
Pheochromocytomas and Abdominal Paragangliomas: A Practical Guidance



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Epidemiology
3.2. Clinical Presentation
3.3. Biochemical Diagnosis
3.4. Imaging
3.5. Histopathology
3.6. Genetics and Molecular Immunohistochemistry
3.7. Management
3.8. Follow-Up
3.9. Metastatic Disease
3.10. Pregnancy in Patients with Pheochromocytomas and Paragangliomas
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Neumann, H.P.H.; Young, W.F., Jr.; Eng, C. Pheochromocytoma and Paraganglioma. N. Engl. J. Med. 2019, 381, 552–565. [Google Scholar] [CrossRef]
- Falhammar, H.; Kjellman, M.; Calissendorff, J. Initial clinical presentation and spectrum of pheochromocytoma: A study of 94 cases from a single center. Endocr. Connect. 2018, 7, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Gruber, L.M.; Hartman, R.P.; Thompson, G.B.; McKenzie, T.J.; Lyden, M.L.; Dy, B.M.; Young, W.F.; Bancos, I. Pheochromocytoma Characteristics and Behavior Differ Depending on Method of Discovery. J. Clin. Endocrinol. Metab. 2019, 104, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.; Murad, M.H.; Naruse, M.; Pacak, K.; Young, W.F., Jr.; Endocrine, S. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014, 99, 1915–1942. [Google Scholar] [CrossRef]
- Lam, A.K. Update on Adrenal Tumours in 2017 World Health Organization (WHO) of Endocrine Tumours. Endocr. Pathol. 2017, 28, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Granberg, D.; Juhlin, C.C.; Falhammar, H. Metastatic Pheochromocytomas and Abdominal Paragangliomas. J. Clin. Endocrinol. Metab. 2021, 106, e1937–e1952. [Google Scholar] [CrossRef]
- Taieb, D.; Timmers, H.; Pacak, K. Diagnostic Investigation of Lesions Associated with Succinate Dehydrogenase Defects. Horm. Metab. Res. 2019, 51, 414–418. [Google Scholar] [CrossRef]
- Martin, T.P.; Irving, R.M.; Maher, E.R. The genetics of paragangliomas: A review. Clin. Otolaryngol. 2007, 32, 7–11. [Google Scholar] [CrossRef]
- Muth, A.; Crona, J.; Gimm, O.; Elmgren, A.; Filipsson, K.; Stenmark Askmalm, M.; Sandstedt, J.; Tengvar, M.; Tham, E. Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J. Intern. Med. 2019, 285, 187–204. [Google Scholar] [CrossRef] [PubMed]
- Amar, L.; Fassnacht, M.; Gimenez-Roqueplo, A.P.; Januszewicz, A.; Prejbisz, A.; Timmers, H.; Plouin, P.F. Long-term postoperative follow-up in patients with apparently benign pheochromocytoma and paraganglioma. Horm. Metab. Res. 2012, 44, 385–389. [Google Scholar] [CrossRef]
- Khatami, F.; Mohammadamoli, M.; Tavangar, S.M. Genetic and epigenetic differences of benign and malignant pheochromocytomas and paragangliomas (PPGLs). Endocr. Regul. 2018, 52, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Zhikrivetskaya, S.O.; Snezhkina, A.V.; Zaretsky, A.R.; Alekseev, B.Y.; Pokrovsky, A.V.; Golovyuk, A.L.; Melnikova, N.V.; Stepanov, O.A.; Kalinin, D.V.; Moskalev, A.A.; et al. Molecular markers of paragangliomas/pheochromocytomas. Oncotarget 2017, 8, 25756–25782. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.V.; Osamura, R.Y.; Klöppel, G.; Rosai, J. WHO Classification of Tumours of Endocrine Organs, 4th ed.; Llyod, R.V., Osamura, R.Y., Klöppel, G., Rosai, J., Eds.; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- Ebbehoj, A.; Stochholm, K.; Jacobsen, S.F.; Trolle, C.; Jepsen, P.; Robaczyk, M.G.; Rasmussen, A.K.; Feldt-Rasmussen, U.; Thomsen, R.W.; Sondergaard, E.; et al. Incidence and Clinical Presentation of Pheochromocytoma and Sympathetic Paraganglioma: A Population-based Study. J. Clin. Endocrinol. Metab. 2021, 106, e2251–e2261. [Google Scholar] [CrossRef] [PubMed]
- Berends, A.M.A.; Buitenwerf, E.; de Krijger, R.R.; Veeger, N.; van der Horst-Schrivers, A.N.A.; Links, T.P.; Kerstens, M.N. Incidence of pheochromocytoma and sympathetic paraganglioma in the Netherlands: A nationwide study and systematic review. Eur. J. Intern. Med. 2018, 51, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Ebbehoj, A.; Li, D.; Kaur, R.J.; Zhang, C.; Singh, S.; Li, T.; Atkinson, E.; Achenbach, S.; Khosla, S.; Arlt, W.; et al. Epidemiology of adrenal tumours in Olmsted County, Minnesota, USA: A population-based cohort study. Lancet Diabetes Endocrinol. 2020, 8, 894–902. [Google Scholar] [CrossRef]
- Falhammar, H.; Kjellman, M.; Calissendorff, J. Treatment and outcomes in pheochromocytomas and paragangliomas: A study of 110 cases from a single center. Endocrine 2018, 62, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Moon, H.; Noh, J.; Lee, J.; Kim, S.G. Epidemiology and Prognosis of Pheochromocytoma/Paraganglioma in Korea: A Nationwide Study Based on the National Health Insurance Service. Endocrinol. Metab. 2020, 35, 157–164. [Google Scholar] [CrossRef]
- Gruber, L.M.; Erickson, D.; Babovic-Vuksanovic, D.; Thompson, G.B.; Young, W.F., Jr.; Bancos, I. Pheochromocytoma and paraganglioma in patients with neurofibromatosis type 1. Clin. Endocrinol. 2017, 86, 141–149. [Google Scholar] [CrossRef]
- Kittah, N.E.; Gruber, L.M.; Bancos, I.; Hamidi, O.; Tamhane, S.; Iniguez-Ariza, N.; Babovic-Vuksanovic, D.; Thompson, G.B.; Lteif, A.; Young, W.F.; et al. Bilateral pheochromocytoma: Clinical characteristics, treatment and longitudinal follow-up. Clin. Endocrinol. 2020, 93, 288–295. [Google Scholar] [CrossRef]
- Canu, L.; Van Hemert, J.A.W.; Kerstens, M.N.; Hartman, R.P.; Khanna, A.; Kraljevic, I.; Kastelan, D.; Badiu, C.; Ambroziak, U.; Tabarin, A.; et al. CT Characteristics of Pheochromocytoma: Relevance for the Evaluation of Adrenal Incidentaloma. J. Clin. Endocrinol. Metab. 2019, 104, 312–318. [Google Scholar] [CrossRef]
- Neumann, H.P.H.; Tsoy, U.; Bancos, I.; Amodru, V.; Walz, M.K.; Tirosh, A.; Kaur, R.J.; McKenzie, T.; Qi, X.; Bandgar, T.; et al. Comparison of Pheochromocytoma-Specific Morbidity and Mortality Among Adults with Bilateral Pheochromocytomas Undergoing Total Adrenalectomy vs Cortical-Sparing Adrenalectomy. JAMA Netw. Open 2019, 2, e198898. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Bancos, I.; Gruber, L.M.; Bancos, C.; McKenzie, T.J.; Babovic-Vuksanovic, D.; Young, W.F., Jr. When Biochemical Phenotype Predicts Genotype: Pheochromocytoma and Paraganglioma. Am. J. Med. 2018, 131, 506–509. [Google Scholar] [CrossRef]
- Manger, W.M. The protean manifestations of pheochromocytoma. Horm. Metab. Res. 2009, 41, 658–663. [Google Scholar] [CrossRef]
- Amar, L.; Servais, A.; Gimenez-Roqueplo, A.P.; Zinzindohoue, F.; Chatellier, G.; Plouin, P.F. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J. Clin. Endocrinol. Metab. 2005, 90, 2110–2116. [Google Scholar] [CrossRef]
- Elliott, P.F.; Berhane, T.; Ragnarsson, O.; Falhammar, H. Ectopic ACTH- and/or CRH-Producing Pheochromocytomas. J. Clin. Endocrinol. Metab. 2021, 106, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Falhammar, H.; Stenman, A.; Calissendorff, J.; Juhlin, C.C. Presentation, Treatment, Histology, and Outcomes in Adrenal Medullary Hyperplasia Compared with Pheochromocytoma. J. Endocr. Soc. 2019, 3, 1518–1530. [Google Scholar] [CrossRef] [PubMed]
- Falhammar, H.; Calissendorff, J.; Hoybye, C. Frequency of Cushing’s syndrome due to ACTH-secreting adrenal medullary lesions: A retrospective study over 10 years from a single center. Endocrine 2017, 55, 296–302. [Google Scholar] [CrossRef]
- Mao, J.J.; Baker, J.E.; Rainey, W.E.; Young, W.F., Jr.; Bancos, I. Concomitant Pheochromocytoma and Primary Aldosteronism: A Case Series and Literature Review. J. Endocr. Soc. 2021, 5, bvab107. [Google Scholar] [CrossRef]
- Y-Hassan, S.; Falhammar, H. Cardiovascular Manifestations and Complications of Pheochromocytomas and Paragangliomas. J. Clin. Med. 2020, 9, 2435. [Google Scholar] [CrossRef]
- Zelinka, T.; Petrak, O.; Turkova, H.; Holaj, R.; Strauch, B.; Krsek, M.; Vrankova, A.B.; Musil, Z.; Duskova, J.; Kubinyi, J.; et al. High incidence of cardiovascular complications in pheochromocytoma. Horm. Metab. Res. 2012, 44, 379–384. [Google Scholar] [CrossRef]
- Pappachan, J.M.; Tun, N.N.; Arunagirinathan, G.; Sodi, R.; Hanna, F.W.F. Pheochromocytomas and Hypertension. Curr. Hypertens Rep. 2018, 20, 3. [Google Scholar] [CrossRef] [PubMed]
- Pacak, K.; Linehan, W.M.; Eisenhofer, G.; Walther, M.M.; Goldstein, D.S. Recent advances in genetics, diagnosis, localization, and treatment of pheochromocytoma. Ann. Intern. Med. 2001, 134, 315–329. [Google Scholar] [CrossRef]
- Greenleaf, C.E.; Griffin, L.A.; Shake, J.G.; Orr, W.S. Hypertensive crisis secondary to pheochromocytoma. Bayl. Univ. Med. Cent. Proc. 2017, 30, 314–315. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yeoh, C.J.; Ng, S.Y.; Goh, B.K. Pheochromocytoma Multisystem Crisis Triggered by Glucocorticoid Administration and Aggravated by Citrate Dialysis. A A Case Rep. 2017, 8, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Riester, A.; Weismann, D.; Quinkler, M.; Lichtenauer, U.D.; Sommerey, S.; Halbritter, R.; Penning, R.; Spitzweg, C.; Schopohl, J.; Beuschlein, F.; et al. Life-threatening events in patients with pheochromocytoma. Eur. J. Endocrinol. 2015, 173, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Y-Hassan, S.; Falhammar, H. Stumbling broke the spleen and unveiled pheochromocytoma, which in turn broke the heart. Endocrine 2020, 67, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Y-Hassan, S.; Falhammar, H. Clinical features, complications, and outcomes of exogenous and endogenous catecholamine-triggered Takotsubo syndrome: A systematic review and meta-analysis of 156 published cases. Clin. Cardiol. 2020, 43, 459–467. [Google Scholar] [CrossRef]
- Y-Hassan, S.; Falhammar, H. Pheochromocytoma- and paraganglioma-triggered Takotsubo syndrome. Endocrine 2019, 65, 483–493. [Google Scholar] [CrossRef]
- Giavarini, A.; Chedid, A.; Bobrie, G.; Plouin, P.F.; Hagege, A.; Amar, L. Acute catecholamine cardiomyopathy in patients with phaeochromocytoma or functional paraganglioma. Heart 2013, 99, 1438–1444. [Google Scholar] [CrossRef]
- Gatzoulis, K.A.; Tolis, G.; Theopistou, A.; Gialafos, J.H.; Toutouzas, P.K. Cardiomyopathy due to a pheochromocytoma. A reversible entity. Acta Cardiol. 1998, 53, 227–229. [Google Scholar]
- Wilkenfeld, C.; Cohen, M.; Lansman, S.L.; Courtney, M.; Dische, M.R.; Pertsemlidis, D.; Krakoff, L.R. Heart transplantation for end-stage cardiomyopathy caused by an occult pheochromocytoma. J. Heart Lung Transpl. 1992, 11, 363–366. [Google Scholar]
- Sardesai, S.H.; Mourant, A.J.; Sivathandon, Y.; Farrow, R.; Gibbons, D.O. Phaeochromocytoma and catecholamine induced cardiomyopathy presenting as heart failure. Br. Heart J. 1990, 63, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Darr, R.; Kuhn, M.; Bode, C.; Bornstein, S.R.; Pacak, K.; Lenders, J.W.M.; Eisenhofer, G. Accuracy of recommended sampling and assay methods for the determination of plasma-free and urinary fractionated metanephrines in the diagnosis of pheochromocytoma and paraganglioma: A systematic review. Endocrine 2017, 56, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Goldstein, D.S.; Walther, M.M.; Friberg, P.; Lenders, J.W.; Keiser, H.R.; Pacak, K. Biochemical diagnosis of pheochromocytoma: How to distinguish true- from false-positive test results. J. Clin. Endocrinol. Metab. 2003, 88, 2656–2666. [Google Scholar] [CrossRef] [PubMed]
- Galati, S.J.; Said, M.; Gospin, R.; Babic, N.; Brown, K.; Geer, E.B.; Kostakoglu, L.; Krakoff, L.R.; Leibowitz, A.B.; Mehta, L.; et al. The Mount Sinai clinical pathway for the management of pheochromocytoma. Endocr. Pract. 2015, 21, 368–382. [Google Scholar] [CrossRef]
- Eisenhofer, G.; Klink, B.; Richter, S.; Lenders, J.W.; Robledo, M. Metabologenomics of Phaeochromocytoma and Paraganglioma: An Integrated Approach for Personalised Biochemical and Genetic Testing. Clin. Biochem. Rev. 2017, 38, 69–100. [Google Scholar]
- Nolting, S.; Bechmann, N.; Taieb, D.; Beuschlein, F.; Fassnacht, M.; Kroiss, M.; Eisenhofer, G.; Grossman, A.; Pacak, K. Personalized management of pheochromocytoma and paraganglioma. Endocr. Rev. 2021, 20, 1–41. [Google Scholar] [CrossRef]
- Proye, C.; Fossati, P.; Fontaine, P.; Lefebvre, J.; Decoulx, M.; Wemeau, J.L.; Dewailly, D.; Rwamasirabo, E.; Cecat, P. Dopamine-secreting pheochromocytoma: An unrecognized entity? Classification of pheochromocytomas according to their type of secretion. Surgery 1986, 100, 1154–1162. [Google Scholar]
- Bozin, M.; Lamb, A.; Putra, L.J. Pheochromocytoma with Negative Metanephrines: A Rarity and the Significance of Dopamine Secreting Tumors. Urol. Case Rep. 2017, 12, 51–53. [Google Scholar] [CrossRef]
- van Berkel, A.; Lenders, J.W.; Timmers, H.J. Diagnosis of endocrine disease: Biochemical diagnosis of phaeochromocytoma and paraganglioma. Eur. J. Endocrinol. 2014, 170, R109–R119. [Google Scholar] [CrossRef]
- Plouin, P.F.; Amar, L.; Dekkers, O.M.; Fassnacht, M.; Gimenez-Roqueplo, A.P.; Lenders, J.W.; Lussey-Lepoutre, C.; Steichen, O. Guideline Working Group European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur. J. Endocrinol. 2016, 174, G1–G10. [Google Scholar] [CrossRef]
- Butler, O.L.; Mekhael, M.M.; Ahmed, A.; Cuthbertson, D.J.; Pritchard, D.M. Frequency and Causes of False-Positive Elevated Plasma Concentrations of Fasting Gut Hormones in a Specialist Neuroendocrine Tumor Center. Front. Endocrinol. 2020, 11, 606264. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Lenders, J.W.; Siegert, G.; Bornstein, S.R.; Friberg, P.; Milosevic, D.; Mannelli, M.; Linehan, W.M.; Adams, K.; Timmers, H.J.; et al. Plasma methoxytyramine: A novel biomarker of metastatic pheochromocytoma and paraganglioma in relation to established risk factors of tumour size, location and SDHB mutation status. Eur. J. Cancer 2012, 48, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Eisenhofer, G.; Lenders, J.W.; Timmers, H.; Mannelli, M.; Grebe, S.K.; Hofbauer, L.C.; Bornstein, S.R.; Tiebel, O.; Adams, K.; Bratslavsky, G.; et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin. Chem. 2011, 57, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Miyake, H.; Maeda, H.; Tashiro, M.; Suzuki, K.; Nagatomo, H.; Aikawa, H.; Ashizawa, A.; Iechika, S.; Moriuchi, A. CT of adrenal tumors: Frequency and clinical significance of low-attenuation lesions. AJR Am. J. Roentgenol. 1989, 152, 1005–1007. [Google Scholar] [CrossRef]
- Gruber, L.M.; Strajina, V.; Bancos, I.; Murad, M.H.; Dy, B.M.; Young, W.F.; Farley, D.R.; Lyden, M.L.; Thompson, G.B.; McKenzie, T.J. Not all adrenal incidentalomas require biochemical testing to exclude pheochromocytoma: Mayo clinic experience and a meta-analysis. Gland Surg. 2020, 9, 362–371. [Google Scholar] [CrossRef]
- Bancos, I.; Prete, A. Approach to the Patient with Adrenal Incidentaloma. J. Clin. Endocrinol. Metab. 2021, 106, 3331–3353. [Google Scholar] [CrossRef]
- Hasassri, M.E.; Pandian, T.K.; Bobr, A.A.; Bancos, I.; Young, W.F., Jr.; Richards, M.L.; Farley, D.R.; Thompson, G.B.; McKenzie, T.J. Pheochromocytoma with Synchronous Ipsilateral Adrenal Cortical Adenoma. World J. Surg. 2017, 41, 3147–3153. [Google Scholar] [CrossRef]
- Dages, K.N.; Kohlenberg, J.D.; Young, W.F., Jr.; Murad, M.H.; Prokop, L.; Rivera, M.; Dy, B.; Foster, T.; Lyden, M.; McKenzie, T.; et al. Presentation and outcomes of adrenal ganglioneuromas: A cohort study and a systematic review of literature. Clin. Endocrinol. 2021, 95, 47–57. [Google Scholar] [CrossRef]
- Taieb, D.; Hicks, R.J.; Hindie, E.; Guillet, B.A.; Avram, A.; Ghedini, P.; Timmers, H.J.; Scott, A.T.; Elojeimy, S.; Rubello, D.; et al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur. J. Nucl. Med. Mol. Imaging. 2019, 46, 2112–2137. [Google Scholar] [CrossRef]
- Reginelli, A.; Vacca, G.; Belfiore, M.; Sangiovanni, A.; Nardone, V.; Vanzulli, A.; Grassi, R.; Cappabianca, S. Pitfalls and differential diagnosis on adrenal lesions: Current concepts in CT/MR imaging: A narrative review. Gland Surg. 2020, 9, 2331–2342. [Google Scholar] [CrossRef]
- Schteingart, D.E.; Doherty, G.M.; Gauger, P.G.; Giordano, T.J.; Hammer, G.D.; Korobkin, M.; Worden, F.P. Management of patients with adrenal cancer: Recommendations of an international consensus conference. Endocr.-Relat. Cancer 2005, 12, 667–680. [Google Scholar] [CrossRef]
- Kong, G.; Schenberg, T.; Yates, C.J.; Trainer, A.; Sachithanandan, N.; Iravani, A.; Ravi Kumar, A.; Hofman, M.S.; Akhurst, T.; Michael, M.; et al. The Role of 68Ga-DOTA-Octreotate PET/CT in Follow-Up of SDH-Associated Pheochromocytoma and Paraganglioma. J. Clin. Endocrinol. Metab. 2019, 104, 5091–5099. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.; van Berkel, A.; Piscaer, I.; Young, W.F.; Gruber, L.; Deutschbein, T.; Fassnacht, M.; Beuschlein, F.; Spyroglou, A.; Prejbisz, A.; et al. Impact of 123 I-MIBG scintigraphy on clinical decision making in pheochromocytoma and paraganglioma. J. Clin. Endocrinol. Metab. 2019, 104, 3812–3820. [Google Scholar] [CrossRef] [PubMed]
- Timmers, H.J.; Chen, C.C.; Carrasquillo, J.A.; Whatley, M.; Ling, A.; Eisenhofer, G.; King, K.S.; Rao, J.U.; Wesley, R.A.; Adams, K.T.; et al. Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. J. Natl. Cancer Inst. 2012, 104, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.A.; Pattison, D.A.; Tothill, R.W.; Kong, G.; Akhurst, T.J.; Hicks, R.J.; Hofman, M.S. (68)Ga-DOTATATE and (18)F-FDG PET/CT in Paraganglioma and Pheochromocytoma: Utility, patterns and heterogeneity. Cancer Imaging 2016, 16, 22. [Google Scholar] [CrossRef]
- Ilias, I.; Chen, C.C.; Carrasquillo, J.A.; Whatley, M.; Ling, A.; Lazurova, I.; Adams, K.T.; Perera, S.; Pacak, K. Comparison of 6-18F-fluorodopamine PET with 123I-metaiodobenzylguanidine and 111in-pentetreotide scintigraphy in localization of nonmetastatic and metastatic pheochromocytoma. J. Nucl. Med. 2008, 49, 1613–1619. [Google Scholar] [CrossRef]
- Timmers, H.J.; Chen, C.C.; Carrasquillo, J.A.; Whatley, M.; Ling, A.; Havekes, B.; Eisenhofer, G.; Martiniova, L.; Adams, K.T.; Pacak, K. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J. Clin. Endocrinol. Metab. 2009, 94, 4757–4767. [Google Scholar] [CrossRef]
- Shulkin, B.L.; Ilias, I.; Sisson, J.C.; Pacak, K. Current trends in functional imaging of pheochromocytomas and paragangliomas. Ann. N. Y. Acad. Sci. 2006, 1073, 374–382. [Google Scholar] [CrossRef]
- Mazzaglia, P.J.; Monchik, J.M. Limited value of adrenal biopsy in the evaluation of adrenal neoplasm: A decade of experience. Arch. Surg. 2009, 144, 465–470. [Google Scholar] [CrossRef]
- Vanderveen, K.A.; Thompson, S.M.; Callstrom, M.R.; Young, W.F., Jr.; Grant, C.S.; Farley, D.R.; Richards, M.L.; Thompson, G.B. Biopsy of pheochromocytomas and paragangliomas: Potential for disaster. Surgery 2009, 146, 1158–1166. [Google Scholar] [CrossRef]
- Juhlin, C.C. Challenges in Paragangliomas and Pheochromocytomas: From Histology to Molecular Immunohistochemistry. Endocr. Pathol. 2021, 32, 228–244. [Google Scholar] [CrossRef]
- Dong, J.; Asa, S.L.; Drucker, D.J. Islet cell and extrapancreatic expression of the LIM domain homeobox gene isl-1. Mol. Endocrinol. 1991, 5, 1633–1641. [Google Scholar] [CrossRef][Green Version]
- Juhlin, C.C.; Zedenius, J.; Hoog, A. Clinical Routine Application of the Second-generation Neuroendocrine Markers ISL1, INSM1, and Secretagogin in Neuroendocrine Neoplasia: Staining Outcomes and Potential Clues for Determining Tumor Origin. Endocr. Pathol. 2020, 31, 401–410. [Google Scholar] [CrossRef]
- Agaimy, A.; Erlenbach-Wunsch, K.; Konukiewitz, B.; Schmitt, A.M.; Rieker, R.J.; Vieth, M.; Kiesewetter, F.; Hartmann, A.; Zamboni, G.; Perren, A.; et al. ISL1 expression is not restricted to pancreatic well-differentiated neuroendocrine neoplasms, but is also commonly found in well and poorly differentiated neuroendocrine neoplasms of extrapancreatic origin. Mod. Pathol. 2013, 26, 995–1003. [Google Scholar] [CrossRef]
- Nonaka, D.; Wang, B.Y.; Edmondson, D.; Beckett, E.; Sun, C.C. A study of gata3 and phox2b expression in tumors of the autonomic nervous system. Am. J. Surg. Pathol. 2013, 37, 1236–1241. [Google Scholar] [CrossRef]
- Lloyd, R.V.; Blaivas, M.; Wilson, B.S. Distribution of chromogranin and S100 protein in normal and abnormal adrenal medullary tissues. Arch. Pathol. Lab. Med. 1985, 109, 633–635. [Google Scholar] [PubMed]
- Iwanaga, T.; Fujita, T. Sustentacular cells in the fetal human adrenal medulla are immunoreactive with antibodies to brain S-100 protein. Cell Tissue Res. 1984, 236, 733–735. [Google Scholar] [CrossRef]
- Thompson, L.D. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: A clinicopathologic and immunophenotypic study of 100 cases. Am. J. Surg. Pathol. 2002, 26, 551–566. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Meng, F.; Bian, W.; Chen, J.; Zhao, H.; Ma, G.; Shi, B.; Zhang, J.; Liu, Y.; Xu, Z. Development and validation of pheochromocytoma of the adrenal gland scaled score for predicting malignant pheochromocytomas. Urology 2006, 68, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Stenman, A.; Svahn, F.; Hojjat-Farsangi, M.; Zedenius, J.; Soderkvist, P.; Gimm, O.; Larsson, C.; Juhlin, C.C. Molecular Profiling of Pheochromocytoma and Abdominal Paraganglioma Stratified by the PASS Algorithm Reveals Chromogranin B as Associated with Histologic Prediction of Malignant Behavior. Am. J. Surg. Pathol. 2019, 43, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Wachtel, H.; Hutchens, T.; Baraban, E.; Schwartz, L.E.; Montone, K.; Baloch, Z.; LiVolsi, V.; Krumeich, L.; Fraker, D.L.; Nathanson, K.L.; et al. Predicting Metastatic Potential in Pheochromocytoma and Paraganglioma: A Comparison of PASS and GAPP Scoring Systems. J. Clin. Endocrinol. Metab. 2020, 105, e4661–e4670. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Tischler, A.S.; Lloyd, R.V.; DeLellis, R.A.; de Krijger, R.; van Nederveen, F.; Nose, V. Observer variation in the application of the Pheochromocytoma of the Adrenal Gland Scaled Score. Am. J. Surg. Pathol. 2009, 33, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N.; Takayanagi, R.; Takizawa, N.; Itagaki, E.; Katabami, T.; Kakoi, N.; Rakugi, H.; Ikeda, Y.; Tanabe, A.; Nigawara, T.; et al. Pathological grading for predicting metastasis in phaeochromocytoma and paraganglioma. Endocr.-Relat. Cancer 2014, 21, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Stenman, A.; Zedenius, J.; Juhlin, C.C. The Value of Histological Algorithms to Predict the Malignancy Potential of Pheochromocytomas and Abdominal Paragangliomas-A Meta-Analysis and Systematic Review of the Literature. Cancers 2019, 11, 225. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N.; Abermil, N.; Buffet, A.; Favier, J.; Gimenez-Roqueplo, A.P. The genetics of paragangliomas. Eur. Ann. Otorhinolaryngol. Head Neck Dis. 2012, 129, 315–318. [Google Scholar] [CrossRef]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Jochmanová, I.; Yang, C.; Zhuang, Z.; Pacak, K. Hypoxia-inducible factor signaling in pheochromocytoma: Turning the rudder in the right direction. J. Natl. Cancer Inst. 2013, 105, 1270–1283. [Google Scholar] [CrossRef]
- Crona, J.; Lamarca, A.; Ghosal, S.; Welin, S.; Skogseid, B.; Pacak, K. Genotype-phenotype correlations in pheochromocytoma and paraganglioma: A systematic review and individual patient meta-analysis. Endocr.-Relat. Cancer 2019, 26, 539–550. [Google Scholar] [CrossRef]
- Jochmanova, I.; Pacak, K. Genomic Landscape of Pheochromocytoma and Paraganglioma. Trends Cancer 2018, 4, 6–9. [Google Scholar] [CrossRef]
- Alzofon, N.; Koc, K.; Panwell, K.; Pozdeyev, N.; Marshall, C.B.; Albuja-Cruz, M.; Raeburn, C.D.; Nathanson, K.L.; Cohen, D.L.; Wierman, M.E.; et al. Mastermind Like Transcriptional Coactivator 3 (MAML3) Drives Neuroendocrine Tumor Progression. Mol. Cancer Res. 2021, 19, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
- Papathomas, T.G.; Oudijk, L.; Persu, A.; Gill, A.J.; van Nederveen, F.; Tischler, A.S.; Tissier, F.; Volante, M.; Matias-Guiu, X.; Smid, M.; et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: A multicenter interobserver variation analysis using virtual microscopy: A Multinational Study of the European Network for the Study of Adrenal Tumors (ENS@T). Mod. Pathol. 2015, 28, 807–821. [Google Scholar] [CrossRef]
- Grabmaier, K.; MC, A.d.W.; Verhaegh, G.W.; Schalken, J.A.; Oosterwijk, E. Strict regulation of CAIX(G250/MN) by HIF-1alpha in clear cell renal cell carcinoma. Oncogene 2004, 23, 5624–5631. [Google Scholar] [CrossRef]
- Pinato, D.J.; Ramachandran, R.; Toussi, S.T.; Vergine, M.; Ngo, N.; Sharma, R.; Lloyd, T.; Meeran, K.; Palazzo, F.; Martin, N.; et al. Immunohistochemical markers of the hypoxic response can identify malignancy in phaeochromocytomas and paragangliomas and optimize the detection of tumours with VHL germline mutations. Br. J. Cancer 2013, 108, 429–437. [Google Scholar] [CrossRef]
- Mete, O.; Pakbaz, S.; Lerario, A.M.; Giordano, T.J.; Asa, S.L. Significance of Alpha-inhibin Expression in Pheochromocytomas and Paragangliomas. Am. J. Surg. Pathol. 2021, 45, 1264–1273. [Google Scholar] [CrossRef] [PubMed]
- Dwight, T.; Flynn, A.; Amarasinghe, K.; Benn, D.E.; Lupat, R.; Li, J.; Cameron, D.L.; Hogg, A.; Balachander, S.; Candiloro, I.L.M.; et al. TERT structural rearrangements in metastatic pheochromocytomas. Endocr.-Relat. Cancer 2018, 25, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Brown, T.C.; Juhlin, C.C.; Andreasson, A.; Wang, N.; Backdahl, M.; Healy, J.M.; Prasad, M.L.; Korah, R.; Carling, T.; et al. The activating TERT promoter mutation C228T is recurrent in subsets of adrenal tumors. Endocr.-Relat. Cancer 2014, 21, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Gurrieri, C.; Butz, J.J.; Weingarten, T.N.; Bancos, I.; Young, W.F., Jr.; Cassivi, S.D.; Said, S.M.; McKenzie, T.J.; Barbara, D.W.; Sprung, J. Resection of Intrathoracic Paraganglioma with and without Cardiopulmonary Bypass. Ann. Thorac. Surg. 2018, 105, 1160–1167. [Google Scholar] [CrossRef]
- Gruber, L.M.; Jasim, S.; Ducharme-Smith, A.; Weingarten, T.; Young, W.F.; Bancos, I. The Role for Metyrosine in the Treatment of Patients with Pheochromocytoma and Paraganglioma. J. Clin. Endocrinol. Metab. 2021, 106, e2393–e2401. [Google Scholar] [CrossRef]
- Butz, J.J.; Weingarten, T.N.; Cavalcante, A.N.; Bancos, I.; Young, W.F., Jr.; McKenzie, T.J.; Schroeder, D.R.; Martin, D.P.; Sprung, J. Perioperative hemodynamics and outcomes of patients on metyrosine undergoing resection of pheochromocytoma or paraganglioma. Int. J. Surg. 2017, 46, 1–6. [Google Scholar] [CrossRef]
- Buitenwerf, E.; Osinga, T.E.; Timmers, H.; Lenders, J.W.M.; Feelders, R.A.; Eekhoff, E.M.W.; Haak, H.R.; Corssmit, E.P.M.; Bisschop, P.; Valk, G.D.; et al. Efficacy of alpha-Blockers on Hemodynamic Control during Pheochromocytoma Resection: A Randomized Controlled Trial. J. Clin. Endocrinol. Metab. 2020, 105, 2381–2391. [Google Scholar] [CrossRef]
- Tamaki, R.; Yamasaki, M.; Nishi, H.; Yoshino, K.; Abe, K.; Misumi, H. Emergency aortic valve replacement complicated by unmanaged pheochromocytoma. J. Card. Surg. 2021, 36, 3425–3428. [Google Scholar] [CrossRef]
- Crona, J.; Taieb, D.; Pacak, K. New Perspectives on Pheochromocytoma and Paraganglioma: Toward a Molecular Classification. Endocr. Rev. 2017, 38, 489–515. [Google Scholar] [CrossRef]
- Buffet, A.; Ben Aim, L.; Leboulleux, S.; Drui, D.; Vezzosi, D.; Libe, R.; Ajzenberg, C.; Bernardeschi, D.; Cariou, B.; Chabolle, F.; et al. Positive Impact of Genetic Test on the Management and Outcome of Patients with Paraganglioma and/or Pheochromocytoma. J. Clin. Endocrinol. Metab. 2019, 104, 1109–1118. [Google Scholar] [CrossRef]
- Glasker, S.; Vergauwen, E.; Koch, C.A.; Kutikov, A.; Vortmeyer, A.O. Von Hippel-Lindau Disease: Current Challenges and Future Prospects. OncoTargets Ther. 2020, 13, 5669–5690. [Google Scholar] [CrossRef]
- Lenders, J.W.M.; Kerstens, M.N.; Amar, L.; Prejbisz, A.; Robledo, M.; Taieb, D.; Pacak, K.; Crona, J.; Zelinka, T.; Mannelli, M.; et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: A position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J. Hypertens. 2020, 38, 1443–1456. [Google Scholar] [CrossRef]
- Al-Sharefi, A.; Javaid, U.; Perros, P.; Ealing, J.; Truran, P.; Nag, S.; Kamaruddin, S.; Abouglila, K.; Cains, F.; Lewis, L.; et al. Clinical Presentation and Outcomes of Phaeochromocytomas/Paragangliomas in Neurofibromatosis Type 1. Eur. Endocrinol. 2019, 15, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Phay, J.E.; Yen, T.W.F.; Dickson, P.V.; Wang, T.S.; Garcia, R.; Yang, A.D.; Solorzano, C.C.; Kim, L.T. Update on Pheochromocytoma and Paraganglioma from the SSO Endocrine/Head and Neck Disease-Site Work Group. Part 1 of 2: Advances in Pathogenesis and Diagnosis of Pheochromocytoma and Paraganglioma. Ann. Surg. Oncol. 2020, 27, 1329–1337. [Google Scholar] [CrossRef]
- Hescot, S.; Curras-Freixes, M.; Deutschbein, T.; van Berkel, A.; Vezzosi, D.; Amar, L.; de la Fouchardière, C.; Valdes, N.; Riccardi, F.; Do Cao, C.; et al. Prognosis of Malignant Pheochromocytoma and Paraganglioma (MAPP-Prono Study): A European Network for the Study of Adrenal Tumors Retrospective Study. J. Clin. Endocrinol. Metab. 2019, 104, 2367–2374. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ramirez, M.; Feng, L.; Johnson, M.M.; Ejaz, S.; Habra, M.A.; Rich, T.; Busaidy, N.; Cote, G.J.; Perrier, N.; Phan, A.; et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: Primary tumor size and primary tumor location as prognostic indicators. J. Clin. Endocrinol. Metab. 2011, 96, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Turkova, H.; Prodanov, T.; Maly, M.; Martucci, V.; Adams, K.; Widimsky, J., Jr.; Chen, C.C.; Ling, A.; Kebebew, E.; Stratakis, C.A.; et al. Characteristics and outcomes of metastatic SDHB and sporadic pheochromocytoma/paraganglioma: An national institutes of health study. Endocr. Pract. 2016, 22, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, O.; Young, W.F., Jr.; Iniguez-Ariza, N.M.; Kittah, N.E.; Gruber, L.; Bancos, C.; Tamhane, S.; Bancos, I. Malignant Pheochromocytoma and Paraganglioma: 272 Patients Over 55 Years. J. Clin. Endocrinol. Metab. 2017, 102, 3296–3305. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, O.; Young, W.F., Jr.; Gruber, L.; Smestad, J.; Yan, Q.; Ponce, O.J.; Prokop, L.; Murad, M.H.; Bancos, I. Outcomes of patients with metastatic phaeochromocytoma and paraganglioma: A systematic review and meta-analysis. Clin. Endocrinol. 2017, 87, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Kohlenberg, J.; Welch, B.; Hamidi, O.; Callstrom, M.; Morris, J.; Sprung, J.; Bancos, I.; Young, W., Jr. Efficacy and Safety of Ablative Therapy in the Treatment of Patients with Metastatic Pheochromocytoma and Paraganglioma. Cancers 2019, 11, 195. [Google Scholar] [CrossRef]
- Deljou, A.; Kohlenberg, J.D.; Weingarten, T.N.; Bancos, I.; Young, W.F., Jr.; Schroeder, D.R.; Martin, D.P.; Sprung, J. Hemodynamic instability during percutaneous ablation of extra-adrenal metastases of pheochromocytoma and paragangliomas: A case series. BMC Anesthesiol. 2018, 18, 158. [Google Scholar] [CrossRef] [PubMed]
- Breen, W.; Bancos, I.; Young, W.F., Jr.; Bible, K.C.; Laack, N.N.; Foote, R.L.; Hallemeier, C.L. External beam radiation therapy for advanced/unresectable malignant paraganglioma and pheochromocytoma. Adv. Radiat. Oncol. 2018, 3, 25–29. [Google Scholar] [CrossRef]
- Roman-Gonzalez, A.; Zhou, S.; Ayala-Ramirez, M.; Shen, C.; Waguespack, S.G.; Habra, M.A.; Karam, J.A.; Perrier, N.; Wood, C.G.; Jimenez, C. Impact of Surgical Resection of the Primary Tumor on Overall Survival in Patients with Metastatic Pheochromocytoma or Sympathetic Paraganglioma. Ann. Surg. 2018, 268, 172–178. [Google Scholar] [CrossRef]
- Ellis, R.J.; Patel, D.; Prodanov, T.; Sadowski, S.; Nilubol, N.; Adams, K.; Steinberg, S.M.; Pacak, K.; Kebebew, E. Response after surgical resection of metastatic pheochromocytoma and paraganglioma: Can postoperative biochemical remission be predicted? J. Am. Coll. Surg. 2013, 217, 489–496. [Google Scholar] [CrossRef]
- Rafat, C.; Zinzindohoue, F.; Hernigou, A.; Hignette, C.; Favier, J.; Tenenbaum, F.; Gimenez-Roqueplo, A.P.; Plouin, P.F.; Amar, L. Peritoneal implantation of pheochromocytoma following tumor capsule rupture during surgery. J. Clin. Endocrinol. Metab. 2014, 99, E2681–E2685. [Google Scholar] [CrossRef]
- De Filpo, G.; Maggi, M.; Mannelli, M.; Canu, L. Management and outcome of metastatic pheochromocytomas/paragangliomas: An overview. J. Endocrinol. Investig. 2021, 44, 15–25. [Google Scholar] [CrossRef]
- Ayala-Ramirez, M.; Palmer, J.L.; Hofmann, M.C.; de la Cruz, M.; Moon, B.S.; Waguespack, S.G.; Habra, M.A.; Jimenez, C. Bone metastases and skeletal-related events in patients with malignant pheochromocytoma and sympathetic paraganglioma. J. Clin. Endocrinol. Metab. 2013, 98, 1492–1497. [Google Scholar] [CrossRef]
- Ilanchezhian, M.; Jha, A.; Pacak, K.; Del Rivero, J. Emerging Treatments for Advanced/Metastatic Pheochromocytoma and Paraganglioma. Curr. Treat. Options Oncol. 2020, 21, 85. [Google Scholar] [CrossRef] [PubMed]
- Loh, K.C.; Fitzgerald, P.A.; Matthay, K.K.; Yeo, P.P.; Price, D.C. The treatment of malignant pheochromocytoma with iodine-131 metaiodobenzylguanidine (131I-MIBG): A comprehensive review of 116 reported patients. J. Endocrinol. Investig. 1997, 20, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Noto, R.B.; Pryma, D.A.; Jensen, J.; Lin, T.; Stambler, N.; Strack, T.; Wong, V.; Goldsmith, S.J. Phase 1 Study of High-Specific-Activity I-131 MIBG for Metastatic and/or Recurrent Pheochromocytoma or Paraganglioma. J. Clin. Endocrinol. Metab. 2018, 103, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, M.P.; Kane, A.; Zhu, J.; Morse, M.A.; Wong, T.; Borges-Neto, S. Long-Term Outcomes of 125 Patients with Metastatic Pheochromocytoma or Paraganglioma Treated with 131-I MIBG. J. Clin. Endocrinol. Metab. 2020, 105, e494–e501. [Google Scholar] [CrossRef] [PubMed]
- Zandee, W.T.; Feelders, R.A.; Smit Duijzentkunst, D.A.; Hofland, J.; Metselaar, R.M.; Oldenburg, R.A.; van Linge, A.; Kam, B.L.R.; Teunissen, J.J.M.; Korpershoek, E.; et al. Treatment of inoperable or metastatic paragangliomas and pheochromocytomas with peptide receptor radionuclide therapy using 177Lu-DOTATATE. Eur. J. Endocrinol. 2019, 181, 45–53. [Google Scholar] [CrossRef]
- Vyakaranam, A.R.; Crona, J.; Norlen, O.; Granberg, D.; Garske-Roman, U.; Sandstrom, M.; Fross-Baron, K.; Thiis-Evensen, E.; Hellman, P.; Sundin, A. Favorable Outcome in Patients with Pheochromocytoma and Paraganglioma Treated with (177) Lu-DOTATATE. Cancers 2019, 11, 909. [Google Scholar] [CrossRef]
- Satapathy, S.; Mittal, B.R.; Bhansali, A. Peptide receptor radionuclide therapy in the management of advanced pheochromocytoma and paraganglioma: A systematic review and meta-analysis. Clin. Endocrinol. 2019, 91, 718–727. [Google Scholar] [CrossRef]
- Jha, A.; Taïeb, D.; Carrasquillo, J.A.; Pryma, D.A.; Patel, M.; Millo, C.; de Herder, W.W.; Del Rivero, J.; Crona, J.; Shulkin, B.L.; et al. High-Specific-Activity-131I-MIBG versus 177Lu-DOTATATE Targeted Radionuclide Therapy for Metastatic Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2021, 27, 2989–2995. [Google Scholar] [CrossRef]
- Nastos, K.; Cheung, V.T.F.; Toumpanakis, C.; Navalkissoor, S.; Quigley, A.M.; Caplin, M.; Khoo, B. Peptide Receptor Radionuclide Treatment and (131)I-MIBG in the management of patients with metastatic/progressive phaeochromocytomas and paragangliomas. J. Surg. Oncol. 2017, 115, 425–434. [Google Scholar] [CrossRef]
- Taïeb, D.; Jha, A.; Treglia, G.; Pacak, K. Molecular imaging and radionuclide therapy of pheochromocytoma and paraganglioma in the era of genomic characterization of disease subgroups. Endocr. Relat. Cancer 2019, 26, R627–R652. [Google Scholar] [CrossRef]
- Huang, H.; Abraham, J.; Hung, E.; Averbuch, S.; Merino, M.; Steinberg, S.M.; Pacak, K.; Fojo, T. Treatment of malignant pheochromocytoma/paraganglioma with cyclophosphamide, vincristine, and dacarbazine: Recommendation from a 22-year follow-up of 18 patients. Cancer 2008, 113, 2020–2028. [Google Scholar] [CrossRef]
- Jawed, I.; Velarde, M.; Darr, R.; Wolf, K.I.; Adams, K.; Venkatesan, A.M.; Balasubramaniam, S.; Poruchynsky, M.S.; Reynolds, J.C.; Pacak, K.; et al. Continued Tumor Reduction of Metastatic Pheochromocytoma/Paraganglioma Harboring Succinate Dehydrogenase Subunit B Mutations with Cyclical Chemotherapy. Cell. Mol. Neurobiol. 2018, 38, 1099–1106. [Google Scholar] [CrossRef]
- Hadoux, J.; Favier, J.; Scoazec, J.Y.; Leboulleux, S.; Al Ghuzlan, A.; Caramella, C.; Deandreis, D.; Borget, I.; Loriot, C.; Chougnet, C.; et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int. J. Cancer 2014, 135, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- O’Kane, G.M.; Ezzat, S.; Joshua, A.M.; Bourdeau, I.; Leibowitz-Amit, R.; Olney, H.J.; Krzyzanowska, M.; Reuther, D.; Chin, S.; Wang, L.; et al. A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: The SNIPP trial. Br. J. Cancer 2019, 120, 1113–1119. [Google Scholar] [CrossRef]
- Oh, D.Y.; Kim, T.W.; Park, Y.S.; Shin, S.J.; Shin, S.H.; Song, E.K.; Lee, H.J.; Lee, K.W.; Bang, Y.J. Phase 2 study of everolimus monotherapy in patients with nonfunctioning neuroendocrine tumors or pheochromocytomas/paragangliomas. Cancer 2012, 118, 6162–6170. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, C.; Subbiah, V.; Stephen, B.; Ma, J.; Milton, D.; Xu, M.; Zarifa, A.; Akhmedzhanov, F.O.; Tsimberidou, A.; Habra, M.A.; et al. Phase II Clinical Trial of Pembrolizumab in Patients with Progressive Metastatic Pheochromocytomas and Paragangliomas. Cancers 2020, 12, 2307. [Google Scholar] [CrossRef] [PubMed]
- Gruber, L.M.; Young, W.F., Jr.; Bancos, I. Pheochromocytoma and Paraganglioma in Pregnancy: A New Era. Curr. Cardiol. Rep. 2021, 23, 60. [Google Scholar] [CrossRef]
- Bancos, I.; Atkinson, E.; Eng, C.; Young, W.F., Jr.; Neumann, H.P.H.; International, P.; Pregnancy Study, G. Maternal and fetal outcomes in phaeochromocytoma and pregnancy: A multicentre retrospective cohort study and systematic review of literature. Lancet Diabetes Endocrinol. 2021, 9, 13–21. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
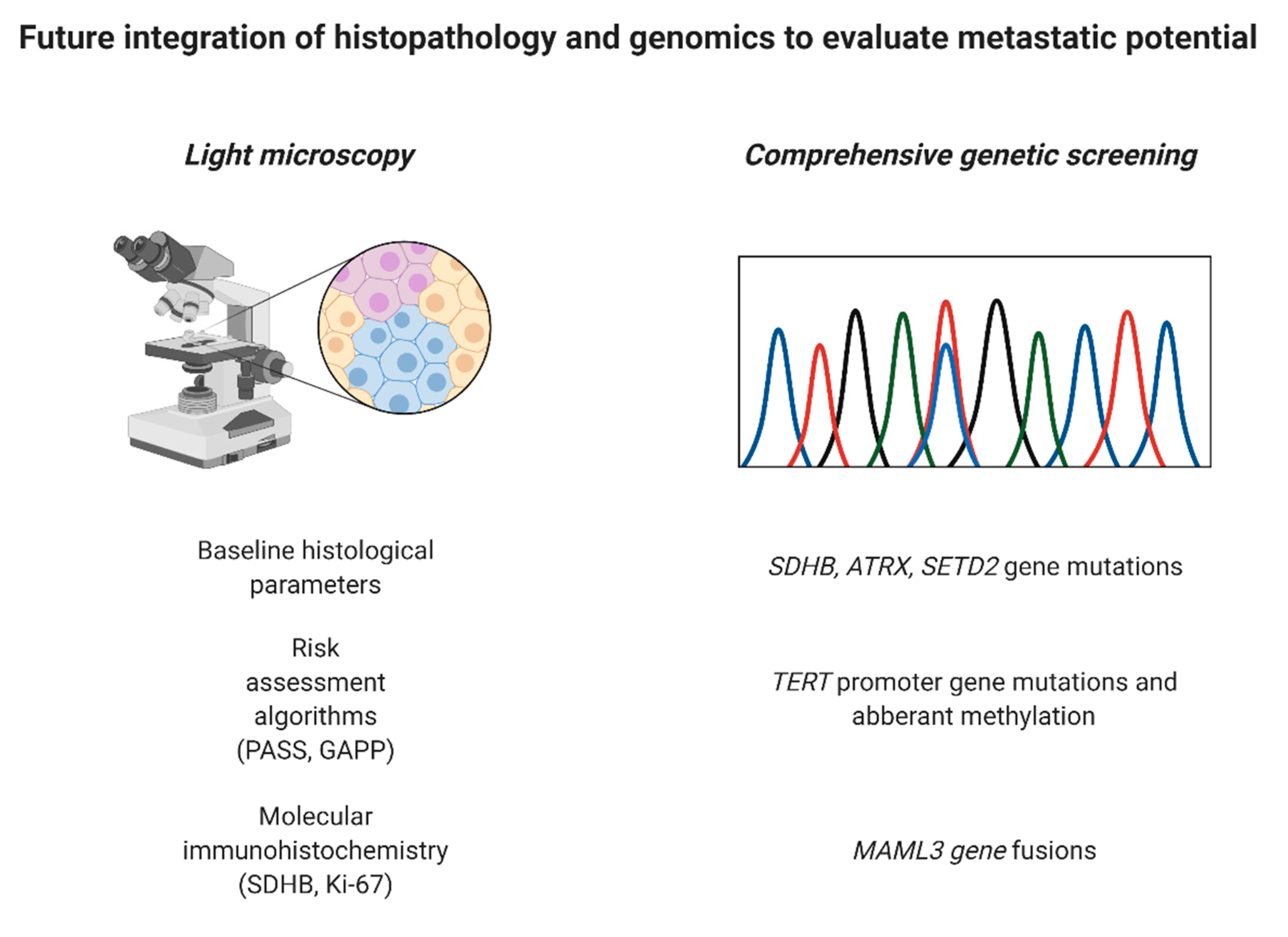{kind=link}
{kind=link}
| Class of Medication | Medication Name | Approach to Titration | Dosing | Monitoring/Goals of Therapy | Side Effects and Counseling |
|---|---|---|---|---|---|
| Alpha-adrenergic blockade | Phenoxybenzamine Doxazosin | Start at least 10–14 days prior to procedure. Titrate daily based on orthostatic blood pressure. | Starting dose: usually 10 mg once or twice daily, gradually increased. Final dose varies (60–120 mg total daily dose in divided doses). Starting dose: usually 1 mg once or twice daily, or 4 mg once daily, gradually increased. Final dose varies (6–40 mg) * total daily dose in divided doses. | Monitoring includes: daily orthostatic vitals, side effects. The goal is low normal blood pressure. | Fatigue, lightheadedness, tachycardia, nasal congestion, diarrhea Counseling: Optimal hydration Increase salt intake. Avoid driving if lightheaded. |
| Beta-adrenergic blockade | Propranolol Metoprolol succinate Atenolol | Start 3–7 days prior to procedure. Start after alpha-adrenergic blockade. Titrate daily based on heart rate. | Starting dose: 10 mg every 6–8 h, gradually increased. Final dose varies (30-90 mg total daily dose). Starting dose: 25 mg daily. Final dose varies (50–200 mg) daily in divided doses. Starting dose: 25 mg daily. Final dose varies. | Absence of tachycardia, with a baseline heart rate <80–90 beats/minute | Usually none if started after alpha-adrenergic blockade and close monitoring as well as treatment of short duration. |
| Calcium channel blockade | Amlodipine | Usually used as an additive agent when blood pressure is uncontrolled with alpha- and beta-blockade. | Starting dose: 5 mg, increase to 10 mg if needed. | Monitoring includes blood pressure measurements. | Usually none with close monitoring and treatment of short duration. |
| Catecholamine synthesis inhibitor | Metyrosine | Usually used when inadequate or intolerant to alpha blockade, when difficult resection is anticipated. Titrated based on the Mayo Clinic protocol. Day 1: 250 mg every 6 h Day 2: 500 mg every 6 h Day 3: 500 mg every 6 h Day 4: 750 mg every 6 h Day 5: 1000 mg every 6 h, last dose of 1000 mg on the morning of procedure | Monitor for side effects | Fatigue Sedation Dizziness Depressed mood Diarrhea, anorexia Extrapyramidal side effects Counseling: Optimal hydration. Avoid driving. Contact physician if extra-pyramidal side effects occur. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calissendorff, J.; Juhlin, C.C.; Bancos, I.; Falhammar, H. Pheochromocytomas and Abdominal Paragangliomas: A Practical Guidance. Cancers 2022, 14, 917. https://doi.org/10.3390/cancers14040917
Calissendorff J, Juhlin CC, Bancos I, Falhammar H. Pheochromocytomas and Abdominal Paragangliomas: A Practical Guidance. Cancers. 2022; 14(4):917. https://doi.org/10.3390/cancers14040917
Chicago/Turabian StyleCalissendorff, Jan, Carl Christofer Juhlin, Irina Bancos, and Henrik Falhammar. 2022. "Pheochromocytomas and Abdominal Paragangliomas: A Practical Guidance" Cancers 14, no. 4: 917. https://doi.org/10.3390/cancers14040917
APA StyleCalissendorff, J., Juhlin, C. C., Bancos, I., & Falhammar, H. (2022). Pheochromocytomas and Abdominal Paragangliomas: A Practical Guidance. Cancers, 14(4), 917. https://doi.org/10.3390/cancers14040917