
Genetic Alterations Predict Long-Term Survival in Ductal Adenocarcinoma of the Pancreatic Head

 , , , ,
, , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
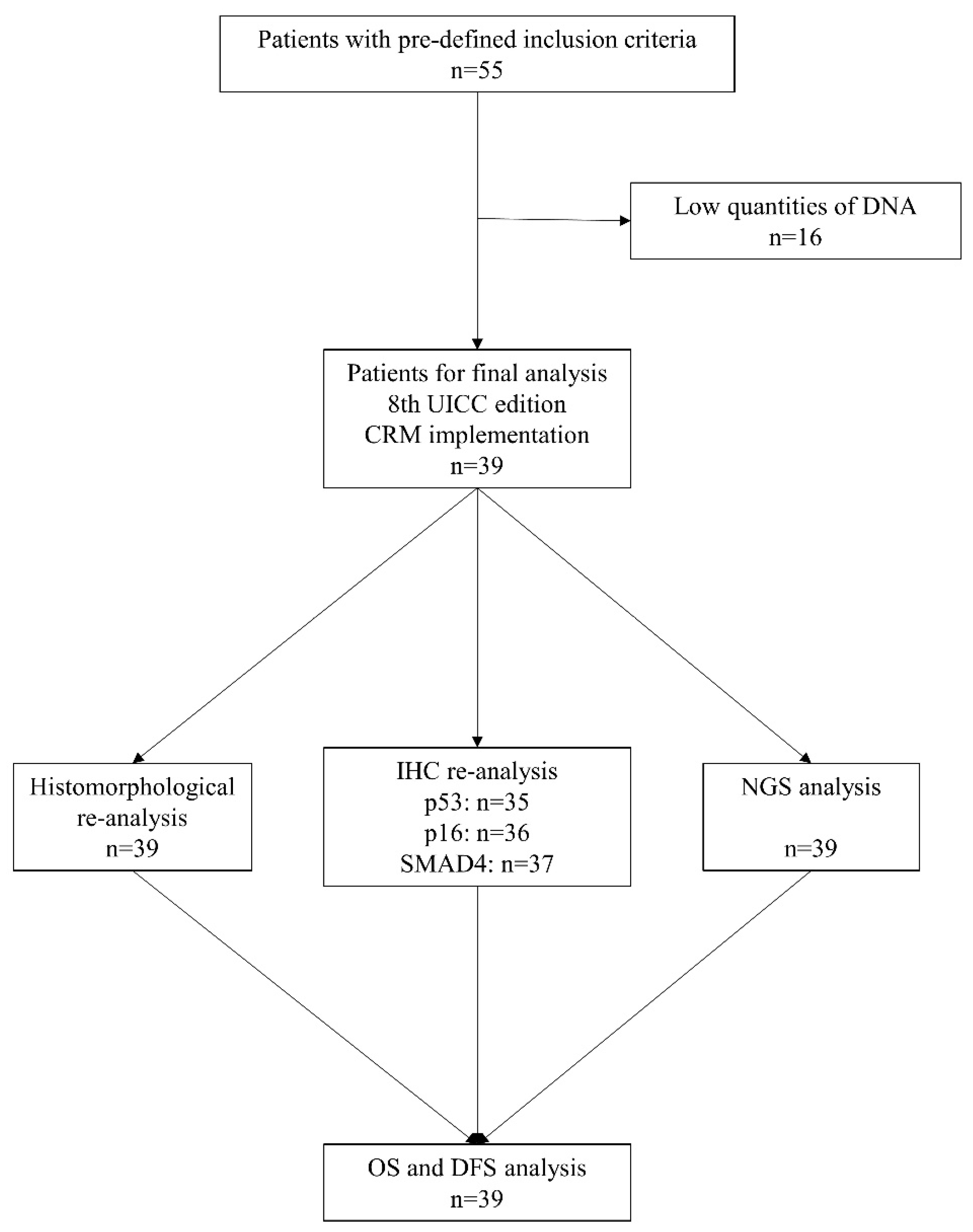
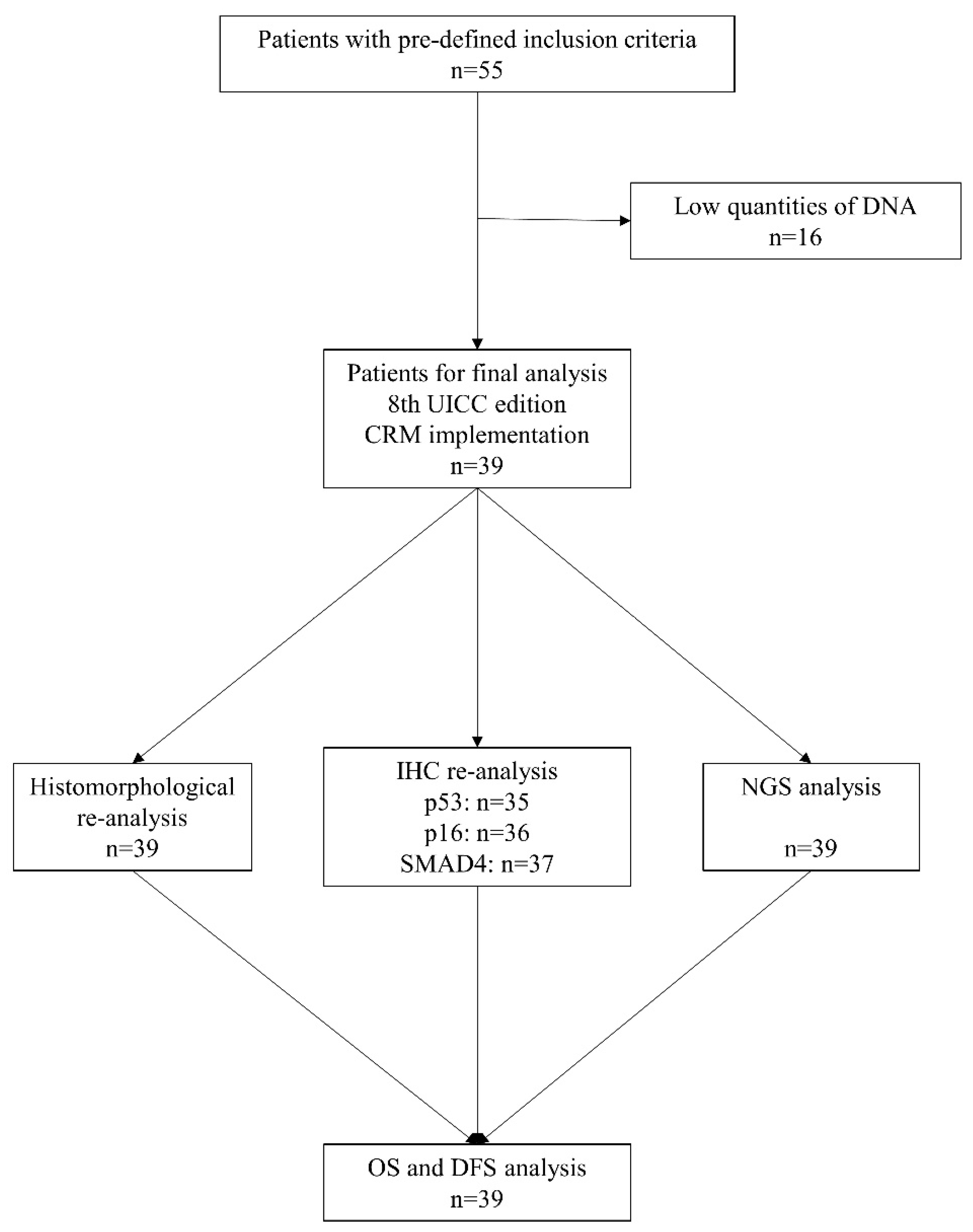
2.1. Patient Selection and Clinicopathological Data
2.2. Histopathological Slide Preparation and DNA Isolation
2.3. DNA Sequencing
2.4. Library Preparation and Massive Parallel Sequencing
2.5. Immunohistochemistry
2.6. Statistical Analysis
3. Results
3.1. Histopathological and Genetic Analysis
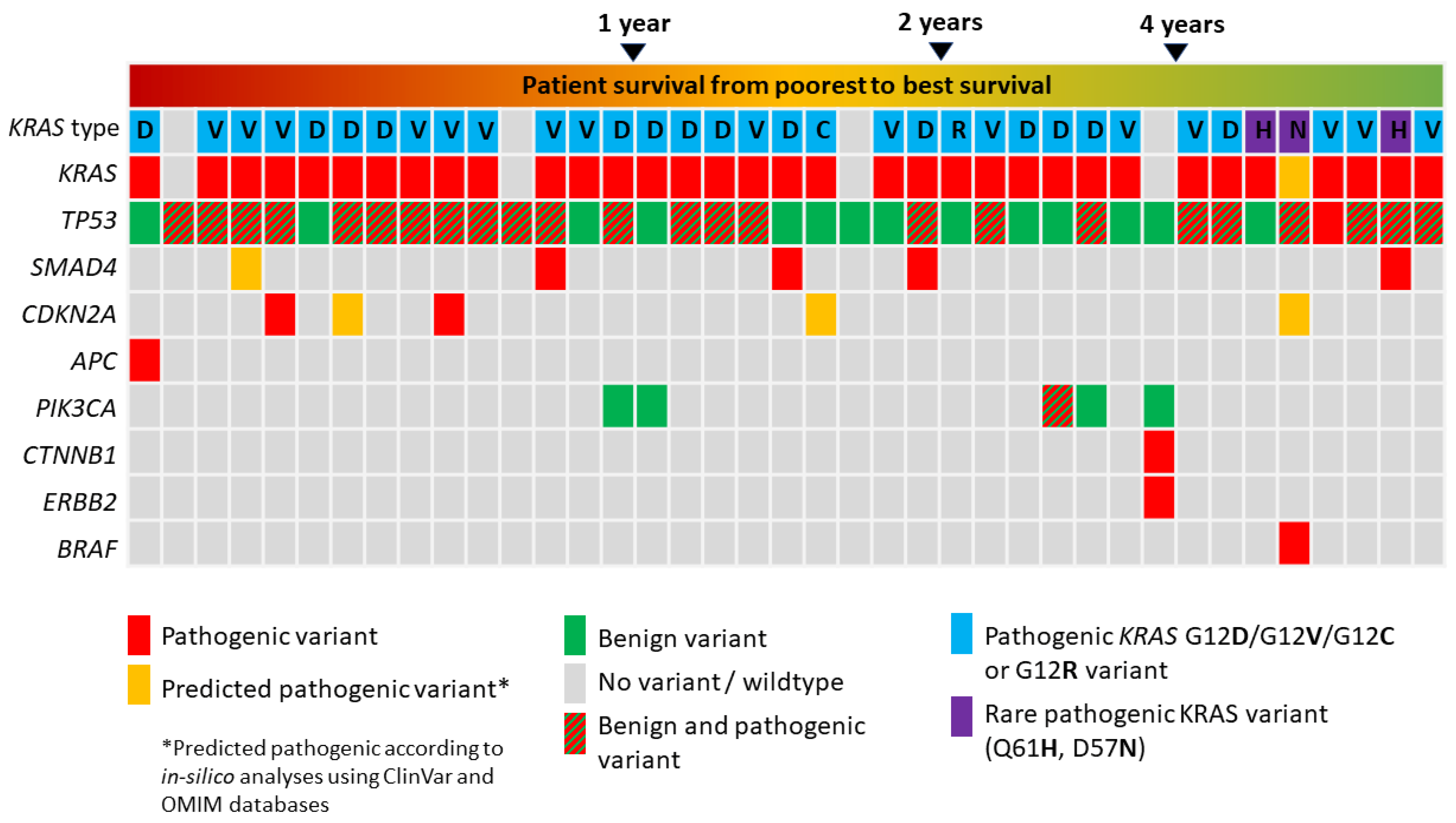
3.2. Panel Next Generation Sequencing
3.3. Allele Frequency Rates of Pathogenic KRAS and TP53 Variants
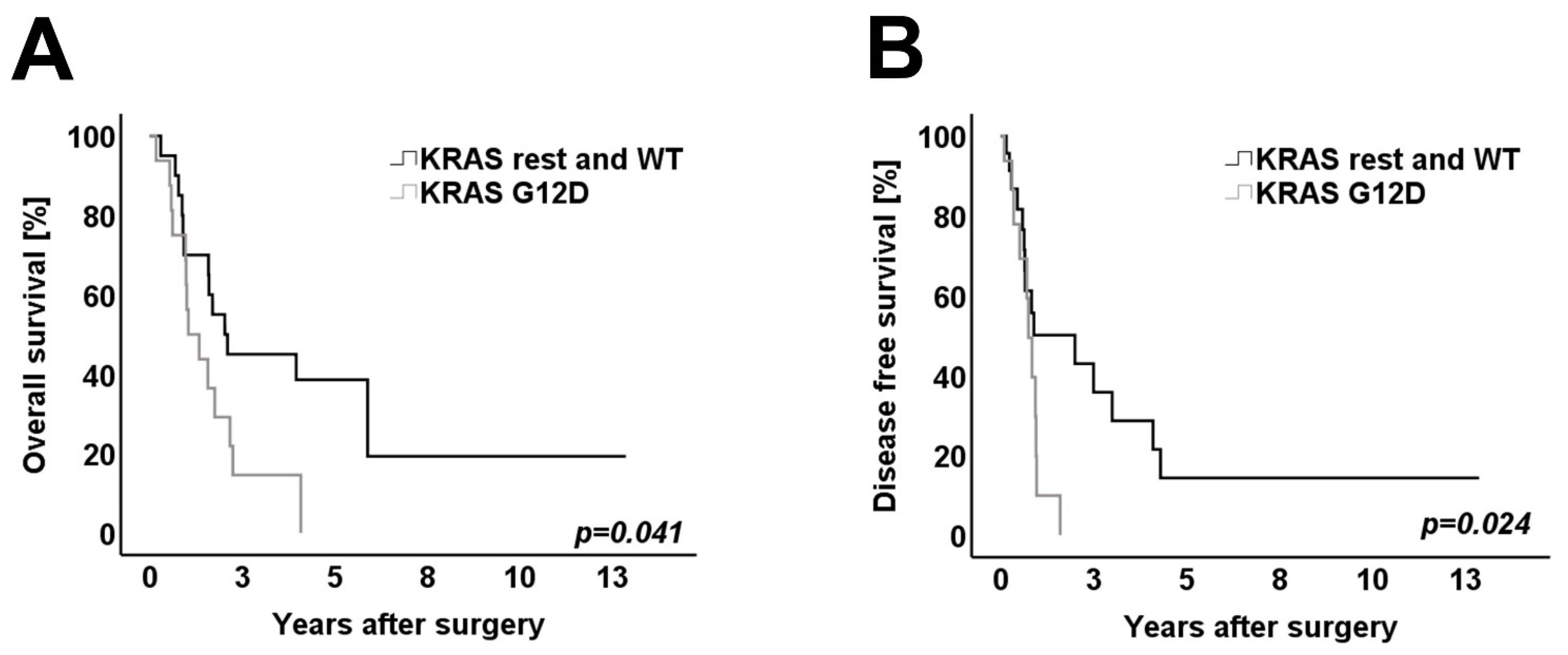
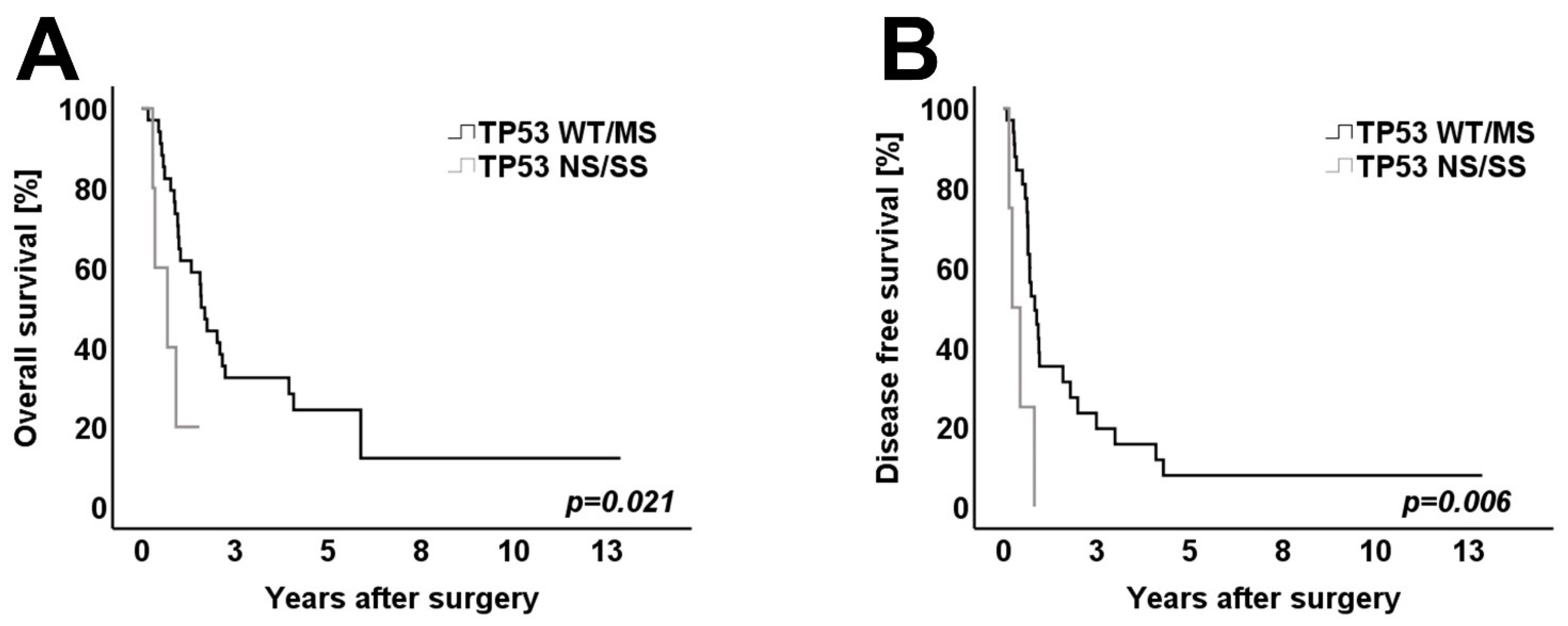
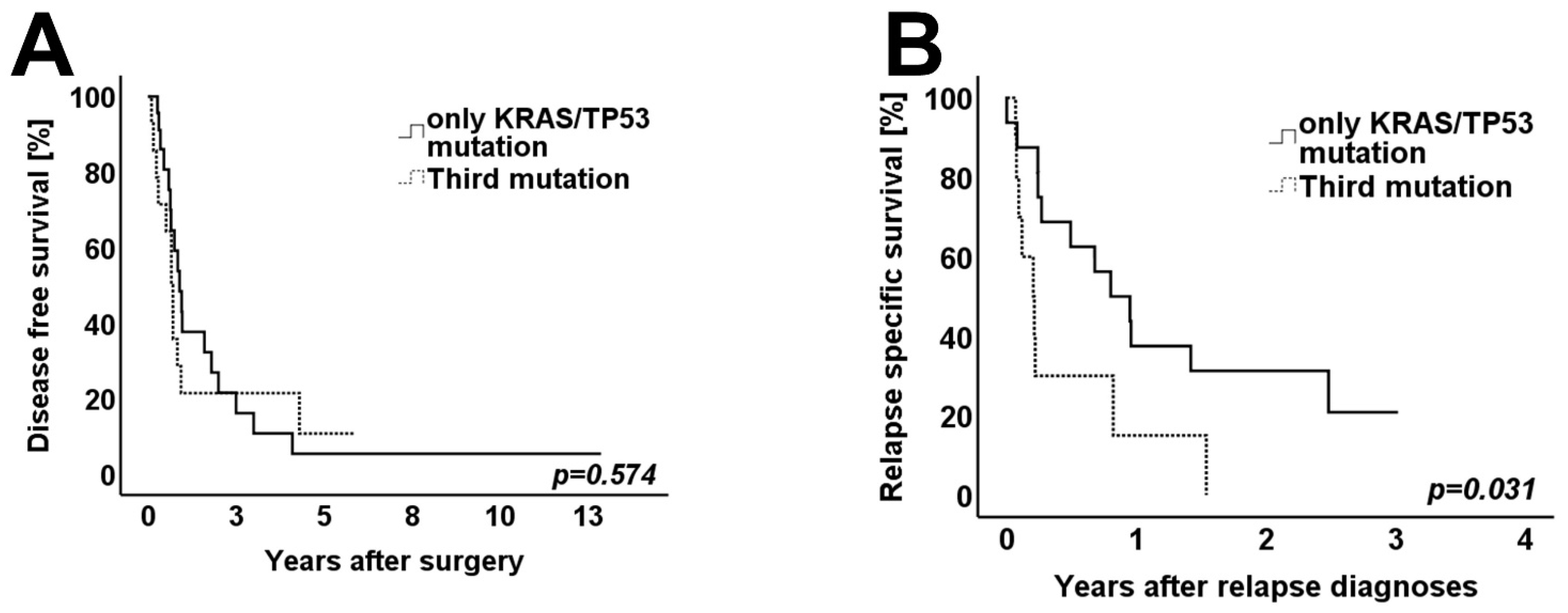
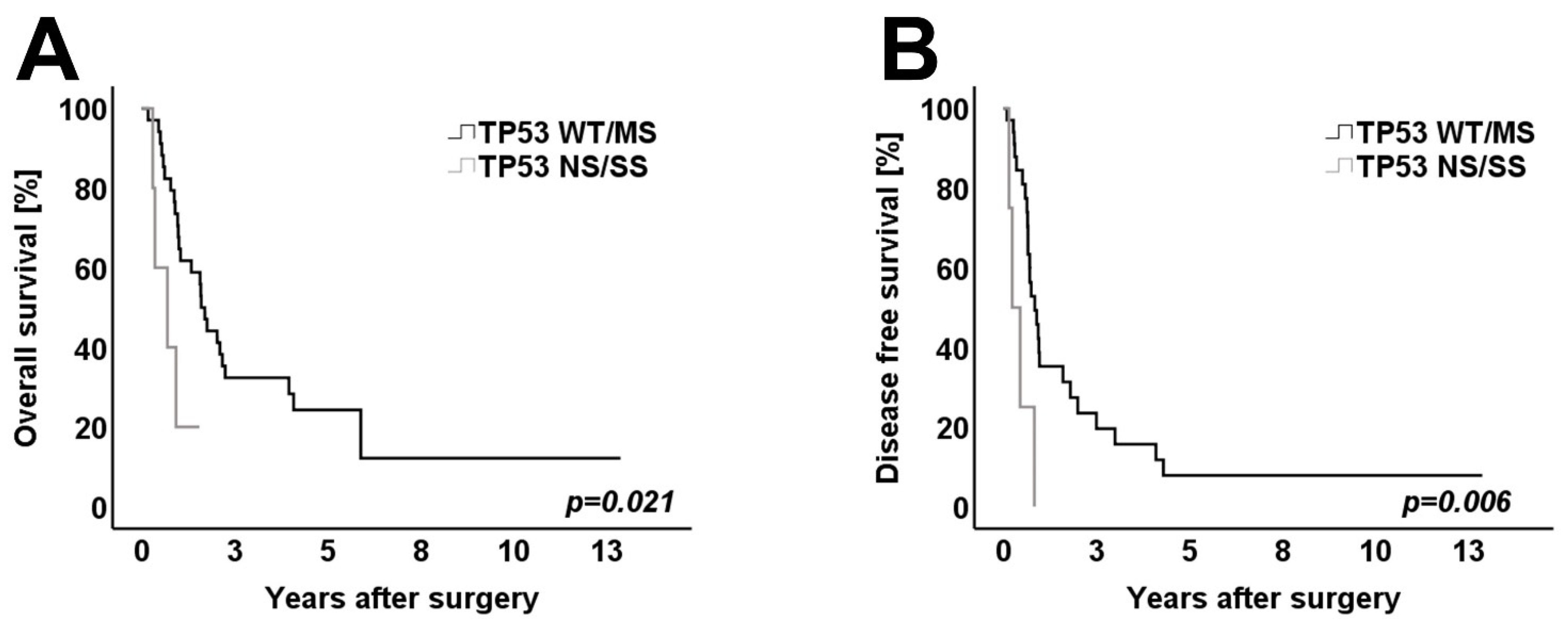
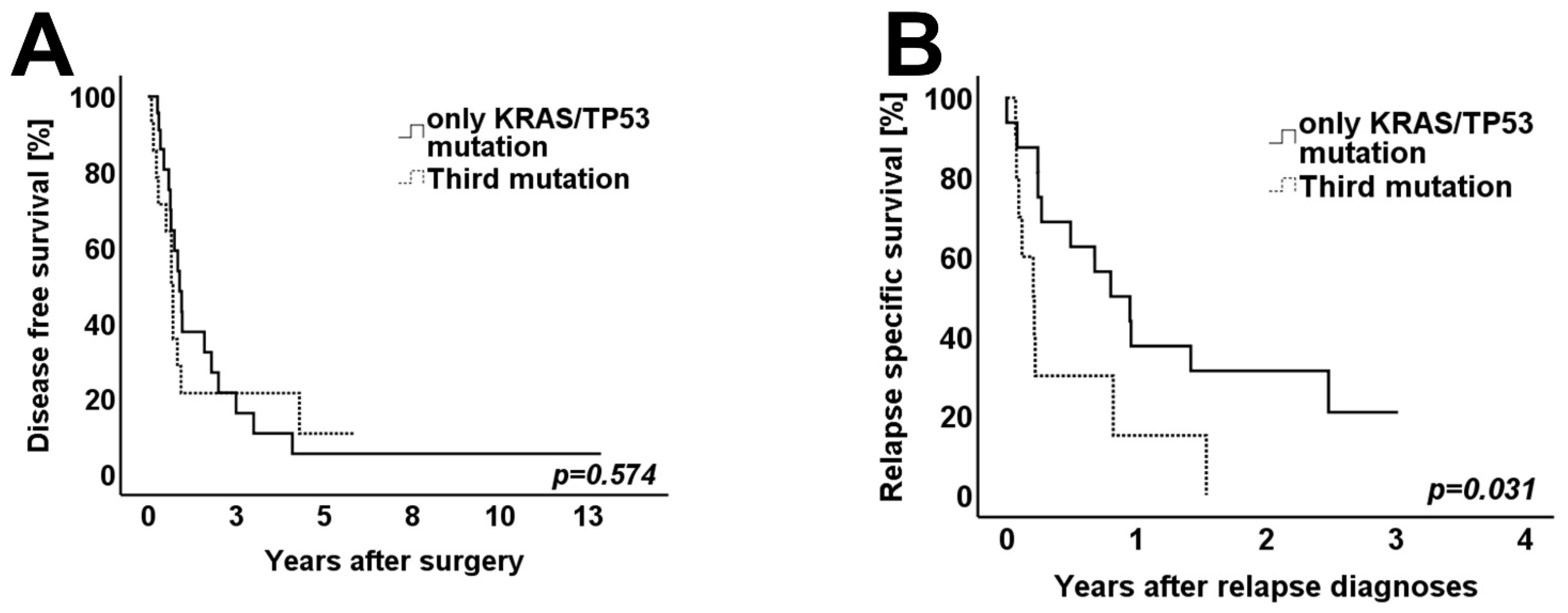
3.4. Survival Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Quante, A.S.; Ming, C.; Rottmann, M.; Engel, J.; Boeck, S.; Heinemann, V.; Westphalen, C.B.; Strauch, K. Projections of cancer incidence and cancer-related deaths in Germany by 2020 and 2030. Cancer Med. 2016, 5, 2649–2656. [Google Scholar] [CrossRef]
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant Chemotherapy With Gemcitabine vs Observation in Patients Undergoing Curative-Intent Resection of Pancreatic Cancer. JAMA 2007, 297, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 2140–2141. [Google Scholar] [CrossRef]
- Safi, S.A.; Lehwald-Tywuschik, N.; Rehders, A.; Fluegen, G.; Haeberle, L.; Keitel, V.; Knoefel, W.T. Site of relapse of ductal adenocarcinoma of the pancreas affects survival after multimodal therapy. BMC Surg. 2021, 21, 1–9. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Werner, J.; Combs, S.; Springfeld, C.; Hartwig, W.; Hackert, T.; Büchler, M.W. Advanced-stage pancreatic cancer: Therapy options. Nat. Rev. Clin. Oncol. 2013, 10, 323–333. [Google Scholar] [CrossRef]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Haeberle, L.; Esposito, I. Pathology of pancreatic cancer. Transl. Gastroenterol. Hepatol. 2019, 4, 50. [Google Scholar] [CrossRef]
- Demir, I.E.; Jäger, C.; Schlitter, A.M.; Konukiewitz, B.; Stecher, L.; Schorn, S.; Tieftrunk, E.; Scheufele, F.; Calavrezos, L.; Schirren, R.; et al. R0 Versus R1 Resection Matters after Pancreaticoduodenectomy, and Less after Distal or Total Pancreatectomy for Pancreatic Cancer. Ann. Surg. 2018, 268, 1058–1068. [Google Scholar] [CrossRef]
- Safi, S.-A.; Haeberle, L.; Fluegen, G.; Lehwald-Tywuschik, N.; Krieg, A.; Keitel, V.; Luedde, T.; Esposito, I.; Rehders, A.; Knoefel, W. Mesopancreatic excision for pancreatic ductal adenocarcinoma improves local disease control and survival. Pancreatology 2021, 21, 787–795. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Isobe, M.; Emanuel, B.S.; Givol, D.; Oren, M.; Croce, C.M. Localization of gene for human p53 tumour antigen to band 17p13. Nature 1986, 320, 84–85. [Google Scholar] [CrossRef]
- Masetti, M.; Acquaviva, G.; Visani, M.; Tallini, G.; Fornelli, A.; Ragazzi, M.; Vasuri, F.; Grifoni, D.; Di Giacomo, S.; Fiorino, S.; et al. Long-term survivors of pancreatic adenocarcinoma show low rates of genetic alterations in KRAS, TP53 and SMAD4. Cancer Biomarkers 2018, 21, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Schlitter, A.M.; Segler, A.; Steiger, K.; Michalski, C.W.; Jäger, C.; Konukiewitz, B.; Pfarr, N.; Endris, V.; Bettstetter, M.; Kong, B.; et al. Molecular, morphological and survival analysis of 177 resected pancreatic ductal adenocarcinomas (PDACs): Identification of prognostic subtypes. Sci. Rep. 2017, 7, srep41064. [Google Scholar] [CrossRef] [Green Version]
- Adham, M.; Jaeck, D.; Le Borgne, J.; Oussoultzouglou, E.; Chenard-Neu, M.-P.; Mosnier, J.-F.; Scoazec, J.-Y.; Mornex, F.; Partensky, C. Long-Term Survival (5–20 Years) After Pancreatectomy for Pancreatic Ductal Adenocarcinoma. Pancreas 2008, 37, 352–357. [Google Scholar] [CrossRef]
- Ferrone, C.R.; Brennan, M.F.; Gonen, M.; Coit, D.G.; Fong, Y.; Chung, S.; Tang, L.; Klimstra, D.; Allen, P.J. Pancreatic adenocarcinoma: The actual 5-year survivors. J. Gastrointest. Surg. 2008, 12, 701–706. [Google Scholar] [CrossRef]
- Ferrone, C.R.; Pieretti-Vanmarcke, R.; Bloom, J.P.; Zheng, H.; Szymonifka, J.; Wargo, J.A.; Thayer, S.P.; Lauwers, G.Y.; Deshpande, V.; Mino-Kenudson, M.; et al. Pancreatic ductal adenocarcinoma: Long-term survival does not equal cure. Surgery 2012, 152, S43–S49. [Google Scholar] [CrossRef] [Green Version]
- El Nakeeb, A.; El Sorogy, M.; Ezzat, H.; Said, R.; El Dosoky, M.; El Gawad, M.A.; Elsabagh, A.M.; El Hanafy, E. Predictors of long-term survival after pancreaticoduodenectomy for peri-ampullary adenocarcinoma: A retrospective study of 5-year survivors. Hepatobiliary Pancreat. Dis. Int. 2018, 17, 443–449. [Google Scholar] [CrossRef]
- Nakagawa, K.; Akahori, T.; Nishiwada, S.; Nagai, M.; Nakamura, K.; Tanaka, T.; Tamamoto, T.; Ohbayashi, C.; Hasegawa, M.; Kichikawa, K.; et al. Prognostic factors for actual long-term survival in the era of multidisciplinary treatment for pancreatic ductal adenocarcinoma. Langenbeck’s Arch. Surg. 2018, 403, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Sendabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef]
- Bertero, L.; Massa, F.; Metovic, J.; Zanetti, R.; Castellano, I.; Ricardi, U.; Papotti, M.; Cassoni, P. Eighth Edition of the UICC Classification of Malignant Tumours: An overview of the changes in the pathological TNM classification criteria-What has changed and why? Virchows Arch. 2018, 472, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A.; The WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Collisson, E.A.; Sadanandam, A.; Olson, P.; Gibb, W.J.; Truitt, M.; Gu, S.; Cooc, J.; Weinkle, J.; Kim, G.E.; Jakkula, L.; et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat. Med. 2011, 17, 500–503. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef]
- Molin, M.D.; Zhang, M.; De Wilde, R.F.; Ottenhof, N.A.; Rezaee, N.; Wolfgang, C.L.; Blackford, A.; Vogelstein, B.; Kinzler, K.W.; Papadopoulos, N.; et al. Very Long-term Survival Following Resection for Pancreatic Cancer Is Not Explained by Commonly Mutated Genes: Results of Whole-Exome Sequencing Analysis. Clin. Cancer Res. 2015, 21, 1944–1950. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.B.; Zhao, L.; Wang, X.; Ghelman, Y.; Overman, M.J.; Javle, M.M.; Shroff, R.T.; Varadhachary, G.R.; Wolff, R.A.; McAllister, F.; et al. Germline DNA Sequencing Reveals Novel Mutations Predictive of Overall Survival in a Cohort of Patients with Pancreatic Cancer. Clin. Cancer Res. 2019, 26, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Srinivasan, R.; Wig, J.D. SMAD4 Genetic Alterations Predict a Worse Prognosis in Patients With Pancreatic Ductal Adenocarcinoma. Pancreas 2012, 41, 541–546. [Google Scholar] [CrossRef]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A.; Cicenas, J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Yokose, T.; Kitago, M.; Matsuda, S.; Sasaki, Y.; Masugi, Y.; Nakamura, Y.; Shinoda, M.; Yagi, H.; Abe, Y.; Oshima, G.; et al. Combination of KRAS and SMAD4 mutations in formalin-fixed paraffin-embedded tissues as a biomarker for pancreatic cancer. Cancer Sci. 2020, 111, 2174–2182. [Google Scholar] [CrossRef] [PubMed]
- Esposito, I.; Kleeff, J.; Bergmann, F.; Reiser, C.; Herpel, E.; Friess, H.; Schirmacher, P.; Büchler, M.W. Most Pancreatic Cancer Resections are R1 Resections. Ann. Surg. Oncol. 2008, 15, 1651–1660. [Google Scholar] [CrossRef]
- Häberle, L.; Esposito, I. Circumferential resection margin (CRM) in pancreatic cancer. Surg. Pract. Sci. 2020, 1, 100006. [Google Scholar] [CrossRef]
- Cook, J.H.; Melloni, G.E.M.; Gulhan, D.C.; Park, P.J.; Haigis, K.M. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.-C.; Mansour, J.; Mollaee, M.; Wagner, K.-U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef]
- Bournet, B.; Muscari, F.; Buscail, C.; Assenat, E.; Barthet, M.; Hammel, P.; Selves, J.; Guimbaud, R.; Cordelier, P.; Buscail, L. KRAS G12D Mutation Subtype Is A Prognostic Factor for Advanced Pancreatic Adenocarcinoma. Clin. Transl. Gastroenterol. 2016, 7, e157. [Google Scholar] [CrossRef]
- Strom, S.P. Current practices and guidelines for clinical next-generation sequencing oncology testing. Cancer Biol. Med. 2016, 13, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berrino, E.; Balsamo, A.; Pisacane, A.; Gallo, S.; Becco, P.; Miglio, U.; Caravelli, D.; Poletto, S.; Paruzzo, L.; Debernardi, C.; et al. High BRAF variant allele frequencies are associated with distinct pathological features and responsiveness to target therapy in melanoma patients. ESMO Open 2021, 6, 100133. [Google Scholar] [CrossRef] [PubMed]
- Sinn, M.; Sinn, B.V.; Treue, D.; Keilholz, U.; Damm, F.; Schmuck, R.B.; Lohneis, P.; Klauschen, F.; Striefler, J.K.; Bahra, M.; et al. TP53 mutations predict sensitivity to adjuvant gemcitabine in patients with pancreatic ductal adenocarcinoma: Next-generation sequencing results from the CONKO-001 trial. Clin. Cancer Res. 2020, 26, 3732–3739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freed-Pastor, W.A.; Prives, C. Mutant p53: One name, many proteins. Genes Dev. 2012, 26, 1268–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P., IV; Wagenblast, E.; Davis, C.A.; Moon, S.-H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 Drives Pancreatic Cancer Metastasis through Cell-Autonomous PDGF Receptor β Signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [Green Version]
- Yachida, S.; White, C.M.; Naito, Y.; Zhong, Y.; Brosnan, J.A.; Macgregor-Das, A.M.; Morgan, R.A.; Saunders, T.; Laheru, D.A.; Herman, J.M.; et al. Clinical Significance of the Genetic Landscape of Pancreatic Cancer and Implications for Identification of Potential Long-term Survivors. Clin. Cancer Res. 2012, 18, 6339–6347. [Google Scholar] [CrossRef] [Green Version]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 Gene Status of the Primary Carcinoma Correlates With Patterns of Failure in Patients With Pancreatic Cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age in Years | Patients (Total) | Patients (Percentage) |
|---|---|---|
| Median (Range) | 66 (47–88) | |
| n | % | |
| Gender | ||
| Male | 24 | 61.5 |
| Female | 15 | 38.5 |
| T stage | ||
| T1 | 3 | 7.7 |
| T2 | 18 | 46.2 |
| T3 | 16 | 41.0 |
| T4 | 2 | 5.1 |
| N stage | ||
| N0 | 7 | 17.9 |
| N1 | 23 | 59.0 |
| N2 | 9 | 23.1 |
| Grading | ||
| G2 | 27 | 69.2 |
| G3 | 12 | 30.8 |
| Pn | ||
| Pn0 | 10 | 25.6 |
| Pn1 | 29 | 74.4 |
| L | ||
| L0 | 16 | 41.0 |
| L1 | 23 | 59.0 |
| V | ||
| V0 | 27 | 69.2 |
| V1 | 12 | 30.8 |
| R status | ||
| R1 | 16 | 41.0 |
| R0CRM+ | 6 | 15.4 |
| R0CRM− | 17 | 43.6 |
| NGS KRAS G12D vs. Rest and WT | NGS TP53 NS/FS/SS vs. Rest and WT | 3rd Mutation vs. Only KRAS/TP53 Mutation | |
|---|---|---|---|
| n = 39 | n = 39 | n = 39 | |
| Chi-squared test | p-value | p-value | p-value |
| Median Age | 0.471 | 0.823 | 1.000 |
| Gender | 0.944 | 0.401 | 0.740 |
| T stage | 0.921 | 0.887 | 0.337 |
| N stage | 0.921 | 0.669 | 0.864 |
| G | 0.288 | 0.239 | 0.824 |
| Pn | 0.326 | 0.066 | 0.052 |
| L | 0.492 | 0.801 | 0.510 |
| V | 0.609 | 0.535 | 0.728 |
| Resection status | 0.275 | 0.951 | 0.855 |
| Relapse location | 0.114 | 0.184 | 0.733 |
| Histomorphology | 0.673 | 0.533 | 0.643 |
| p53 IHC | 0.561 | 0.369 | 0.489 |
| p16 IHC | 0.924 | 0.625 | 0.716 |
| Smad4 IHC | 0.161 | 0.714 | 0.143 |
| Mutational Status | OS | OS | DFS | DFS | ||
|---|---|---|---|---|---|---|
| <4 Years | ≥4 Years | p-Value | <2 Years | ≥2 Years | p-Value | |
| n = 31 | n = 8 | n = 32 | n = 7 | |||
| KRAS NGS | ||||||
| G12D | 13 | 1 | 0.015 | 14 | 0 | 0.005 |
| G12V/R/C and WT | 18 | 4 | 18 | 4 | ||
| Q61H/D57N | 0 | 3 | 0 | 3 | ||
| TP53 NGS | ||||||
| NS + SS | 5 | 0 | 0.563 | 5 | 0 | 0.563 |
| WT + MS | 26 | 8 | 27 | 7 | ||
| SMAD4 NGS | ||||||
| Yes | 4 | 1 | 1.000 | 5 | 0 | 0.898 |
| No | 27 | 7 | - | - | ||
| Other 3rd driver mutation | ||||||
| Yes | 11 | 2 | 0.751 | 11 | 2 | 0.884 |
| No | 20 | 6 | 21 | 5 | ||
| p53 IHC | ||||||
| Aberrant | 15 | 5 | 1.000 | 16 | 4 | 1.000 |
| Normal | 12 | 3 | 12 | 3 | ||
| p16 IHC | ||||||
| Normal | 8 | 2 | 1.000 | 8 | 2 | 1.000 |
| Loss | 20 | 6 | 21 | 5 | ||
| Smad4 IHC | ||||||
| Normal | 8 | 3 | 0.672 | 7 | 4 | 0.163 |
| Loss | 21 | 5 | 23 | 3 | ||
| OS | OS | DFS | DFS | |||
|---|---|---|---|---|---|---|
| <4 Years | ≥4 Years | p-Value | <2 Years | ≥2 Years | p-Value | |
| n = 31 | n = 8 | n = 32 | n = 7 | |||
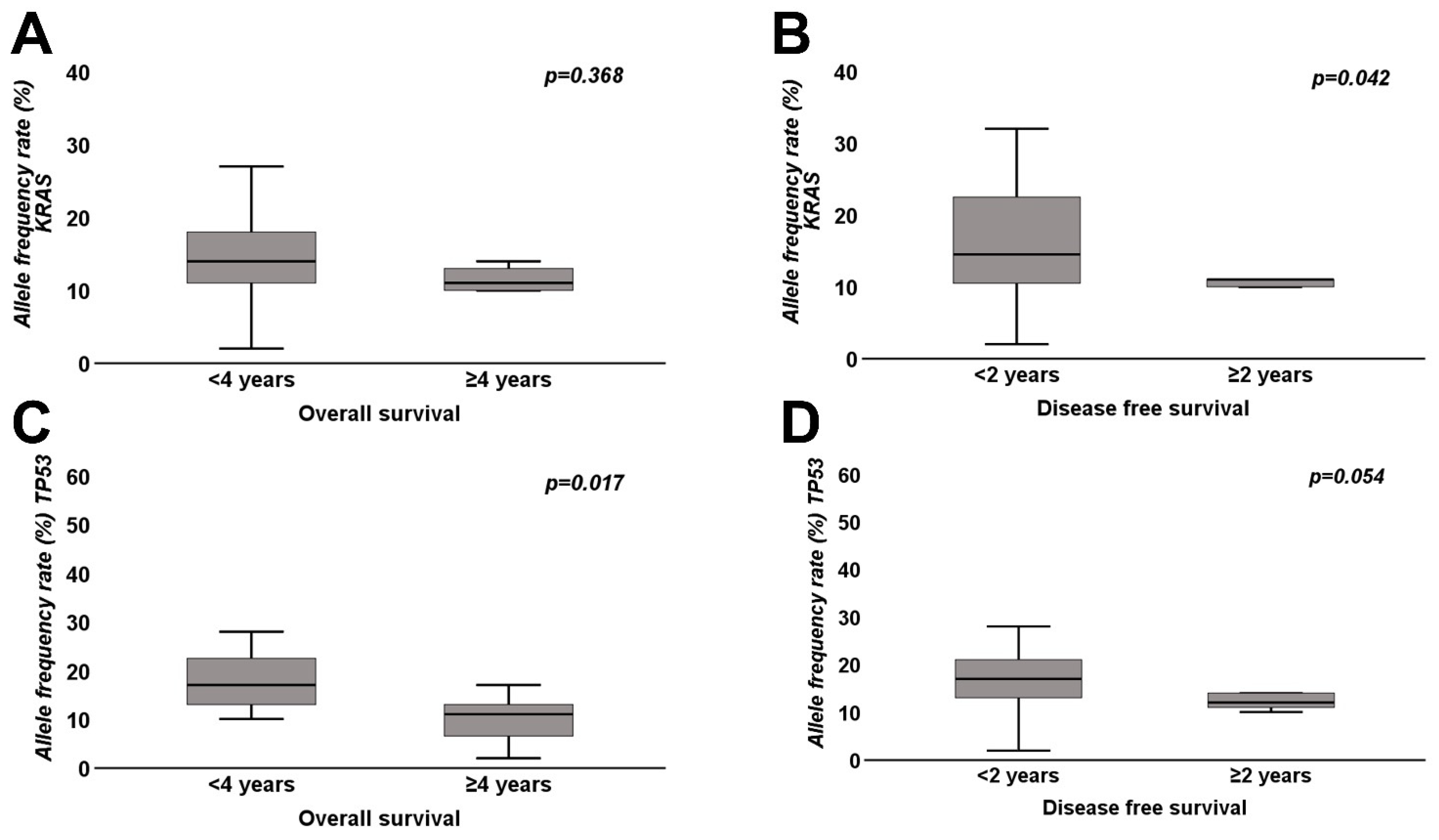
| Median (range) allele frequency rate KRAS | 14% (2–32%) | 11% (4–30%) | 0.368 | 14.5% (2–32%) | 11.0% (4–13%) | 0.042 |
| Median (range) allele frequency rate TP53 | 17% (10–54%) | 11% (2–17%) | 0.017 | 17% (2–54%) | 12% (10–14%) | 0.054 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Safi, S.-A.; Haeberle, L.; Goering, W.; Keitel, V.; Fluegen, G.; Stoecklein, N.; Rehders, A.; Knoefel, W.T.; Esposito, I. Genetic Alterations Predict Long-Term Survival in Ductal Adenocarcinoma of the Pancreatic Head. Cancers 2022, 14, 850. https://doi.org/10.3390/cancers14030850
Safi S-A, Haeberle L, Goering W, Keitel V, Fluegen G, Stoecklein N, Rehders A, Knoefel WT, Esposito I. Genetic Alterations Predict Long-Term Survival in Ductal Adenocarcinoma of the Pancreatic Head. Cancers. 2022; 14(3):850. https://doi.org/10.3390/cancers14030850
Chicago/Turabian StyleSafi, Sami-Alexander, Lena Haeberle, Wolfgang Goering, Verena Keitel, Georg Fluegen, Nikolas Stoecklein, Alexander Rehders, Wolfram Trudo Knoefel, and Irene Esposito. 2022. "Genetic Alterations Predict Long-Term Survival in Ductal Adenocarcinoma of the Pancreatic Head" Cancers 14, no. 3: 850. https://doi.org/10.3390/cancers14030850
APA StyleSafi, S.-A., Haeberle, L., Goering, W., Keitel, V., Fluegen, G., Stoecklein, N., Rehders, A., Knoefel, W. T., & Esposito, I. (2022). Genetic Alterations Predict Long-Term Survival in Ductal Adenocarcinoma of the Pancreatic Head. Cancers, 14(3), 850. https://doi.org/10.3390/cancers14030850