Res. 2018, 78, 4059–4072. [Google Scholar] [CrossRef] [Green Version]
- Bierie, B.; Moses, H.L. Tumour Microenvironment: TGFbeta: The Molecular Jekyll and Hyde of Cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The Roles of TGFβ in the Tumour Microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Zeisberg, M. Fibroblasts in Cancer. Nat. Rev. Cancer 2006, 6, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Riechelmann, H.; Sauter, A.; Golze, W.; Hanft, G.; Schroen, C.; Hoermann, K.; Erhardt, T.; Gronau, S. Phase I Trial with the CD44v6-Targeting Immunoconjugate Bivatuzumab Mertansine in Head and Neck Squamous Cell Carcinoma. Oral Oncol. 2008, 44, 823–829. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Becker, J.C.; Stang, A.; DeCaprio, J.A.; Cerroni, L.; Lebbé, C.; Veness, M.; Nghiem, P. Merkel Cell Carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17077. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Bhatia, S.; Brohl, A.S.; Hamid, O.; Mehnert, J.M.; Terheyden, P.; Shih, K.C.; Brownell, I.; Lebbé, C.; Lewis, K.D.; et al. Avelumab in Patients with Previously Treated Metastatic Merkel Cell Carcinoma: Long-Term Data and Biomarker Analyses from the Single-Arm Phase 2 JAVELIN Merkel 200 Trial. J. Immunother. Cancer 2020, 8, e000674. [Google Scholar] [CrossRef]
- Nghiem, P.T.; Bhatia, S.; Lipson, E.J.; Kudchadkar, R.R.; Miller, N.J.; Annamalai, L.; Berry, S.; Chartash, E.K.; Daud, A.; Fling, S.P.; et al. PD-1 Blockade with Pembrolizumab in Advanced Merkel-Cell Carcinoma. N. Engl. J. Med. 2016, 374, 2542–2552. [Google Scholar] [CrossRef]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Durable Tumor Regression and Overall Survival in Patients with Advanced Merkel Cell Carcinoma Receiving Pembrolizumab as First-Line Therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 693–702. [Google Scholar] [CrossRef]
- Ollier, J.; Kervarrec, T.; Samimi, M.; Benlalam, H.; Aumont, P.; Vivien, R.; Touzé, A.; Labarrière, N.; Vié, H.; Clémenceau, B. Merkel Cell Carcinoma and Cellular Cytotoxicity: Sensitivity to Cellular Lysis and Screening for Potential Target Antigens Suitable for Antibody-Dependent Cellular Cytotoxicity. Cancer Immunol. Immunother. 2018, 67, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.H.; Lorigan, P.; O’Brien, M.E.R.; Fossella, F.V.; Moore, K.N.; Bhatia, S.; Kirby, M.; Woll, P.J. Phase I Study of IMGN901, a CD56-Targeting Antibody-Drug Conjugate, in Patients with CD56-Positive Solid Tumors. Investig. New Drugs 2016, 34, 290–299. [Google Scholar] [CrossRef]
- Ailawadhi, S.; Kelly, K.R.; Vescio, R.A.; Jagannath, S.; Wolf, J.; Gharibo, M.; Sher, T.; Bojanini, L.; Kirby, M.; Chanan-Khan, A. A Phase I Study to Assess the Safety and Pharmacokinetics of Single-Agent Lorvotuzumab Mertansine (IMGN901) in Patients with Relapsed and/or Refractory CD-56-Positive Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Kaye, F.J.; Spigel, D.R.; Kudrik, F.J.; Ponce, S.; Ellis, P.M.; Majem, M.; Lorigan, P.; Gandhi, L.; Gutierrez, M.E.; et al. Phase 1/2 Study of the CD56-Targeting Antibody-Drug Conjugate Lorvotuzumab Mertansine (IMGN901) in Combination With Carboplatin/Etoposide in Small-Cell Lung Cancer Patients With Extensive-Stage Disease. Clin. Lung Cancer 2017, 18, 68–76.e2. [Google Scholar] [CrossRef]
- Esnault, C.; Leblond, V.; Martin, C.; Desgranges, A.; Baltus, C.B.; Aubrey, N.; Lakhrif, Z.; Lajoie, L.; Lantier, L.; Clémenceau, B.; et al. Adcitmer®, a New CD56-Targeting Monomethyl Auristatin E-Conjugated Antibody, Is a Potential Therapeutic Approach in Merkel Cell Carcinoma. Br. J. Dermatol. 2021. [Google Scholar] [CrossRef]
- Lashari, B.H.; Vallatharasu, Y.; Kolandra, L.; Hamid, M.; Uprety, D. Rovalpituzumab Tesirine: A Novel DLL3-Targeting Antibody-Drug Conjugate. Drugs RD 2018, 18, 255–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rand, J.; Balzer, B.L.; Frishberg, D.P.; Essner, R.; Shon, W. Prevalence of Delta-Like Protein 3 Expression in Merkel Cell Carcinoma. J. Am. Acad. Dermatol. 2019, 85, 749–750. [Google Scholar] [CrossRef]
- Xie, H.; Kaye, F.J.; Isse, K.; Sun, Y.; Ramoth, J.; French, D.M.; Flotte, T.J.; Luo, Y.; Saunders, L.R.; Mansfield, A.S. Delta-like Protein 3 Expression and Targeting in Merkel Cell Carcinoma. Oncol. 2020, 25, 810–817. [Google Scholar] [CrossRef]
- Morgensztern, D.; Besse, B.; Greillier, L.; Santana-Davila, R.; Ready, N.; Hann, C.L.; Glisson, B.S.; Farago, A.F.; Dowlati, A.; Rudin, C.M.; et al. Efficacy and Safety of Rovalpituzumab Tesirine in Third-Line and Beyond Patients with DLL3-Expressing, Relapsed/Refractory Small-Cell Lung Cancer: Results From the Phase II TRINITY Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6958–6966. [Google Scholar] [CrossRef] [Green Version]
- Szijj, P.A.; Bahou, C.; Chudasama, V. Minireview: Addressing the Retro-Michael Instability of Maleimide Bioconjugates. Drug Discov. Today Technol. 2018, 30, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Parslow, A.C.; Parakh, S.; Lee, F.-T.; Gan, H.K.; Scott, A.M. Antibody-Drug Conjugates for Cancer Therapy. Biomedicines 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Patel, S.; Goyal, K.; Morgan, E.A.; Foreman, R.K. Variable Loss of CD30 Expression by Immunohistochemistry in Recurrent Cutaneous CD30+ Lymphoid Neoplasms Treated with Brentuximab Vedotin. J. Cutan. Pathol. 2019, 46, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Desnoyers, L.R.; Vasiljeva, O.; Richardson, J.H.; Yang, A.; Menendez, E.E.M.; Liang, T.W.; Wong, C.; Bessette, P.H.; Kamath, K.; Moore, S.J.; et al. Tumor-Specific Activation of an EGFR-Targeting Probody Enhances Therapeutic Index. Sci. Transl. Med. 2013, 5, 207ra144. [Google Scholar] [CrossRef] [PubMed]
- Autio, K.A.; Boni, V.; Humphrey, R.W.; Naing, A. Probody Therapeutics: An Emerging Class of Therapies Designed to Enhance On-Target Effects with Reduced Off-Tumor Toxicity for Use in Immuno-Oncology. Clin. Cancer Res. 2020, 26, 984–989. [Google Scholar] [CrossRef] [Green Version]
- Chomet, M.; Schreurs, M.; Nguyen, M.; Howng, B.; Villanueva, R.; Krimm, M.; Vasiljeva, O.; van Dongen, G.A.M.S.; Vugts, D.J. The Tumor Targeting Performance of Anti-CD166 Probody Drug Conjugate CX-2009 and Its Parental Derivatives as Monitored by 89Zr-Immuno-PET in Xenograft Bearing Mice. Theranostics 2020, 10, 5815–5828. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, H. Effects of Antibody, Drug and Linker on the Preclinical and Clinical Toxicities of Antibody-Drug Conjugates. mAbs 2016, 8, 659–671. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Hematological Malignancies | |||||||
|---|---|---|---|---|---|---|---|
| Commercial Name | International Non-Proprietary Names (INN) | Target | Antibody Isotype | Bioconjugation Head (Antibody Amino Acid) | Linker | Drug (Therapeutic Class) | Indication |
| Adcetris® | brentuximab vedotin | CD30 | Chimeric IgG1 | Maleimidocaproyl (Cysteine) | Cleavable/proteolytic (cathepsin B) | auristatin | anaplastic large cell lymphoma + Hodgkin’s lymphoma |
| Polivy® | polatuzumab vedotin-piiq | CD79b | Humanized IgG1 | Maleimidocaproyl (Cysteine) | Cleavable/proteolytic (cathepsin B) | auristatin | relapsed or refractory diffuse large B-cell lymphoma |
| Mylotarg® | gemtuzumab ozogamicin | CD33 | Humanized IgG4 | Acetyl butyrate (Lysine) | Cleavable/hydrazone | calicheamicin | CD33-positive acute myeloid leukemia |
| Beponsa® | inotuzumab ozogamicin | CD22 | Humanized IgG4 | Acetyl butyrate (Lysine) | Cleavable/hydrazone | calicheamicin | lymphoblastic leukemia |
| Lumoxiti® | moxetumomab pasudotox | CD22 | N/A | N/A | Cleavable/proteolytic (furin) | Pseudomonas endotoxin A | hairy cell leukemia |
| Zynlonta® | loncastuximab tesirine-lpyl | CD19 | Humanized IgG1 | Maleimidocaproyl (Cysteine) | Cleavable/proteolytic (cathepsin B) | pyrrolobenzodiazepine dimer (PBD) | relapsed/refractory diffuse large B-cell lymphoma |
| Blenrep® | belantamab mafodotin | CD38 | Humanized IgG1k | Maleimidocaproyl (Cysteine) | Uncleavable | auristatin | relapsed/refractory multiple myeloma |
| Solid Tumors | |||||||
| Commercial Name | International Non-Proprietary Names (INN) | Target | Antibody Isotype | Bioconjugation Head (Antibody Amino Acid) | Linker | Drug | Indication |
| Kadcyla® | ado-trastuzumab emtansine | HER2 | Humanized IgG1 | Maleimidocaproyl (Lysine) | Uncleavable | maytansine | HER2/neu positive breast cancer |
| Padcev® | enfortumab vedotin | Nectin-4 | Human IgG1 | Maleimidocaproyl (Cysteine) | Cleavable/proteolytic (cathepsin B) | auristatin | locally advanced or metastatic urothelial cancer |
| Enhertu® | trastuzumab deruxtecan | HER2 | Humanized IgG1 | Maleimidocaproyl (Cysteine) | Cleavable/proteolytic (cathepsin B) | deruxtecan (topoisomerase inhibitor) | breast cancer HER2 positive after two or more lines of anti-HER2 therapy |
| Trodelvy® | sacituzumab govitecan | TROP-2 | Humanized IgG1 | Maleimidocaproyl (Cysteine) | Cleavable/hydrazone | topoisomerase inhibitor | metastatic triple-negative breast cancer |
| Tivdak® | tisotumab vedotin | Tissue factor | Human IgG1 | Maleimidocaproyl (Cysteine) | Cleavable/proteolytic (cathepsin B) | auristatin | cervical cancer |
| Commercial Name | Target | Percentage of Positivity | Expression in Healthy Tissues | Antibody | Antibody Isotype | Bioconjugation Head (Antibody Amino Acid) | Linker | Drug | Phase |
|---|---|---|---|---|---|---|---|---|---|
| Brentuximab vedotin | CD30 | 75% | Activated T, B and NK cells | Brentuximab | Chimeric IgG1 | Maleimidocaproyl (Cysteine) | Cleavable/proteolytic (cathepsin B) | MMAE | Approved |
| F0002ADC | CD30 | 75% | Activated T, B and NK cells | Brentuximab | Chimeric IgG1 | Ester NHS (Lysine) | Uncleavable | DM1 | Phase I |
| N/A | Inducible co-stimulator | MF: 61% (n = 23), SS: 88% (n = 17) | Lymph node, kidney, liver | Mogamulizumab | Murine monoclonal antibody | N/A | N/A | MMAE | Preclinical |
| N/A | Cell surface heat shock protein 70 | N/A | N/A | 239-87 | N/A | N/A | N/A | MMAE | Preclinical |
| Study | Phase | Patient Number | Response Rate | Survival |
|---|---|---|---|---|
| Duvic et al. | II | 48 (28 with MF, 9 with LyP, 2 with pcALCL) | ORR 73% ORR 54% in MF group ORR 100% in other subgroups CR 2/28, PR 13/28 in MF subgroup | PFS 1.1 years (95% CI 0.9−1.4) |
| Kim et al. | II | 32 (MF and SS) | ORR in 21/30 (70%, 90% CI 53−83) CR in 1/30 PR in 20/30SD in 4/30 | Median PFS not reached at 12 months Median EFS > 6 months 61% event free at 6 months 28% event free at 12 months |
| Prince et al. | III Comparison of BV with physician’s choice of either bexarotene or methotrexate | 123 (97 with MF, 31 with pcALCL) with 64 in BV group, 64 in PC group | ORR 56.3% (BV group) versus 12.5% (PC group), with p < 0.0001 ORR 67% in BV group CR 16% ORR 20% in PC group CR 2% | Median PFS 16.7 months (BV group) versus 3.5 months (PC group), with p < 0.0001 |
| Commercial Name | Target | Percentage of Positivity | Expression in Healthy Tissues | Antibody | Antibody Isotype | Bioconjugation Head (Antibody Amino Acid) | Linker | Drug | Phase |
|---|---|---|---|---|---|---|---|---|---|
| Glembatumumab vedotin | gpNMB | 86% (n = 21) | Skin, bones | Glembatumumab | Human IgG2 | Maleimidocaproyl (Cysteine) | Cleavable/proteolytic | MMAE | Phase I/II |
| N/A | PMEL17 | 64% (n = 58) | Melanocytes | 17A9 | Mouse N/A | N/A | Cleavable/proteolytic | MMAE | Preclinical |
| EV20/MMAF ADC | HER3 | 65% (n = 130) | Liver, pancreas, epithelial cells | EV20 | Humanized IgG1 | Maleimidocaproyl (Cysteine) | Uncleavable | MMAF | Preclinical |
| DEDN6526A | ETBR | Majority of tumors (% N/A) | Liver, cortex, medulla | MEDN6000A | Humanized IgG1 | Maleimidocaproyl (Cysteine) | Cleavable/proteolytic | MMAE | Phase I |
| LOP628 | c-KIT | 66% to 88% | Skin epithelial cells, breast, neurons | LMJ | Humanized IgG1 | Ester NHS (Lysine) | Uncleavable | DM1 | Phase I (stopped) |
| Enapotamab vedotin | AXL | N/A | Muscle, testis | Enapotamab | Human IgG1 | Maleimidocaproyl (Cysteine) | Cleavable/proteolytic | MMAE | Phase I/II |
| Squamous Cell Carcinoma | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Commercial Name | Target | Percentage of Positivity | Expression in Healthy Tissues | Antibody | Antibody Isotype | Bioconjugation Head (Antibody Amino Acid) | Linker | Drug | Phase |
| Tisotumab vedotin | TF | 75% (n = 20) | Brain, heart, intestine, kidney, lung, placenta, uterus | Tisotumab | Human IgG1 | Maleimidocaproyl (Cysteine) | Cleavable/proteolytic | MMAE | Phase I/II |
| Bivatuzumab mertansine | CD44v6 | 100% (n = 5) | Skin keratinocytes, cervix, cornea, tonsil | Bivatuzumab (or BIWA 4) | Humanized IgG1 | Disulfide linker SPP (Lysine) | Cleavable/hydrazone | DM1 | Phase I (stopped) |
| Samrotamab vedotin (ABBV-085) | LRRC15 | 64% (n = 115) | Hair follicles, tonsil, stomach, spleen, osteoblasts | Ab1 | Humanized IgG1 | Maleimidocaproyl (Cysteine) | Cleavable/proteolytic | MMAE | Phase I |
| Merkel Cell Carcinoma | |||||||||
| Commercial Name | Target | Percentage of Positivity | Expression in Healthy Tissues | Antibody | Antibody Isotype | Bioconjugation Head (Antibody Amino Acid) | Linker | Drug | Phase |
| Lorvotuzumab mertansine (IMGN901) | CD56 | 88% (n = 64) | NK cells, neuroendocrine cells, neurons | Lorvotuzumab (huN901) | Humanized IgG1 | Disulfide linker SPP (Lysine) | Cleavable/hydrazone | DM1 | Phase I |
| Adcitmer® | CD56 | 66% (n = 90) | NK cells, neuroendocrine cells, neurons | m906 | Human IgG1 | Maleimidocaproyl (Cysteine) | Cleavable proteolytic (cathepsin B) | MMAE | Preclincal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esnault, C.; Schrama, D.; Houben, R.; Guyétant, S.; Desgranges, A.; Martin, C.; Berthon, P.; Viaud-Massuard, M.-C.; Touzé, A.; Kervarrec, T.; et al. Antibody–Drug Conjugates as an Emerging Therapy in Oncodermatology. Cancers 2022, 14, 778. https://doi.org/10.3390/cancers14030778
Esnault C, Schrama D, Houben R, Guyétant S, Desgranges A, Martin C, Berthon P, Viaud-Massuard M-C, Touzé A, Kervarrec T, et al. Antibody–Drug Conjugates as an Emerging Therapy in Oncodermatology. Cancers. 2022; 14(3):778. https://doi.org/10.3390/cancers14030778
Chicago/Turabian StyleEsnault, Clara, David Schrama, Roland Houben, Serge Guyétant, Audrey Desgranges, Camille Martin, Patricia Berthon, Marie-Claude Viaud-Massuard, Antoine Touzé, Thibault Kervarrec, and et al. 2022. "Antibody–Drug Conjugates as an Emerging Therapy in Oncodermatology" Cancers 14, no. 3: 778. https://doi.org/10.3390/cancers14030778
APA StyleEsnault, C., Schrama, D., Houben, R., Guyétant, S., Desgranges, A., Martin, C., Berthon, P., Viaud-Massuard, M.-C., Touzé, A., Kervarrec, T., & Samimi, M. (2022). Antibody–Drug Conjugates as an Emerging Therapy in Oncodermatology. Cancers, 14(3), 778. https://doi.org/10.3390/cancers14030778