
Slow-Cycling Cells in Glioblastoma: A Specific Population in the Cellular Mosaic of Cancer Stem Cells

 , , ,
, , , 
Abstract
:Simple Summary
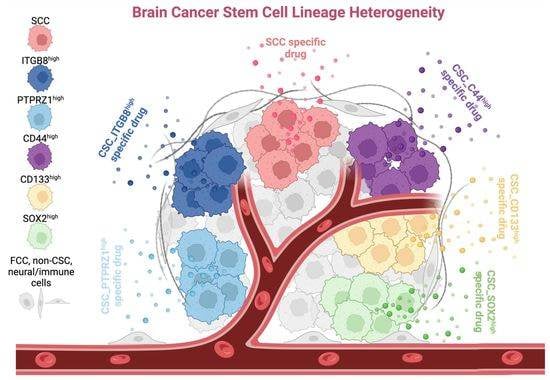
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Material and Methods
2.1. GBM Patient-Derived Cell Lines
2.2. Isolation of Slow-Cycling Cells
2.3. Antibodies
2.4. TMZ Treatment
2.5. TMZ Sensitivity Assays
2.6. Lentivirus Transduction
2.7. Flow Cytometry
2.8. Bulk RNA Sequencing
2.9. Single-Cell RNA Sequencing
2.10. Digital Cytometry for SCC and FCC Deconvolution
2.11. Hierarchical Clustering of CSC
2.12. Statistical Tests
3. Results
3.1. Slow-Cycling Cells Express a Wide Range of CSC Markers
3.2. Single-Cell Transcriptomics Identify Multiple Populations of CSCs
3.3. Heterogeneity between CSC Populations
3.4. Correlation with Clinicopathological Characters and Patient Survival
3.5. Functional Profiling of TMZ Sensitivity
3.6. Genomic Profile Predicting Heterogeneity of Drug-Sensitivity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Deleyrolle, L.P.; Harding, A.; Cato, K.; Siebzehnrubl, F.A.; Rahman, M.; Azari, H.; Olson, S.; Gabrielli, B.; Osborne, G.; Vescovi, A.; et al. Evidence for label-retaining tumour-initiating cells in human glioblastoma. Brain 2011, 134, 1331–1343. [Google Scholar] [CrossRef] [Green Version]
- Hoang-Minh, L.B.; Siebzehnrubl, F.A.; Yang, C.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Schmoll Massari, M.; Patel, J.; Amin, K.; et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018, 37, e98772. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma Stem Cells: Driving Resilience through Chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef] [Green Version]
- Bhaduri, A.; Di Lullo, E.; Jung, D.; Muller, S.; Crouch, E.E.; Espinosa, C.S.; Ozawa, T.; Alvarado, B.; Spatazza, J.; Cadwell, C.R.; et al. Outer Radial Glia-like Cancer Stem Cells Contribute to Heterogeneity of Glioblastoma. Cell Stem Cell 2020, 26, 48–63. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangemi, R.M.; Griffero, F.; Marubbi, D.; Perera, M.; Capra, M.C.; Malatesta, P.; Ravetti, G.L.; Zona, G.L.; Daga, A.; Corte, G. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells 2009, 27, 40–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berezovsky, A.D.; Poisson, L.M.; Cherba, D.; Webb, C.P.; Transou, A.D.; Lemke, N.W.; Hong, X.; Hasselbach, L.A.; Irtenkauf, S.M.; Mikkelsen, T.; et al. Sox2 promotes malignancy in glioblastoma by regulating plasticity and astrocytic differentiation. Neoplasia 2014, 16, 193–206.e25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Ping, Y.F.; Zhou, W.; He, Z.C.; Chen, C.; Bian, B.S.; Zhang, L.; Chen, L.; Lan, X.; Zhang, X.C.; et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 2017, 8, 15080. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Ouyang, D.; Li, Q.; Li, M.; Zou, Q.; Li, L.; Yi, W.; Zhou, E. The Expression, Functions, Interactions and Prognostic Values of PTPRZ1: A Review and Bioinformatic Analysis. J. Cancer 2019, 10, 1663–1674. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, P.A.; Tchaicha, J.H.; Chen, Z.; Morales, J.E.; McCarty, N.; Wang, Q.; Sulman, E.P.; Fuller, G.; Lang, F.F.; Rao, G.; et al. Glioblastoma stem cells exploit the alphavbeta8 integrin-TGFbeta1 signaling axis to drive tumor initiation and progression. Oncogene 2017, 36, 6568–6580. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, K.; Troike, K.; Silver, D.J.; Lathia, J.D. The evolution of the cancer stem cell state in glioblastoma: Emerging insights into the next generation of functional interactions. Neuro Oncol. 2021, 23, 199–213. [Google Scholar] [CrossRef]
- Deleyrolle, L.P.; Rohaus, M.R.; Fortin, J.M.; Reynolds, B.A.; Azari, H. Identification and isolation of slow-dividing cells in human glioblastoma using carboxy fluorescein succinimidyl ester (CFSE). J. Vis. Exp. 2012, 3918. [Google Scholar] [CrossRef] [Green Version]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef]
- Si, D.; Yin, F.; Peng, J.; Zhang, G. High Expression of CD44 Predicts a Poor Prognosis in Glioblastomas. Cancer Manag. Res. 2020, 12, 769–775. [Google Scholar] [CrossRef] [Green Version]
- Mobley, A.K.; Tchaicha, J.H.; Shin, J.; Hossain, M.G.; McCarty, J.H. Beta8 integrin regulates neurogenesis and neurovascular homeostasis in the adult brain. J. Cell Sci. 2009, 122, 1842–1851. [Google Scholar] [CrossRef] [Green Version]
- Roth, P.; Silginer, M.; Goodman, S.L.; Hasenbach, K.; Thies, S.; Maurer, G.; Schraml, P.; Tabatabai, G.; Moch, H.; Tritschler, I.; et al. Integrin control of the transforming growth factor-beta pathway in glioblastoma. Brain J. Neurol. 2013, 136, 564–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, S.B.; Narayanan, A.S.; Lee, H.S.; Tchaicha, J.H.; Aldape, K.D.; Lang, F.F.; Tolias, K.F.; McCarty, J.H. alphavbeta8 integrin interacts with RhoGDI1 to regulate Rac1 and Cdc42 activation and drive glioblastoma cell invasion. Mol. Biol. Cell 2013, 24, 474–482. [Google Scholar] [CrossRef]
- Fujikawa, A.; Sugawara, H.; Tanaka, T.; Matsumoto, M.; Kuboyama, K.; Suzuki, R.; Tanga, N.; Ogata, A.; Masumura, M.; Noda, M. Targeting PTPRZ inhibits stem cell-like properties and tumorigenicity in glioblastoma cells. Sci. Rep. 2017, 7, 5609. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.H.; Yin, Y.H.; Chen, H.Z.; Feng, S.Y.; Liu, J.L.; Chen, L.; Fu, W.L.; Sun, G.C.; Yu, X.G.; Xu, D.G. TRIM24 promotes stemness and invasiveness of glioblastoma cells via activating Sox2 expression. Neuro Oncol. 2020, 22, 1797–1808. [Google Scholar] [CrossRef] [PubMed]
- Sarkisian, M.R.; Siebzehnrubl, D.; Hoang-Minh, L.; Deleyrolle, L.; Silver, D.J.; Siebzehnrubl, F.A.; Guadiana, S.M.; Srivinasan, G.; Semple-Rowland, S.; Harrison, J.K.; et al. Detection of primary cilia in human glioblastoma. J. Neuro-Oncol. 2014, 117, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portnow, J.; Badie, B.; Chen, M.; Liu, A.; Blanchard, S.; Synold, T.W. The neuropharmacokinetics of temozolomide in patients with resectable brain tumors: Potential implications for the current approach to chemoradiation. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 7092–7098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostermann, S.; Csajka, C.; Buclin, T.; Leyvraz, S.; Lejeune, F.; Decosterd, L.A.; Stupp, R. Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 3728–3736. [Google Scholar] [CrossRef] [Green Version]
- Brada, M.; Judson, I.; Beale, P.; Moore, S.; Reidenberg, P.; Statkevich, P.; Dugan, M.; Batra, V.; Cutler, D. Phase I dose-escalation and pharmacokinetic study of temozolomide (SCH 52365) for refractory or relapsing malignancies. Br. J. Cancer 1999, 81, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [Green Version]
- Borcherding, N.; Andrews, J. Escape: Easy Single Cell Analysis Platform for Enrichment, R Package Version 1.3.1. Home Page; Bioconductor: Boston, MA, USA, 2020. Available online: https://bioconductor.org/packages/release/bioc/html/escape.html(accessed on 20 February 2022).
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H., 2nd; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Tirosh, I.; Venteicher, A.S.; Hebert, C.; Escalante, L.E.; Patel, A.P.; Yizhak, K.; Fisher, J.M.; Rodman, C.; Mount, C.; Filbin, M.G.; et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 2016, 539, 309. [Google Scholar] [CrossRef] [Green Version]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveros, J.C. Venny. An interactive tool for comparing lists with Venn’s diagrams. Home Page. Available online: https://bioinfogp.cnb.csic.es/tools/venny/ (accessed on 20 February 2022).
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- McInnes, L.; Healy, J. UMAP: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Le, T.D.; Liu, L.; Su, N.; Wang, R.; Sun, B.; Colaprico, A.; Bontempi, G.; Li, J. CancerSubtypes: An R/Bioconductor package for molecular cancer subtype identification, validation and visualization. Bioinformatics 2017, 33, 3131–3133. [Google Scholar] [CrossRef] [PubMed]
- Kolde, R. Pheatmap: Pretty Heatmaps Home Page. 2015. Available online: https://cran.r-project.org/web/packages/pheatmap/index.html (accessed on 20 February 2022).
- Merico, D.; Isserlin, R.; Stueker, O.; Emili, A.; Bader, G.D. Enrichment map: A network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 2010, 5, e13984. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Badr, C.E.; Silver, D.J.; Siebzehnrubl, F.A.; Deleyrolle, L.P. Metabolic heterogeneity and adaptability in brain tumors. Cell. Mol. Life Sci. 2020, 77, 5101–5119. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Dong, Y.; Kumar, R.; Jeter, C.; Tang, D.G. Slow-cycling (dormant) cancer cells in therapy resistance, cancer relapse and metastasis. Semin. Cancer Biol. 2021, 78, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018, 37, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasper, M.; Schafer, A.; Piontek, G.; Teufel, J.; Brockhoff, G.; Ringel, F.; Heindl, S.; Zimmer, C.; Schlegel, J. Aldehyde dehydrogenase 1 positive glioblastoma cells show brain tumor stem cell capacity. Neuro Oncol. 2010, 12, 1024–1033. [Google Scholar] [CrossRef]
- Jin, X.; Jin, X.; Jung, J.E.; Beck, S.; Kim, H. Cell surface Nestin is a biomarker for glioma stem cells. Biochem. Biophys. Res. Commun. 2013, 433, 496–501. [Google Scholar] [CrossRef]
- Ligon, K.L.; Huillard, E.; Mehta, S.; Kesari, S.; Liu, H.; Alberta, J.A.; Bachoo, R.M.; Kane, M.; Louis, D.N.; Depinho, R.A.; et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 2007, 53, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.N.; Borges, I.; Ruiz i Altaba, A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef] [Green Version]
- Son, M.J.; Woolard, K.; Nam, D.H.; Lee, J.; Fine, H.A. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell 2009, 4, 440–452. [Google Scholar] [CrossRef] [Green Version]
- Reinartz, R.; Wang, S.; Kebir, S.; Silver, D.J.; Wieland, A.; Zheng, T.; Kupper, M.; Rauschenbach, L.; Fimmers, R.; Shepherd, T.M.; et al. Functional Subclone Profiling for Prediction of Treatment-Induced Intratumor Population Shifts and Discovery of Rational Drug Combinations in Human Glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 562–574. [Google Scholar] [CrossRef] [Green Version]
- Oren, Y.; Tsabar, M.; Cuoco, M.S.; Amir-Zilberstein, L.; Cabanos, H.F.; Hutter, J.C.; Hu, B.; Thakore, P.I.; Tabaka, M.; Fulco, C.P.; et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature 2021, 596, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Hussein, A.M.; Wang, Y.; Mathieu, J.; Margaretha, L.; Song, C.; Jones, D.C.; Cavanaugh, C.; Miklas, J.W.; Mahen, E.; Showalter, M.R.; et al. Metabolic Control over mTOR-Dependent Diapause-like State. Dev. Cell 2020, 52, 236–250. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.Q.; Liu, W.; Zhu, F.; Lei, C.L.; Wang, X.P. Fatty acid synthase 2 contributes to diapause preparation in a beetle by regulating lipid accumulation and stress tolerance genes expression. Sci. Rep. 2017, 7, 40509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arena, R.; Bisogno, S.; Gasior, L.; Rudnicka, J.; Bernhardt, L.; Haaf, T.; Zacchini, F.; Bochenek, M.; Fic, K.; Bik, E.; et al. Lipid droplets in mammalian eggs are utilized during embryonic diapause. Proc. Natl. Acad. Sci. USA 2021, 118, e2018362118. [Google Scholar] [CrossRef] [PubMed]
- Dhimolea, E.; de Matos Simoes, R.; Kansara, D.; Al’Khafaji, A.; Bouyssou, J.; Weng, X.; Sharma, S.; Raja, J.; Awate, P.; Shirasaki, R.; et al. An Embryonic Diapause-like Adaptation with Suppressed Myc Activity Enables Tumor Treatment Persistence. Cancer Cell 2021, 39, 240–256. [Google Scholar] [CrossRef]
- Rehman, S.K.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.; Nixon, A.M.L.; Bruce, J.P.; Wintersinger, J.A.; Singh Mer, A.; Lo, E.B.L.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242. [Google Scholar] [CrossRef]
- Lan, X.Y.; Jorg, D.J.; Cavalli, F.M.G.; Richards, L.M.; Nguyen, L.V.; Vanner, R.J.; Guilhamon, P.; Lee, L.; Kushida, M.M.; Pellacani, D.; et al. Fate mapping of human glioblastoma reveals an invariant stem cell hierarchy. Nature 2017, 549, 227. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Tian, G.; Dajac, M.; Doty, A.; Wang, S.; Lee, J.-H.; Rahman, M.; Huang, J.; Reynolds, B.A.; Sarkisian, M.R.; et al. Slow-Cycling Cells in Glioblastoma: A Specific Population in the Cellular Mosaic of Cancer Stem Cells. Cancers 2022, 14, 1126. https://doi.org/10.3390/cancers14051126
Yang C, Tian G, Dajac M, Doty A, Wang S, Lee J-H, Rahman M, Huang J, Reynolds BA, Sarkisian MR, et al. Slow-Cycling Cells in Glioblastoma: A Specific Population in the Cellular Mosaic of Cancer Stem Cells. Cancers. 2022; 14(5):1126. https://doi.org/10.3390/cancers14051126
Chicago/Turabian StyleYang, Changlin, Guimei Tian, Mariana Dajac, Andria Doty, Shu Wang, Ji-Hyun Lee, Maryam Rahman, Jianping Huang, Brent A. Reynolds, Matthew R. Sarkisian, and et al. 2022. "Slow-Cycling Cells in Glioblastoma: A Specific Population in the Cellular Mosaic of Cancer Stem Cells" Cancers 14, no. 5: 1126. https://doi.org/10.3390/cancers14051126
APA StyleYang, C., Tian, G., Dajac, M., Doty, A., Wang, S., Lee, J.-H., Rahman, M., Huang, J., Reynolds, B. A., Sarkisian, M. R., Mitchell, D., & Deleyrolle, L. P. (2022). Slow-Cycling Cells in Glioblastoma: A Specific Population in the Cellular Mosaic of Cancer Stem Cells. Cancers, 14(5), 1126. https://doi.org/10.3390/cancers14051126