
Discovery of a New CaMKII-Targeted Synthetic Lethal Therapy against Glioblastoma Stem-like Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. GSC Culture
2.3. Cell Viability Assay
2.4. Tumorsphere Forming Assay
2.5. Chick Embryo Chorioallantoic Membrane (CAM) Assay
2.6. Western Blot Analysis
2.7. Analysis of Apoptosis
2.8. Measurement of Reactive Oxygen Species (ROS)
2.9. RNA Interference
2.10. Measurement of Calcium
2.11. Statistical Analysis
3. Results
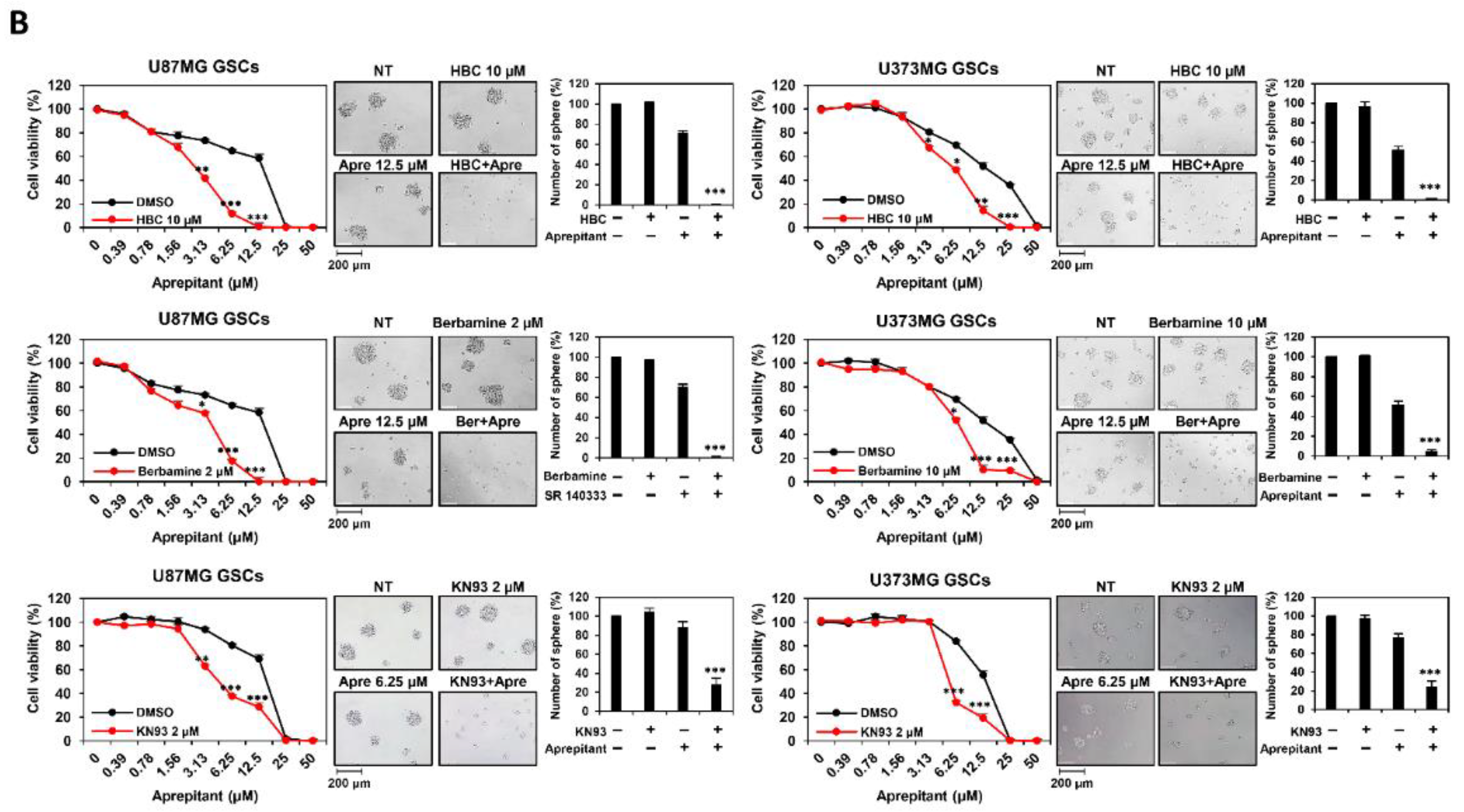
3.1. Combined Treatment of CaMKII and NK1R Inhibitors Increases GSC Lethality
3.2. Combined Treatment of CaMKII and NK1R Inhibitors Potently Suppresses GSC-Derived Tumor Growth In Vivo
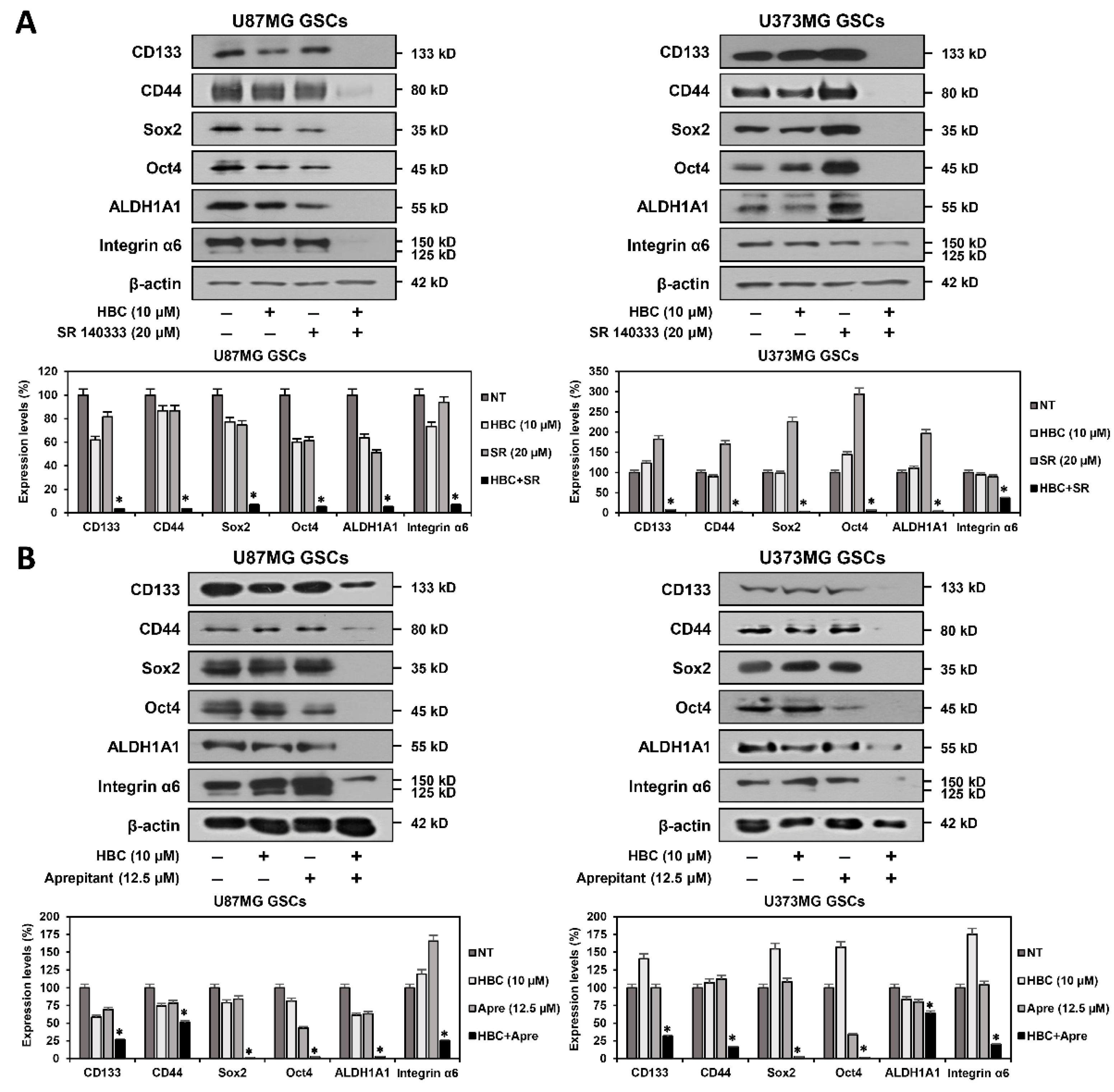
3.3. Combined Treatment of CaMKII and NK1R Inhibitors Synergistically Suppresses Expression of GSC Markers
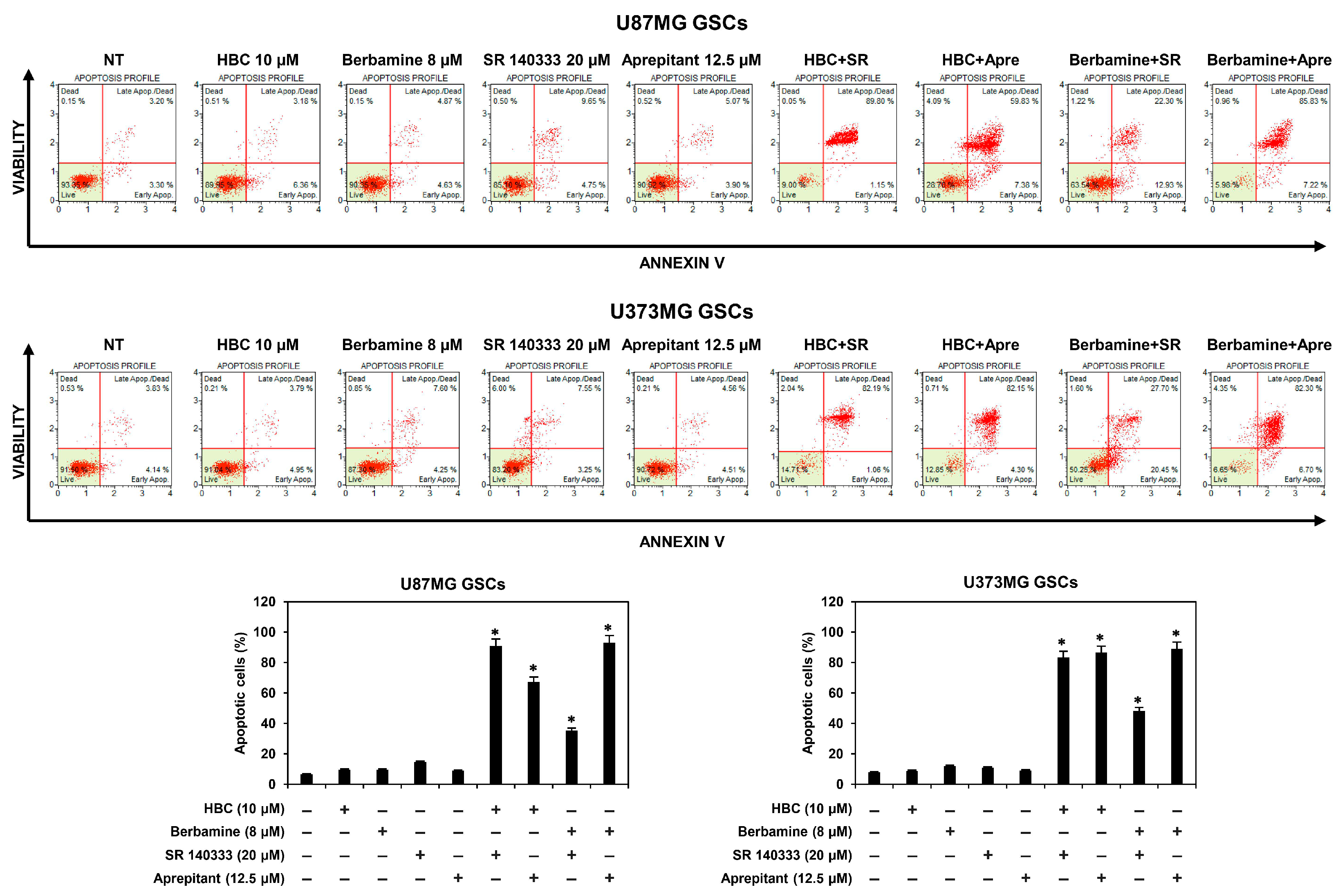
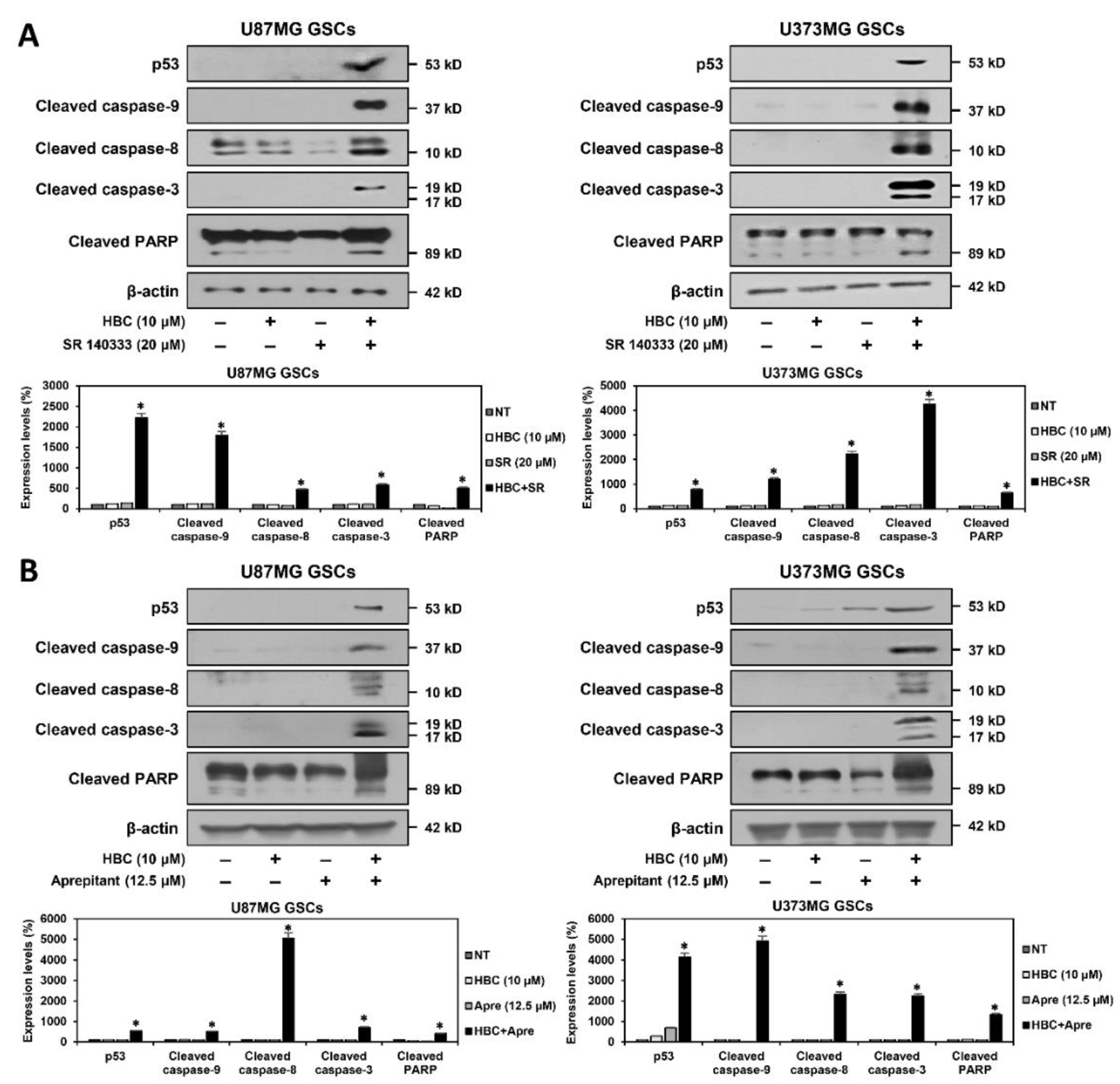
3.4. Combined Treatment of CaMKII and NK1R Inhibitors Strongly Promotes the Apoptotic Cell Death of GSCs
3.5. Combined Treatment of CaMKII and NK1R Inhibitors Increases ROS Generation in GSCs
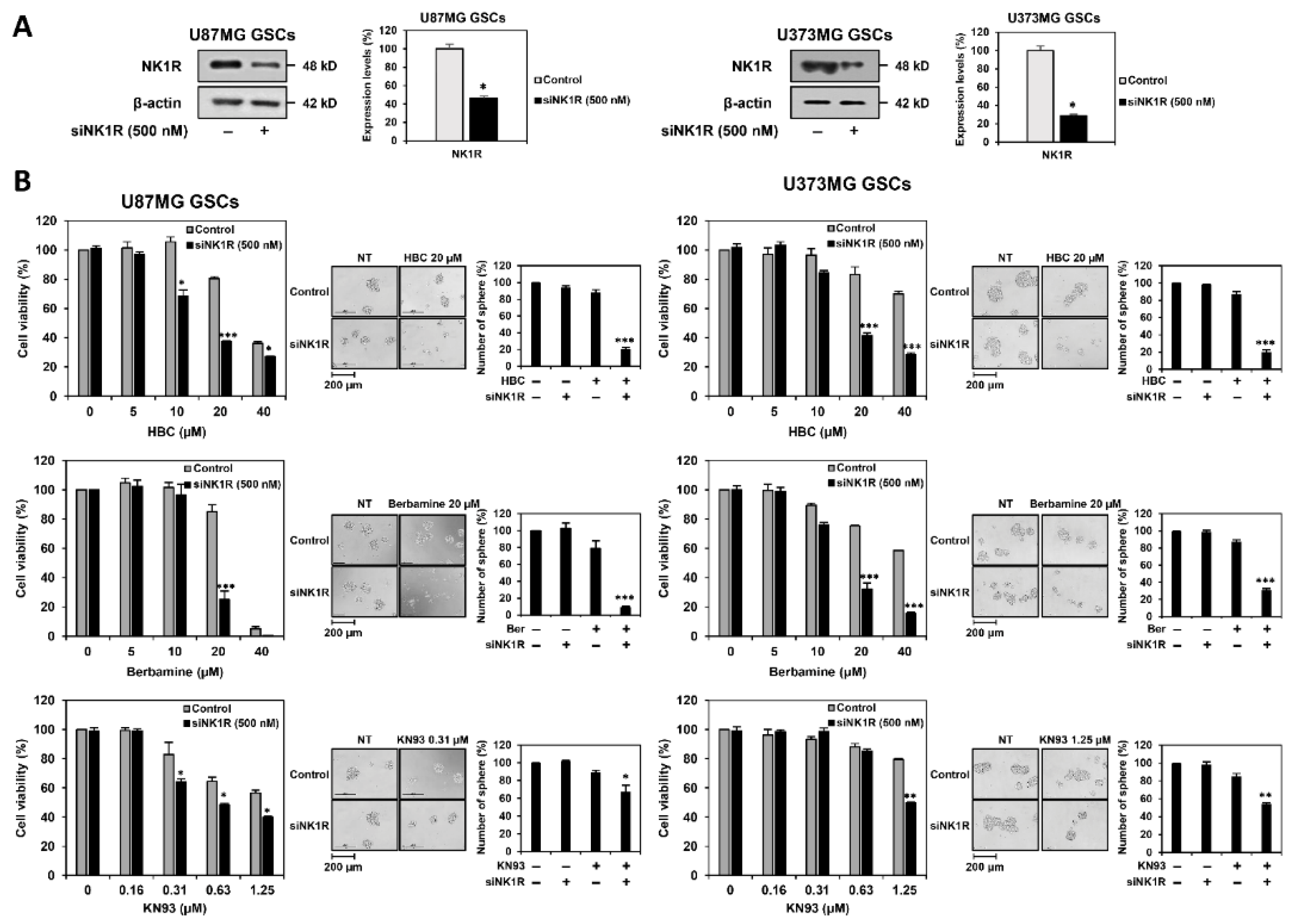
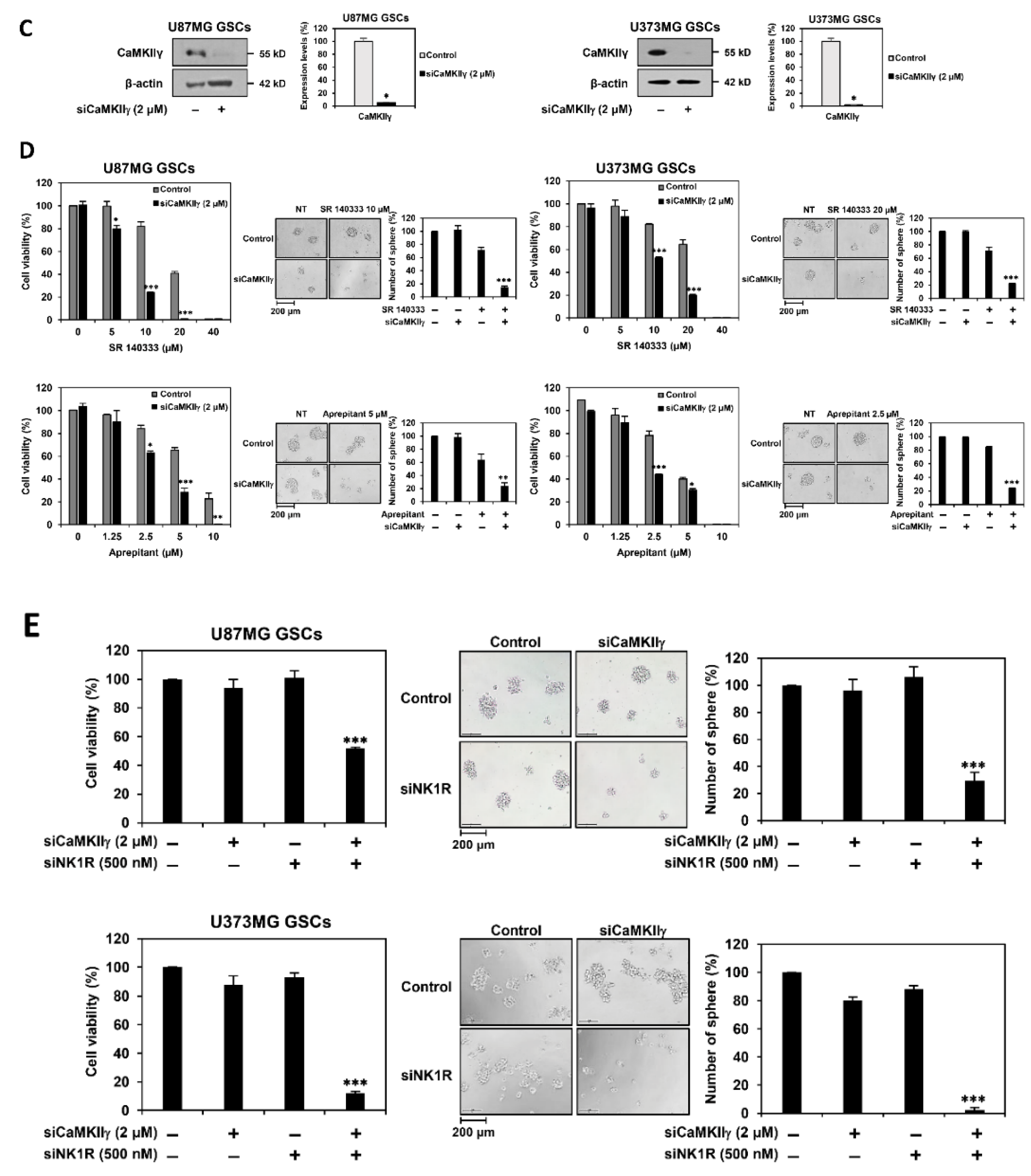
3.6. Identification of Synthetic Lethal Interaction between CaMKII and NK1R by RNA Interference in GSCs
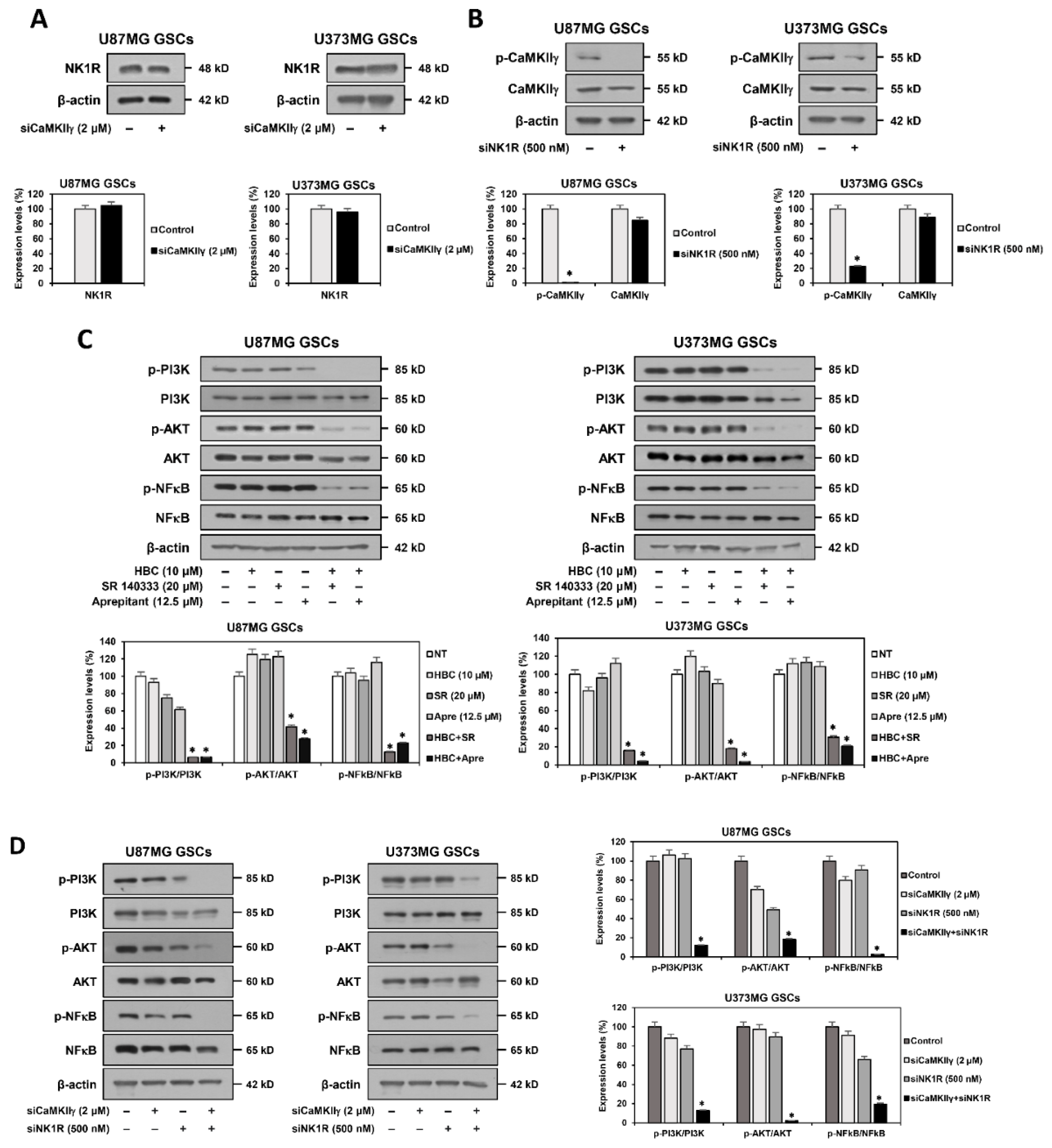
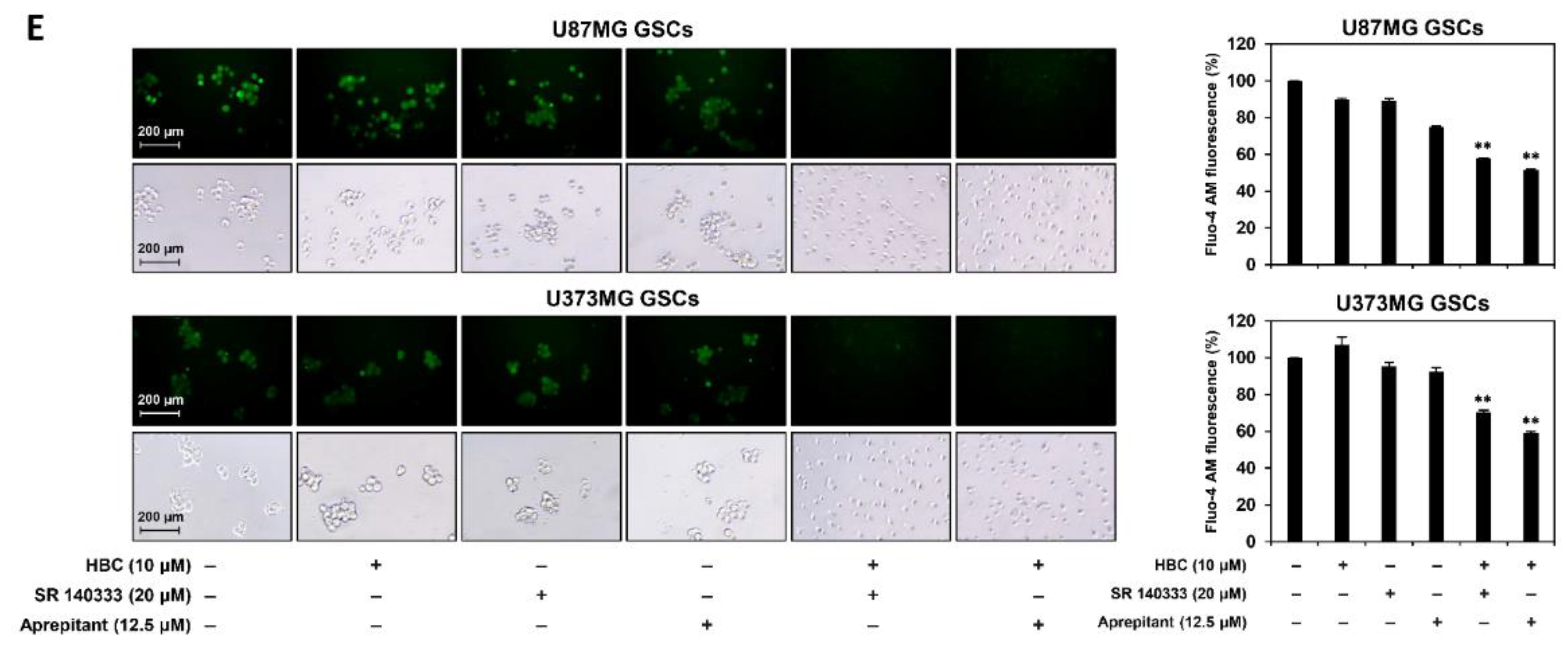
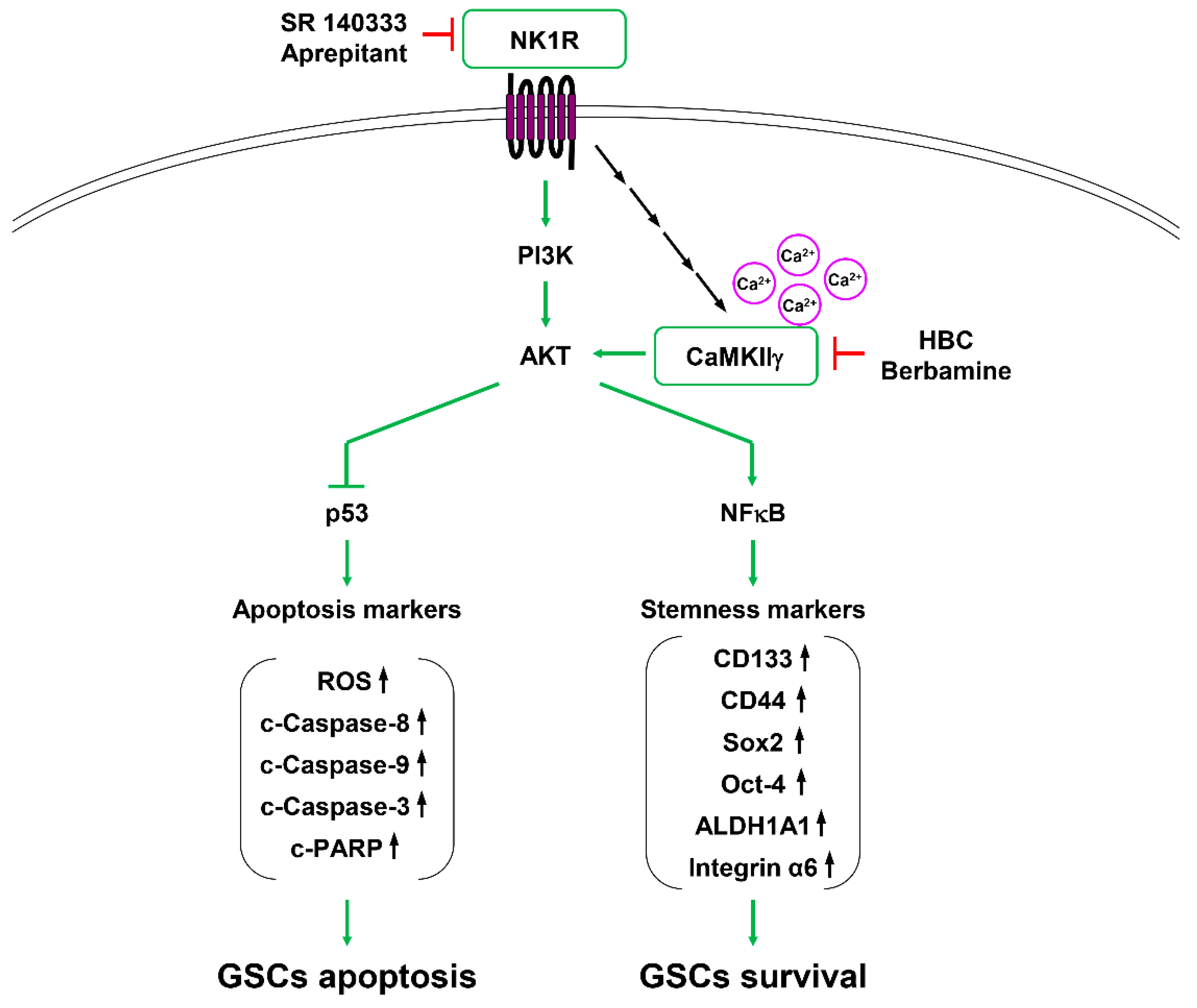
3.7. Inhibition of PI3K/AKT/NF-κB and Calcium Signaling Contributes to Synthetic Lethality between CaMKII and NK1R Inhibitors in GSCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef] [Green Version]
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. Potential strategies overcoming the temozolomide resistance for glioblastoma. Neurol. Med. Chir. 2018, 58, 405–421. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Finocchiaro, G. TLRgeting evasion of immune pathways in glioblastoma. Cell Stem Cell 2017, 20, 422–424. [Google Scholar] [CrossRef] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef]
- Caragher, S.P.; Sachdev, S.; Ahmed, A. Radiotherapy and glioma stem cells: Searching for chinks in cellular armor. Curr. Stem Cell Rep. 2017, 3, 348–357. [Google Scholar] [CrossRef]
- Kaelin, W.G. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Leung, A.W.; de Silva, T.; Bally, M.B.; Lockwood, W.W. Synthetic lethality in lung cancer and translation to clinical therapies. Mol. Cancer 2016, 15, 61. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ishida, C.T.; Shu, C.; Kleiner, G.; Sanchez-Quintero, M.J.; Bianchetti, E.; Quinzii, C.M.; Westhoff, M.A.; Karpel-Massler, G.; Siegelin, M.D. Inhibition of Bcl-2/Bcl-xL and c-MET causes synthetic lethality in model systems of glioblastoma. Sci. Rep. 2018, 8, 7373. [Google Scholar] [CrossRef]
- Gao, S.; Lai, L. Synthetic lethality in drug development: The dawn is coming. Future Med. Chem. 2018, 10, 2129–2132. [Google Scholar] [CrossRef] [Green Version]
- Hook, S.S.; Means, A.R. Ca(2+)/CaM-dependent kinases: From activation to function. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 471–505. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Zhao, R.; Zhe, H. The emerging role of CaMKII in cancer. Oncotarget 2015, 6, 11725–11734. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Guo, S.; Liu, Z.; Wu, L.; Li, M.; Yang, J.; Chen, R.; Liu, X.; Xu, H.; Cai, S.; et al. CAMK2N1 inhibits prostate cancer progression through androgen receptor-dependent signaling. Oncotarget 2014, 5, 10293–10306. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, N.; Liu, X.; Zheng, Y.; Cao, X. A novel endogenous human CaMKII inhibitory protein suppresses tumor growth by inducing cell cycle arrest via p27 stabilization. J. Biol. Chem. 2008, 283, 11565–11574. [Google Scholar] [CrossRef] [Green Version]
- Han, X.C.; Zhang, Y.J.; Dong, X.; Xing, Q.Z.; Li, K.H.; Zhang, L. Sevoflurane modulates the cancer stem cell-like properties and mitochondrial membrane potential of glioma via Ca2+-dependent CaMKII/JNK cascade. Life Sci. 2020, 253, 117675. [Google Scholar] [CrossRef]
- Gu, Y.; Chen, T.; Meng, Z.; Gan, Y.; Xu, X.; Lou, G.; Li, H.; Gan, X.; Zhou, H.; Tang, J.; et al. CaMKIIγ, a critical regulator of CML stem/progenitor cells, is a target of the natural product berbamine. Blood 2012, 120, 4829–4839. [Google Scholar] [CrossRef] [Green Version]
- Chai, S.; Xu, X.; Wang, Y.; Zhou, Y.; Zhang, C.; Yang, Y.; Yang, Y.; Xu, H.; Xu, R.; Wang, K. Ca2+/calmodulin-dependent protein kinase IIγ enhances stem-like traits and tumorigenicity of lung cancer cells. Oncotarget 2015, 6, 16069–16083. [Google Scholar] [CrossRef] [Green Version]
- Meng, Z.; Li, T.; Ma, X.; Wang, X.; Van Ness, C.; Gan, Y.; Zhou, H.; Tang, J.; Lou, G.; Wang, Y.; et al. Berbamine inhibits the growth of liver cancer cells and cancer initiating cells by targeting Ca2+/Calmodulin-dependent protein kinase II. Mol. Cancer Ther. 2013, 12, 2067–2077. [Google Scholar] [CrossRef] [Green Version]
- Chai, S.; Qian, Y.; Tang, J.; Liang, Z.; Zhang, M.; Si, J.; Li, X.; Huang, W.; Xu, R.; Wang, K. RETRACTED: Ca(2+)/calmodulin-dependent protein kinase IIγ, a critical mediator of the NF-κB network, is a novel therapeutic target in non-small cell lung cancer. Cancer Lett. 2014, 344, 119–128. [Google Scholar] [CrossRef]
- Mamaeva, O.A.; Kim, J.; Feng, G.; McDonald, J.M. Calcium/calmodulin-dependent kinase II regulates notch-1 signaling in prostate cancer cells. J. Cell. Biochem. 2009, 106, 25–32. [Google Scholar] [CrossRef]
- Si, J.; Collins, S.J. Activated Ca2+/calmodulin-dependent protein kinase IIgamma is a critical regulator of myeloid leukemia cell proliferation. Cancer Res. 2008, 68, 3733–3742. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.J.; Lee, S.H.; Jung, H.J. A curcumin derivative hydrazinobenzoylcurcumin suppresses stem-like features of glioblastoma cells by targeting Ca2+/calmodulin-dependent protein kinase II. J. Cell. Biochem. 2018, 120, 6741–6752. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H. Cancer stem cells and self-renewal. Clin. Cancer Res. 2010, 16, 3113–3120. [Google Scholar] [CrossRef] [Green Version]
- Dragu, D.L.; Necula, L.G.; Bleotu, C.; Diaconu, C.C.; Chivu-Economescu, M. Therapies targeting cancer stem cells: Current trends and future challenges. World J. Stem Cells 2015, 7, 1185–1201. [Google Scholar]
- Xu, H.S.; Qin, X.L.; Zong, H.L.; He, X.G.; Cao, L. Cancer stem cell markers in glioblastoma—An update. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3207–3211. [Google Scholar]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [Green Version]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signaling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Javid, H.; Asadi, J.; Zahedi Avval, F.; Afshari, A.R.; Hashemy, S.I. The role of substance P/neurokinin 1 receptor in the pathogenesis of esophageal squamous cell carcinoma through constitutively active PI3K/Akt/NF-κB signal transduction pathways. Mol. Biol. Rep. 2020, 47, 2253–2263. [Google Scholar] [CrossRef]
- Xu, B.; Chen, S.; Luo, Y.; Chen, Z.; Liu, L.; Zhou, H.; Chen, W.; Shen, T.; Han, X.; Chen, L.; et al. Calcium signaling is involved in cadmium-induced neuronal apoptosis via induction of reactive oxygen species and activation of MAPK/mTOR network. PLoS ONE 2011, 6, e19052. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, L.; Farley, M.M.; Waxham, M.N. Calcium-calmodulin-dependent protein kinase II isoforms differentially impact the dynamics and structure of the actin cytoskeleton. Biochemistry 2013, 52, 1198–1207. [Google Scholar] [CrossRef]
- Pellicena, P.; Schulman, H. CaMKII inhibitors: From research tools to therapeutic agents. Front. Pharmacol. 2014, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Quartara, L.; Maggi, C.A. The tachykinin NK1 receptor. Part I: Ligands and mechanisms of cellular activation. Neuropeptides 1997, 31, 537–563. [Google Scholar] [CrossRef]
- Muñoz, M.; Coveñas, R. Involvement of substance P and the NK-1 receptor in cancer progression. Peptides 2013, 48, 1–9. [Google Scholar] [CrossRef]
- Ghahremani, F.; Sabbaghzadeh, R.; Ebrahimi, S.; Javid, H.; Ghahremani, J.; Hashemy, S.I. Pathogenic role of the SP/NK1R system in GBM cells through inhibiting the thioredoxin system. Iran. J. Basic Med. Sci. 2021, 24, 499–505. [Google Scholar]
- Takeda, Y.; Chou, K.B.; Takeda, J.; Sachais, B.S.; Krause, J.E. Molecular cloning, structural characterization and functional expression of the human substance P receptor. Biochem. Biophys. Res. Commun. 1991, 179, 1232–1240. [Google Scholar] [CrossRef]
- Afshari, A.R.; Motamed-Sanaye, A.; Sabri, H.; Soltani, A.; Karkon-Shayan, S.; Radvar, S.; Javid, H.; Mollazadeh, H.; Sathyapalan, T.; Sahebkar, A. Neurokinin-1 receptor (NK-1R) antagonists: Potential targets in the treatment of glioblastoma multiforme. Curr. Med. Chem. 2021, 28, 4877–4892. [Google Scholar] [CrossRef]
- Mou, L.; Kang, Y.; Zhou, Y.; Zeng, Q.; Song, H.; Wang, R. Neurokinin-1 receptor directly mediates glioma cell migration by up-regulation of matrix metalloproteinase-2 (MMP-2) and membrane type 1-matrix metalloproteinase (MT1-MMP). J. Biol. Chem. 2013, 288, 306–318. [Google Scholar] [CrossRef] [Green Version]
- Akazawa, T.; Kwatra, S.G.; Goldsmith, L.E.; Richardson, M.D.; Cox, E.A.; Sampson, J.H.; Kwatra, M.M. A constitutively active form of neurokinin 1 receptor and neurokinin 1 receptor-mediated apoptosis in glioblastomas. J. Neurochem. 2009, 109, 1079–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Kanduluru, A.K.; Desai, P.; Ahad, A.; Carlin, S.; Tandon, N.; Weber, W.A.; Low, P.S. Synthesis and evaluation of a novel 64 Cu- and 67 Ga-labeled neurokinin 1 receptor antagonist for in vivo targeting of NK1R-positive tumor xenografts. Bioconjug. Chem. 2018, 29, 1319–1326. [Google Scholar] [CrossRef]
- Kast, R.E. Glioblastoma: Synergy of growth promotion between CCL5 and NK-1R can be thwarted by blocking CCL5 with miraviroc, an FDA approved anti-HIV drug and blocking NK-1R with aprepitant, an FDA approved anti-nausea drug. J. Clin. Pharm. Ther. 2010, 35, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Skaga, E.; Skaga, I.Ø.; Grieg, Z.; Sandberg, C.J.; Langmoen, I.A.; Vik-Mo, E.O. The efficacy of a coordinated pharmacological blockade in glioblastoma stem cells with nine repurposed drugs using the CUSP9 strategy. J. Cancer Res. Clin. Oncol. 2019, 145, 1495–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halatsch, M.E.; Kast, R.E.; Karpel-Massler, G.; Mayer, B.; Zolk, O.; Schmitz, B.; Scheuerle, A.; Maier, L.; Bullinger, L.; Mayer-Steinacker, R.; et al. A phase Ib/IIa trial of 9 repurposed drugs combined with temozolomide for the treatment of recurrent glioblastoma: CUSP9v3. Neuro-Oncol. Adv. 2021, 3, vdab075. [Google Scholar] [CrossRef]
- Javid, H.; Afshari, A.R.; Zahedi Avval, F.; Asadi, J.; Hashemy, S.I. Aprepitant promotes caspase-dependent apoptotic cell death and G2/M arrest through PI3K/Akt/NF-κB axis in cancer stem-like esophageal squamous cell carcinoma spheres. Biomed. Res. Int. 2021, 2021, 8808214. [Google Scholar] [CrossRef]
- Jiao, Y.; Li, H.; Liu, Y.; Guo, A.; Xu, X.; Qu, X.; Wang, S.; Zhao, J.; Li, Y.; Cao, Y. Resveratrol inhibits the invasion of glioblastoma-initiating cells via down-regulation of the PI3K/Akt/NF-κB signaling pathway. Nutrients 2015, 7, 4383–4402. [Google Scholar] [CrossRef] [Green Version]
- Tuluc, F.; Lai, J.P.; Kilpatrick, L.E.; Evans, D.L.; Douglas, S.D. Neurokinin 1 receptor isoforms and the control of innate immunity. Trends Immunol. 2009, 30, 271–276. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.M.; Kim, Y.J.; Jung, H.J. Discovery of a New CaMKII-Targeted Synthetic Lethal Therapy against Glioblastoma Stem-like Cells. Cancers 2022, 14, 1315. https://doi.org/10.3390/cancers14051315
Han JM, Kim YJ, Jung HJ. Discovery of a New CaMKII-Targeted Synthetic Lethal Therapy against Glioblastoma Stem-like Cells. Cancers. 2022; 14(5):1315. https://doi.org/10.3390/cancers14051315
Chicago/Turabian StyleHan, Jang Mi, Yu Jin Kim, and Hye Jin Jung. 2022. "Discovery of a New CaMKII-Targeted Synthetic Lethal Therapy against Glioblastoma Stem-like Cells" Cancers 14, no. 5: 1315. https://doi.org/10.3390/cancers14051315
APA StyleHan, J. M., Kim, Y. J., & Jung, H. J. (2022). Discovery of a New CaMKII-Targeted Synthetic Lethal Therapy against Glioblastoma Stem-like Cells. Cancers, 14(5), 1315. https://doi.org/10.3390/cancers14051315