
Current Limitations and Novel Perspectives in Pancreatic Cancer Treatment


Abstract
:Simple Summary
Abstract
1. Introduction
2. PDAC Standard-of-Care Treatments
2.1. Surgery
2.2. Neoadjuvant Chemotherapy
2.3. Adjuvant Chemotherapy for Resected Pancreatic Cancer
2.4. Treatment of Locally Advanced and Metastatic Disease
3. PDAC TME, Immunotherapies and Therapy Resistance: What to Target Next?
3.1. Pancreatic Cancer Cells
3.2. PDAC Stroma
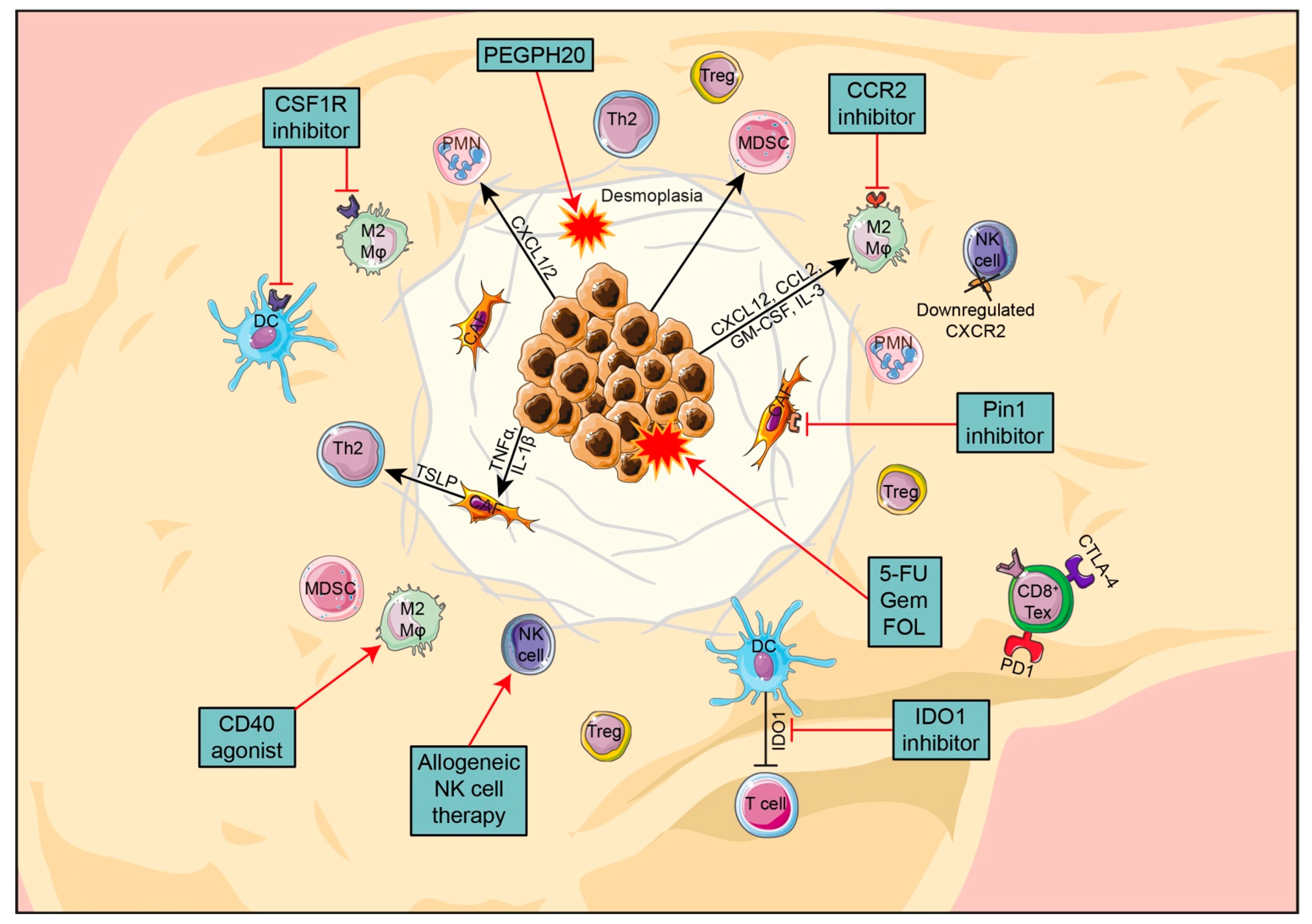
3.3. Treatments Targeting PDAC Stroma
3.4. The Immune Compartment in PDAC
3.4.1. Dendritic Cells
3.4.2. Macrophages
3.4.3. Neutrophils
3.4.4. Myeloid-Derived Suppressor Cells
3.4.5. Natural Killer Cells
3.4.6. Adaptive Immune Cells
3.4.7. CD4+ T Cells
3.4.8. CD8+ T Cells
3.5. Immunotherapy
3.6. Cancer Cell-Based Vaccines
4. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Strobel, O.; Neoptolemos, J.; Jäger, D.; Büchler, M.W. Optimizing the outcomes of pancreatic cancer surgery. Nat. Rev. Clin. Oncol. 2019, 16, 11–26. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, C.L.; Herman, J.M.; Laheru, D.A.; Klein, A.P.; Erdek, M.A.; Fishman, E.K.; Hruban, R.H. Recent progress in pancreatic cancer. CA Cancer J. Clin. 2013, 63, 318–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic Adenocarcinoma. N. Engl. J. Med. 2014, 371, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Lowenfels, A.B. The epidemiology of pancreatitis and pancreatic cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [Green Version]
- Genkinger, J.M.; Spiegelman, D.; Anderson, K.E.; Bergkvist, L.; Bernstein, L.; van den Brandt, P.A.; English, D.R.; Freudenheim, J.L.; Fuchs, C.S.; Giles, G.G.; et al. Alcohol Intake and Pancreatic Cancer Risk: A Pooled Analysis of Fourteen Cohort Studies. Cancer Epidemiol. Prev. Biomark. 2009, 18, 765–776. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic cancer. Lancet 2004, 363, 1049–1057. [Google Scholar] [CrossRef]
- Henley, S.J.; Ward, E.M.; Scott, S.; Ma, J.; Anderson, R.N.; Firth, A.U.; Thomas, C.C.; Islami, F.; Weir, H.K.; Lewis, D.R.; et al. Annual report to the nation on the status of cancer, part I: National cancer statistics. Cancer 2020, 126, 2225–2249. [Google Scholar] [CrossRef]
- Hruban, R.H.; Maitra, A.; Goggins, M. Pancreatic Intraepithelial Neoplasia. Definitions 2020, 28, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Takaori, K. Current understanding of precursors to pancreatic cancer. J. Hepatobiliary Pancreat. Surg. 2007, 14, 217–223. [Google Scholar] [CrossRef]
- Biankin, A.V.; Maitra, A. Subtyping Pancreatic Cancer. Cancer Cell 2015, 28, 411–413. [Google Scholar] [CrossRef] [Green Version]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Edge, S.B.; Byrd, D.R.; Carducci, M.A.; Compton, C.C.; Fritz, A.G.; Greene, F.L. AJCC Cancer Staging Manual; Springer: New York, NY, USA, 2010; Volume 649. [Google Scholar]
- Bilimoria, K.Y.; Bentrem, D.J.; Ko, C.Y.; Ritchey, J.; Stewart, A.K.; Winchester, D.P.; Talamonti, M.S. Validation of the 6th edition AJCC pancreatic cancer staging system: Report from the National Cancer Database. Cancer 2007, 110, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Lopez, N.E.; Prendergast, C.; Lowy, A.M. Borderline resectable pancreatic cancer: Definitions and management. World J. Gastroenterol. 2014, 20, 10740–10751. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network. Practice Guidelines in Oncology for Pancreatic Adenocarcinoma; Version 2; National Comprehensive Cancer Network: Jenkintown, PA, USA, 2021. [Google Scholar]
- Esposito, I.; Kleeff, J.J.; Bergmann, F.; Reiser, C.; Herpel, E.; Friess, H.; Schirmacher, P.; Büchlerbüchler, M.W. Most Pancreatic Cancer Resections are R1 Resections. Ann. Surg. Oncol. 2008, 15, 1651–1660. [Google Scholar] [CrossRef] [PubMed]
- Ferrone, C.R.; Marchegiani, G.; Hong, T.S.; Ryan, D.P.; Deshpande, V.; McDonnell, E.I.; Sabbatino, F.; Santos, D.D.; Allen, J.N.; Blaszkowsky, L.S.; et al. Radiological and surgical implications of neoadjuvant treatment with FOLFIRINOX for locally advanced and borderline resectable pancreatic cancer. Ann. Surg. 2015, 261, 12–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alliance for Clinical Trials in Oncology. Testing the Use of the Usual Chemotherapy before and after Surgery for Removable Pancreatic Cancer; Alliance for Clinical Trials in Oncology: Boston, MA, USA, 2020. [Google Scholar]
- Fatima, J. Pancreatoduodenectomy for Ductal Adenocarcinoma. Arch. Surg. 2010, 145, 167. [Google Scholar] [CrossRef] [Green Version]
- Delpero, J.R.; Boher, J.M.; Sauvanet, A.; Le Treut, Y.P.; Sa-Cunha, A.; Mabrut, J.Y.; Chiche, L.; Turrini, O.; Bachellier, P.; Paye, F. Pancreatic Adenocarcinoma with Venous Involvement: Is Up-Front Synchronous Portal-Superior Mesenteric Vein Resection Still Justified? A Survey of the Association Française de Chirurgie. Ann. Surg. Oncol. 2015, 22, 1874–1883. [Google Scholar] [CrossRef]
- Petrelli, F.; Coinu, A.; Borgonovo, K.; Cabiddu, M.; Ghilardi, M.; Lonati, V.; Aitini, E.; Barni, S. FOLFIRINOX-based neoadjuvant therapy in borderline resectable or unresectable pancreatic cancer: A meta-analytical review of published studies. Pancreas 2015, 44, 515–521. [Google Scholar] [CrossRef]
- Versteijne, E.; Vogel, J.A.; Besselink, M.G.; Busch, O.R.C.; Wilmink, J.W.; Daams, J.G.; van Eijck, C.H.J.; Groot Koerkamp, B.; Rasch, C.R.N.; van Tienhoven, G.; et al. Meta-analysis comparing upfront surgery with neoadjuvant treatment in patients with resectable or borderline resectable pancreatic cancer. Br. J. Surg. 2018, 105, 946–958. [Google Scholar] [CrossRef] [Green Version]
- Neoptolemos, J.P.; Stocken, D.D.; Friess, H.; Bassi, C.; Dunn, J.A.; Hickey, H.; Beger, H.; Fernandez-Cruz, L.; Dervenis, C.; Lacaine, F.; et al. A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N. Engl. J. Med. 2004, 350, 1200–1210. [Google Scholar] [CrossRef] [Green Version]
- Oettle, H.; Post, S.; Neuhaus, P. Vs Observation in Patients Undergoing Curative-Intent Resection of Pancreatic Cancer. JAMA 2007, 297, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Oettle, H.; Neuhaus, P.; Hochhaus, A.; Hartmann, J.T.; Gellert, K.; Ridwelski, K.; Niedergethmann, M.; Zülke, C.; Fahlke, J.; Arning, M.B.; et al. Adjuvant chemotherapy with gemcitabine and long-term outcomes among patients with resected pancreatic cancer: The CONKO-001 randomized trial. JAMA J. Am. Med. Assoc. 2013, 310, 1473–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C. Adjuvant Chemotherapy with Fluorouracil Plus Folinic Acid vs. Gemcitabine Following Pancreatic Cancer Resection. JAMA J. Am. Med. Assoc. 2010, 304, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Turpin, A.; Belletier, C.; Wei, A.; Mitry, E.; Lopez, A.; Francois, E.; Artru, P.; Biagi, J.; et al. LBA57 Unicancer PRODIGE 24/CCTG PA6 trial: Updated results of a multicenter international randomized phase III trial of adjuvant mFOLFIRINOX (mFFX) versus gemcitabine (gem) in patients (pts) with resected pancreatic ductal adenocarcinomas (PDAC). Ann. Oncol. 2021, 32, S1334. [Google Scholar] [CrossRef]
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Behrman, S.W.; Benson, A.B.; Cardin, D.B.; Chiorean, E.G.; Chung, V.; Czito, B.; Del Chiaro, M.; et al. Pancreatic Adenocarcinoma, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 439–457. [Google Scholar] [CrossRef] [PubMed]
- Loehrer Sr, P.J.; Feng, Y.; Cardenes, H.; Wagner, L.; Brell, J.M.; Cella, D.; Flynn, P.; Ramanathan, R.K.; Crane, C.H.; Alberts, S.R.; et al. Gemcitabine Alone Versus Gemcitabine Plus Radiotherapy in Patients with Locally Advanced Pancreatic Cancer: An Eastern Cooperative Oncology Group Trial. J. Clin. Oncol. 2011, 29, 4105–4112. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Wee Ma, W.; Saleh, N.; et al. Increased Survival in Pancreatic Cancer with nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 18, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, D.; El-Maraghi, R.H.; Hammel, P.; Heinemann, V.; Kunzmann, V.; Sastre, J.; Scheithauer, W.; Siena, S.; Tabernero, J.; Teixeira, L.; et al. nab-Paclitaxel Plus Gemcitabine for Metastatic Pancreatic Cancer: Long-Term Survival From a Phase III Trial. JNCI J. Natl. Cancer Inst. 2015, 107, dju413. [Google Scholar] [CrossRef]
- Cho, I.R.; Kang, H.; Jo, J.H.; Lee, H.S.; Chung, M.J.; Park, J.Y.; Park, S.W.; Song, S.Y.; An, C.; Park, M.-S.; et al. FOLFIRINOX vs gemcitabine/nab-paclitaxel for treatment of metastatic pancreatic cancer: Single-center cohort study. World J. Gastrointest. Oncol. 2020, 12, 182–194. [Google Scholar] [CrossRef]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, M.K.; Kapoor, A.; Bagri, P.K.; Narayan, S.; Singh, D.; Nirban, R.K.; Singh, G.; Maharia, S.; Kumari, P.; Jakhar, S.L.; et al. A Phase III Trial Comparing Folfirinox Versus Gemcitabine for Metastatic Pancreatic Cancer. Ann. Oncol. 2014, 25, iv210. [Google Scholar] [CrossRef]
- Cunningham, D.; Chau, I.; Stocken, D.D.; Valle, J.W.; Smith, D.; Steward, W.; Harper, P.G.; Dunn, J.; Tudur-Smith, C.; West, J.; et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J. Clin. Oncol. 2009, 27, 5513–5518. [Google Scholar] [CrossRef]
- Li, Q.; Yan, H.; Liu, W.; Zhen, H.; Yang, Y.; Cao, B. Efficacy and Safety of Gemcitabine-Fluorouracil Combination Therapy in the Management of Advanced Pancreatic Cancer: A Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2014, 9, e104346. [Google Scholar] [CrossRef]
- Heinemann, V.; Boeck, S.; Hinke, A.; Labianca, R.; Louvet, C. Meta-analysis of randomized trials: Evaluation of benefit from gemcitabine-based combination chemotherapy applied in advanced pancreatic cancer. BMC Cancer 2008, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Shelemey, P.T.; Amaro, C.P.; Ng, D.; Falck, V.; Tam, V.C. Metastatic pancreatic cancer with complete response to FOLFIRINOX treatment. BMJ Case Rep. 2021, 14, e238395. [Google Scholar] [CrossRef]
- Ryschich, E.; Nötzel, T.; Hinz, U.; Autschbach, F.; Ferguson, J.; Simon, I.; Weitz, J.; Fröhlich, B.; Klar, E.; Büchler, M.W.; et al. Control of T-Cell-Mediated Immune Response by HLA Class I in Human Pancreatic Carcinoma. Clin. Cancer Res. 2005, 11, 498–504. [Google Scholar]
- Pandha, H.; Rigg, A.; John, J.; Lemoine, N. Loss of expression of antigen-presenting molecules in human pancreatic cancer and pancreatic cancer cell lines. Clin. Exp. Immunol. 2007, 148, 127–135. [Google Scholar] [CrossRef]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory Monocyte Mobilization Decreases Patient Survival in Pancreatic Cancer: A Role for Targeting the CCL2/CCR2 Axis. Clin. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef] [Green Version]
- Tan, M.C.B.; Goedegebuure, P.S.; Belt, B.A.; Flaherty, B.; Sankpal, N.; Gillanders, W.E.; Eberlein, T.J.; Hsieh, C.-S.; Linehan, D.C. Disruption of CCR5-Dependent Homing of Regulatory T Cells Inhibits Tumor Growth in a Murine Model of Pancreatic Cancer. J. Immunol. 2009, 182, 1746–1755. [Google Scholar] [CrossRef] [PubMed]
- Basso, D.; Fogar, P.; Falconi, M.; Fadi, E.; Sperti, C.; Frasson, C.; Greco, E.; Tamburrino, D.; Teolato, S.; Moz, S.; et al. Pancreatic Tumors and Immature Immunosuppressive Myeloid Cells in Blood and Spleen: Role of Inhibitory Co-Stimulatory Molecules PDL1 and CTLA4. An In Vivo and In Vitro Study. PLoS ONE 2013, 8, e54824. [Google Scholar] [CrossRef] [PubMed]
- Uyttenhove, C.; Pilotte, L.; Théate, I.; Stroobant, V.; Colau, D.; Parmentier, N.; Boon, T.; Van den Eynde, B.J. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat. Med. 2003, 9, 1269–1274. [Google Scholar] [CrossRef]
- Witkiewicz, A.; Williams, T.K.; Cozzitorto, J.; Durkan, B.; Showalter, S.L.; Yeo, C.J.; Brody, J.R. Expression of Indoleamine 2,3-Dioxygenase in Metastatic Pancreatic Ductal Adenocarcinoma Recruits Regulatory T Cells to Avoid Immune Detection. J. Am. Coll. Surg. 2008, 206, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; You, S.; McGrath, B.C.; Cavener, D.R.; Vacca, C.; Orabona, C.; Bianchi, R.; Belladonna, M.L.; Volpi, C.; et al. The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells. J. Immunol. 2006, 176, 6752–6761. [Google Scholar] [CrossRef]
- Mahadevan, D.; Von Hoff, D.D. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther. 2007, 6, 1186–1197. [Google Scholar] [CrossRef] [Green Version]
- Masamune, A.; Shimosegawa, T. Signal transduction in pancreatic stellate cells. J. Gastroenterol. 2009, 44, 249–260. [Google Scholar] [CrossRef]
- Neesse, A.; Algül, H.; Tuveson, D.A.; Gress, T.M. Stromal biology and therapy in pancreatic cancer: A changing paradigm. Gut 2015, 64, 1476–1484. [Google Scholar] [CrossRef] [Green Version]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [Green Version]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, P.; Basu, G.D.; Tinder, T.L.; Subramani, D.B.; Bradley, J.M.; Arefayene, M.; Skaar, T.; De Petris, G. Progression of Pancreatic Adenocarcinoma Is Significantly Impeded with a Combination of Vaccine and COX-2 Inhibition. J. Immunol. 2009, 182, 216–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Infante, J.R.; Matsubayashi, H.; Sato, N.; Tonascia, J.; Klein, A.P.; Riall, T.A.; Yeo, C.; Iacobuzio-Donahue, C.; Goggins, M. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J. Clin. Oncol. 2007, 25, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Biffi, G.; Oni, T.E.; Spielman, B.; Hao, Y.; Elyada, E.; Park, Y.; Preall, J.; Tuveson, D.A. IL1-Induced JAK/STAT Signaling Is Antagonized by TGFβ to Shape CAF Heterogeneity in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2019, 9, 282–301. [Google Scholar] [CrossRef] [Green Version]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Bullock, A.J.; Seery, T.E.; Zheng, L.; Sigal, D.; Ritch, P.S.; Braiteh, F.S.; Zalupski, M.; Bahary, N.; Harris, W.P.; et al. Randomized phase II study of PEGPH20 plus nab-paclitaxel/gemcitabine (PAG) vs AG in patients (Pts) with untreated, metastatic pancreatic ductal adenocarcinoma (mPDA). J. Clin. Oncol. 2017, 35, 4008. [Google Scholar] [CrossRef]
- Tempero, M.A.; Van Cutsem, E.; Sigal, D.; Oh, D.-Y.; Fazio, N.; Macarulla, T.; Hitre, E.; Hammel, P.; Hendifar, A.E.; Bates, S.E.; et al. HALO 109-301: A randomized, double-blind, placebo-controlled, phase 3 study of pegvorhyaluronidase alfa (PEGPH20) + nab-paclitaxel/gemcitabine (AG) in patients (pts) with previously untreated hyaluronan (HA)-high metastatic pancreatic ductal adenocarcinom. J. Clin. Oncol. 2020, 38, 638. [Google Scholar] [CrossRef]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef] [Green Version]
- Padoan, A.; Plebani, M.; Basso, D. Inflammation and Pancreatic Cancer: Focus on Metabolism, Cytokines, and Immunity. Int. J. Mol. Sci. 2019, 20, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanţău, A.; Leucuţa, D.-C.; Tanţău, M.; Boţan, E.; Zaharie, R.; Mândruţiu, A.; Tomuleasa, I.-C. Inflammation, Tumoral Markers and Interleukin-17, -10, and -6 Profiles in Pancreatic Adenocarcinoma and Chronic Pancreatitis. Dig. Dis. Sci. 2020, 66, 3427–3438. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Maitra, A.; Ying, H. Recent insights into the biology of pancreatic cancer. EBioMedicine 2020, 53, 102655. [Google Scholar] [CrossRef] [PubMed]
- Saka, D.; Gökalp, M.; Piyade, B.; Cevik, N.C.; Arik Sever, E.; Unutmaz, D.; Ceyhan, G.O.; Demir, I.E.; Asimgil, H. Mechanisms of T-Cell Exhaustion in Pancreatic Cancer. Cancers 2020, 12, 2274. [Google Scholar] [CrossRef]
- Roshani, R.; McCarthy, F.; Hagemann, T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 2014, 345, 157–163. [Google Scholar] [CrossRef]
- Lei, X.; Lei, Y.; Li, J.-K.; Du, W.-X.; Li, R.-G.; Yang, J.; Li, J.; Li, F.; Tan, H.-B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef]
- Jang, J.E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef] [Green Version]
- Bellone, G.; Carbone, A.; Smirne, C.; Scirelli, T.; Buffolino, A.; Novarino, A.; Stacchini, A.; Bertetto, O.; Palestro, G.; Sorio, C.; et al. Cooperative Induction of a Tolerogenic Dendritic Cell Phenotype by Cytokines Secreted by Pancreatic Carcinoma Cells. J. Immunol. 2006, 177, 3448–3460. [Google Scholar] [CrossRef]
- Bharadwaj, U.; Li, M.; Zhang, R.; Chen, C.; Yao, Q. Elevated interleukin-6 and G-CSF in human pancreatic cancer cell conditioned medium suppress dendritic cell differentiation and activation. Cancer Res. 2007, 67, 5479–5488. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Santoni, M.; Bracarda, S.; Nabissi, M.; Massari, F.; Conti, A.; Bria, E.; Tortora, G.; Santoni, G.; Cascinu, S. CXC and CC Chemokines as Angiogenic Modulators in Nonhaematological Tumors. Biomed Res. Int. 2014, 2014, 768758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.-W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338. [Google Scholar] [CrossRef]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Penny, H.L.; Sieow, J.L.; Adriani, G.; Yeap, W.H.; See Chi Ee, P.; San Luis, B.; Lee, B.; Lee, T.; Mak, S.Y.; Ho, Y.S.; et al. Warburg metabolism in tumor-conditioned macrophages promotes metastasis in human pancreatic ductal adenocarcinoma. Oncoimmunology 2016, 5, e1191731. [Google Scholar] [CrossRef] [PubMed]
- Kurahara, H.; Shinchi, H.; Mataki, Y.; Maemura, K.; Noma, H.; Kubo, F.; Sakoda, M.; Ueno, S.; Natsugoe, S.; Takao, S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J. Surg. Res. 2011, 167, e211–e219. [Google Scholar] [CrossRef]
- Zeng, L.; Guo, Y.; Liang, J.; Chen, S.; Peng, P.; Zhang, Q.; Su, H.; Chen, Y.; Huang, K. Perineural invasion and TAMs in pancreatic ductal adenocarcinomas: Review of the original pathology reports using immunohistochemical enhancement and relationships with clinicopathological features. J. Cancer 2014, 5, 754–760. [Google Scholar] [CrossRef]
- Kurahara, H.; Takao, S.; Maemura, K.; Mataki, Y.; Kuwahata, T.; Maeda, K.; Sakoda, M.; Iino, S.; Ishigami, S.; Ueno, S.; et al. M2-Polarized tumor-associated macrophage infiltration of regional lymph nodes is associated with nodal lymphangiogenesis and occult nodal involvement in pn0 pancreatic cancer. Pancreas 2013, 42, 155–159. [Google Scholar] [CrossRef]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef] [Green Version]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2+ neutrophils and CCR2+ macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F. Neutrophil Degranulation, Plasticity, and Cancer Metastasis. Trends Immunol. 2019, 40, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, R.; Kawada, K.; Itatani, Y.; Ogawa, R.; Kiyasu, Y.; Sakai, Y. The Role of Tumor-Associated Neutrophils in Colorectal Cancer. Int. J. Mol. Sci. 2019, 20, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masucci, M.T.; Minopoli, M.; Carriero, M.V. Tumor Associated Neutrophils. Their Role in Tumorigenesis, Metastasis, Prognosis and Therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felix, K.; Gaida, M.M. Neutrophil-Derived Proteases in the Microenvironment of Pancreatic Cancer-Active Players in Tumor Progression. Int. J. Biol. Sci. 2016, 12, 302–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Fang, T.; Huang, L.; Wang, H.; Zhang, L.; Wang, Z.; Cui, Y. Neutrophils infiltrating pancreatic ductal adenocarcinoma indicate higher malignancy and worse prognosis. Biochem. Biophys. Res. Commun. 2018, 501, 313–319. [Google Scholar] [CrossRef]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [Green Version]
- Takesue, S.; Ohuchida, K.; Shinkawa, T.; Otsubo, Y.; Matsumoto, S.; Sagara, A.; Yonenaga, A.; Ando, Y.; Kibe, S.; Nakayama, H.; et al. Neutrophil extracellular traps promote liver micrometastasis in pancreatic ductal adenocarcinoma via the activation of cancer-associated fibroblasts. Int. J. Oncol. 2020, 56, 596–605. [Google Scholar] [CrossRef]
- Thyagarajan, A.; Alshehri, M.S.A.; Miller, K.L.R.; Sherwin, C.M.; Travers, J.B.; Sahu, R.P. Myeloid-Derived Suppressor Cells and Pancreatic Cancer: Implications in Novel Therapeutic Approaches. Cancers 2019, 11, 1627. [Google Scholar] [CrossRef] [Green Version]
- Stromnes, I.M.; Brockenbrough, J.S.; Izeradjene, K.; Carlson, M.A.; Cuevas, C.; Simmons, R.M.; Greenberg, P.D.; Hingorani, S.R. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut 2014, 63, 1769–1781. [Google Scholar] [CrossRef] [Green Version]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Mace, T.A.; Bloomston, M.; Lesinski, G.B. Pancreatic cancer-associated stellate cells: A viable target for reducing immunosuppression in the tumor microenvironment. Oncoimmunology 2013, 2, e24891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinton, L.; Solito, S.; Damuzzo, V.; Francescato, S.; Pozzuoli, A.; Berizzi, A.; Mocellin, S.; Rossi, C.R.; Bronte, V.; Mandruzzato, S. Activated T cells sustain myeloid-derived suppressor cellmediated immune suppression. Oncotarget 2016, 7, 1168–1184. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.; Pan, P.Y.; Li, Q.; Sato, A.I.; Levy, D.E.; Bromberg, J.; Divino, C.M.; Chen, S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006, 66, 1123–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Velez-Delgado, A.; Mathew, E.; Li, D.; Mendez, F.M.; Flannagan, K.; Rhim, A.D.; Simeone, D.M.; Beatty, G.L.; Di Magliano, M.P. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut 2017, 66, 124–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Yu, S.; Kappes, J.; Wang, J.; Grizzle, W.E.; Zinn, K.R.; Zhang, H.G. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood 2007, 109, 4336–4342. [Google Scholar] [CrossRef]
- Sinha, P.; Clements, V.K.; Bunt, S.K.; Albelda, S.M.; Ostrand-Rosenberg, S. Cross-Talk between Myeloid-Derived Suppressor Cells and Macrophages Subverts Tumor Immunity toward a Type 2 Response. J. Immunol. 2007, 179, 977–983. [Google Scholar] [CrossRef]
- Chiossone, L.; Dumas, P.-Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef]
- Lanier, L.L.; Testi, R.; Bindl, J.; Phillips, J.H. Identity of Leu-19 (CD56) leukocyte differentiation antigen and neural cell adhesion molecule. J. Exp. Med. 1989, 169, 2233–2238. [Google Scholar] [CrossRef] [Green Version]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef]
- Liu, S.; Dhar, P.; Wu, J.D. NK Cell Plasticity in Cancer. J. Clin. Med. 2019, 8, 1492. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.; Conlon, K.; Bohac, G.C.; Barcenas, J.; Leslie, W.; Watkins, L.; Lamzabi, I.; Deng, Y.; Li, Y.; Plate, J.M.D. Effect of Pemetrexed on Innate Immune Killer Cells and Adaptive Immune T Cells in Subjects with Adenocarcinoma of the Pancreas. J. Immunother. 2012, 35, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Funa, K.; Nilsson, B.; Jacobsson, G.; Alm, G.V. Decreased natural killer cell activity and interferon production by leucocytes in patients with adenocarcinoma of the pancreas. Br. J. Cancer 1984, 50, 231–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.P.; Zhang, J.J.; Liang, W.B.; Tu, M.; Lu, Z.P.; Wei, J.S.; Jiang, K.R.; Gao, W.T.; Wu, J.L.; Xu, Z.K.; et al. Elevation of MMP-9 and IDO induced by pancreatic cancer cells mediates natural killer cell dysfunction. BMC Cancer 2014, 14, 738. [Google Scholar] [CrossRef] [Green Version]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef] [PubMed]
- De Monte, L.; Reni, M.; Tassi, E.; Clavenna, D.; Papa, I.; Recalde, H.; Braga, M.; Di Carlo, V.; Doglioni, C.; Protti, M.P. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J. Exp. Med. 2011, 208, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Prokopchuk, O.; Liu, Y.; Henne-Bruns, D.; Kornmann, M. Interleukin-4 enhances proliferation of human pancreatic cancer cells: Evidence for autocrine and paracrine actions. Br. J. Cancer 2005, 92, 921–928. [Google Scholar] [CrossRef] [Green Version]
- Brunetto, E.; De Monte, L.; Balzano, G.; Camisa, B.; Laino, V.; Riba, M.; Heltai, S.; Bianchi, M.; Bordignon, C.; Falconi, M.; et al. The IL-1/IL-1 receptor axis and tumor cell released inflammasome adaptor ASC are key regulators of TSLP secretion by cancer associated fibroblasts in pancreatic cancer. J. Immunother. Cancer 2019, 7, 45. [Google Scholar] [CrossRef]
- Protti, M.P.; De Monte, L. Cross-talk within the tumor microenvironment mediates Th2-type inflammation in pancreatic cancer. Oncoimmunology 2012, 1, 89–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Fei, M.; Wu, Y.; Zheng, D.; Wan, D.; Wang, L.; Li, D. Distribution and Clinical Significance of Th17 Cells in the Tumor Microenvironment and Peripheral Blood of Pancreatic Cancer Patients. Int. J. Mol. Sci. 2011, 12, 7424. [Google Scholar] [CrossRef]
- McAllister, F.; Bailey, J.M.; Alsina, J.; Nirschl, C.J.; Sharma, R.; Fan, H.; Rattigan, Y.; Roeser, J.C.; Lankapalli, R.H.; Zhang, H.; et al. Oncogenic Kras Activates a Hematopoietic-to-Epithelial IL-17 Signaling Axis in Preinvasive Pancreatic Neoplasia. Cancer Cell 2014, 25, 621–637. [Google Scholar] [CrossRef] [Green Version]
- Liyanage, U.K.; Moore, T.T.; Joo, H.-G.; Tanaka, Y.; Herrmann, V.; Doherty, G.; Drebin, J.A.; Strasberg, S.M.; Eberlein, T.J.; Goedegebuure, P.S.; et al. Prevalence of Regulatory T Cells Is Increased in Peripheral Blood and Tumor Microenvironment of Patients with Pancreas or Breast Adenocarcinoma. J. Immunol. 2002, 169, 2756–2761. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.-J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef] [Green Version]
- Orhan, A.; Vogelsang, R.P.; Andersen, M.B.; Madsen, M.T.; Hölmich, E.R.; Raskov, H.; Gögenur, I. The prognostic value of tumour-infiltrating lymphocytes in pancreatic cancer: A systematic review and meta-analysis. Eur. J. Cancer 2020, 132, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.; Denizot, F.; Luciani, M.; Roux-Dosseto, M.; Suzan, M.; Mattei, M. A new member of the immunoglobulin superfamily-CTLA-4. Nature 1987, 328, 267–270. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Zou, W.; Chen, L. Inhibitory B7-family molecules in the tumour microenvironment. Nat. Rev. Immunol. 2008, 8, 467–477. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of Antitumor Immunity by CTLA-4 Blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Sunshine, J.; Taube, J.M. PD-1/PD-L1 inhibitors. Curr. Opin. Pharmacol. 2015, 23, 32–38. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.M.; Hwu, W.-J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and Activity of Anti–PD-L1 Antibody in Patients with Advanced Cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [Green Version]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef]
- Stromnes, I.M.; Hulbert, A.; Pierce, R.H.; Greenberg, P.D.; Hingorani, S.R. T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol. Res. 2017, 5, 978–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balli, D.; Rech, A.J.; Stanger, B.Z.; Vonderheide, R.H. Immune cytolytic activity stratifies molecular subsets of human pancreatic cancer. Clin. Cancer Res. 2017, 23, 3129–3138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.M.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Humphris, J.L.; Patch, A.M.; Nones, K.; Bailey, P.J.; Johns, A.L.; McKay, S.; Chang, D.K.; Miller, D.K.; Pajic, M.; Kassahn, K.S.; et al. Hypermutation In Pancreatic Cancer. Gastroenterology 2017, 152, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Koikawa, K.; Kibe, S.; Suizu, F.; Sekino, N.; Kim, N.; Manz, T.D.; Pinch, B.J.; Akshinthala, D.; Verma, A.; Gaglia, G.; et al. Targeting Pin1 renders pancreatic cancer eradicable by synergizing with immunochemotherapy. Cell 2021, 184, 4753–4771. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting Tumor-Infiltrating Macrophages Decreases Tumor-Initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic Responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nywening, T.M.; Wang-Gillam, A.; Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Cusworth, B.M.; Toriola, A.T.; Nieman, R.K.; Worley, L.A.; Yano, M.; et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: A single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet. Oncol. 2016, 17, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; De Nardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [Green Version]
- Wiehagen, K.R.; Girgis, N.M.; Yamada, D.H.; Smith, A.A.; Chan, S.R.; Grewal, I.S.; Quigley, M.; Verona, R.I. Combination of CD40 agonism and CSF-1R blockade reconditions tumor-associated macrophages and drives potent antitumor immunity. Cancer Immunol. Res. 2017, 5, 1109–1121. [Google Scholar] [CrossRef] [Green Version]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Liang, S.; Wang, X.; Liang, Y.; Zhang, M.; Chen, J.; Niu, L.; Xu, K. Percutaneous irreversible electroporation combined with allogeneic natural killer cell immunotherapy for patients with unresectable (stage III/IV) pancreatic cancer: A promising treatment. J. Cancer Res. Clin. Oncol. 2017, 143, 2607–2618. [Google Scholar] [CrossRef] [PubMed]
- Gürlevik, E.; Fleischmann-Mundt, B.; Brooks, J.; Demir, I.E.; Steiger, K.; Ribback, S.; Yevsa, T.; Woller, N.; Kloos, A.; Ostroumov, D.; et al. Administration of Gemcitabine after Pancreatic Tumor Resection in Mice Induces an Antitumor Immune Response Mediated by Natural Killer Cells. Gastroenterology 2016, 151, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manuel, E.R.; Chen, J.; D’Apuzzo, M.; Lampa, M.G.; Kaltcheva, T.I.; Thompson, C.B.; Ludwig, T.; Chung, V.; Diamond, D.J. Salmonella-based therapy targeting indoleamine 2,3-dioxygenase coupled with enzymatic depletion of tumor hyaluronan induces complete regression of aggressive pancreatic tumors. Cancer Immunol. Res. 2015, 3, 1096–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Wang-Gillam, A.; Picozzi, V.; Greten, T.F.; Crocenzi, T.; Springett, G.; Morse, M.; Zeh, H.; Cohen, D.; Fine, R.L.; et al. Safety and survival with GVAX pancreas prime and Listeria monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 1325–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernhardt, S.L.; Gjertsen, M.K.; Trachsel, S.; Møller, M.; Eriksen, J.A.; Meo, M.; Buanes, T.; Gaudernack, G. Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: A dose escalating phase I/II study. Br. J. Cancer 2006, 95, 1474–1482. [Google Scholar] [CrossRef]
- Mayanagi, S.; Kitago, M.; Sakurai, T.; Matsuda, T.; Fujita, T.; Higuchi, H.; Taguchi, J.; Takeuchi, H.; Itano, O.; Aiura, K.; et al. Phase I pilot study of Wilms tumor gene 1 peptide-pulsed dendritic cell vaccination combined with gemcitabine in pancreatic cancer. Cancer Sci. 2015, 106, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Lutz, E.R.; Wu, A.A.; Bigelow, E.; Sharma, R.; Mo, G.; Soares, K.; Solt, S.; Dorman, A.; Wamwea, A.; Yager, A.; et al. Immunotherapy converts nonimmunogenic pancreatic tumors into immunogenic foci of immune regulation. Cancer Immunol. Res. 2014, 2, 616–631. [Google Scholar] [CrossRef] [Green Version]
- Keenan, B.P.; Saenger, Y.; Kafrouni, M.I.; Leubner, A.; Lauer, P.; Maitra, A.; Rucki, A.A.; Gunderson, A.J.; Coussens, L.M.; Brockstedt, D.G.; et al. A listeria vaccine and depletion of t-regulatory cells activate immunity against early stage pancreatic intraepithelial neoplasms and prolong survival of mice. Gastroenterology 2014, 146, 1784–1794. [Google Scholar] [CrossRef] [Green Version]
- Thind, K.; Padrnos, L.J.; Ramanathan, R.K.; Borad, M.J. Immunotherapy in pancreatic cancer treatment: A new frontier. Therap. Adv. Gastroenterol. 2017, 10, 168–194. [Google Scholar] [CrossRef] [Green Version]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.-H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.-B.; Chadwick, D.; Zhang, A.; et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin. Cancer Res. 2018, 24, 1344–1354. [Google Scholar] [CrossRef] [Green Version]
- Holter, S.; Borgida, A.; Dodd, A.; Grant, R.; Semotiuk, K.; Hedley, D.; Dhani, N.; Narod, S.; Akbari, M.; Moore, M.; et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients with Pancreatic Adenocarcinoma. J. Clin. Oncol. 2015, 33, 3124–3129. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Kindler, H.L.; Park, J.O.; Reni, M.; Mercade, T.M.; Hammel, P.; Van Cutsem, E.; Arnold, D.; Hochhauser, D.; Locker, G.Y.; et al. Geographic and ethnic heterogeneity in the BRCA1/2 pre-screening population for the randomized phase III POLO study of olaparib maintenance in metastatic pancreatic cancer (mPC). J. Clin. Oncol. 2018, 36, 4115. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Romero-Calvo, I.; Weber, C.R.; Ray, M.; Brown, M.; Kirby, K.; Nandi, R.K.; Long, T.M.; Sparrow, S.M.; Ugolkov, A.; Qiang, W.; et al. Human Organoids Share Structural and Genetic Features with Primary Pancreatic Adenocarcinoma Tumors. Mol. Cancer Res. 2019, 17, 70–83. [Google Scholar] [CrossRef] [Green Version]
- Frappart, P.-O.; Hofmann, T.G. Pancreatic Ductal Adenocarcinoma (PDAC) Organoids: The Shining Light at the End of the Tunnel for Drug Response Prediction and Personalized Medicine. Cancers 2020, 12, 2750. [Google Scholar] [CrossRef]

{kind=link}
| AJCC Staging | I–II | II–III | II–III | IV |
|---|---|---|---|---|
| Clinical stage | Resectable | Borderline resectable | Locally advanced | Metastatic |
| Vascular involvement | No or <180° contact | <180° contact | >180° contact | N/A |
| Prevalence at diagnosis (~%) a | 10–15 | 30–35 | 30–35 | 50 |
| Treatment intent | Curative | Curative | Palliative | Palliative |
| 5-year survival rate (~%) a | 35–45 | 10–15 | 10–15 | <5 |
| Trial | Therapy Type | Treatment Groups | Number of Patients | Median Survival in Months | Year of Publication |
|---|---|---|---|---|---|
| ESPAC-1 | Adjuvant | 5-FU vs. resection only | 188 | 19.7 vs. 14 | 2001 |
| CONKO-001 | Adjuvant | Gemcitabine vs. resection only | 368 | 13.4 vs. 6.7 | 2007 |
| ESPAC-3 | Adjuvant | 5-FU + folinic acid vs. gemcitabine | 1088 | 23 vs. 23.6 | 2010 |
| PRODIGE | First line for stage IV | FOLFIRINOX vs. gemcitabine | 342 | 11.1 vs. 6.8 | 2011 |
| PRODIGE-24 | Adjuvant | FOLFIRINOX vs. Gemcitabine | 493 | 54.4 vs. 35 | 2017 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robatel, S.; Schenk, M. Current Limitations and Novel Perspectives in Pancreatic Cancer Treatment. Cancers 2022, 14, 985. https://doi.org/10.3390/cancers14040985
Robatel S, Schenk M. Current Limitations and Novel Perspectives in Pancreatic Cancer Treatment. Cancers. 2022; 14(4):985. https://doi.org/10.3390/cancers14040985
Chicago/Turabian StyleRobatel, Steve, and Mirjam Schenk. 2022. "Current Limitations and Novel Perspectives in Pancreatic Cancer Treatment" Cancers 14, no. 4: 985. https://doi.org/10.3390/cancers14040985
APA StyleRobatel, S., & Schenk, M. (2022). Current Limitations and Novel Perspectives in Pancreatic Cancer Treatment. Cancers, 14(4), 985. https://doi.org/10.3390/cancers14040985