
Chemotherapy Resistance: Role of Mitochondrial and Autophagic Components


Abstract
:Simple Summary
Abstract
1. Introduction
2. Potential Conventional Mitochondrial Targets for Chemotherapy
2.1. Mitochondrial Calcium Ion (Ca2+)
2.1.1. The Voltage-Dependent Anion Channel (VDAC)
2.1.2. The Mitochondrial Calcium Uniporter (MCU) Complex
2.2. Mitochondrial Permeability Transition Pore Complex (mPTPC)
2.3. ATP Synthase and Mitochondrial Reactive Oxygen Species (mtROS)
2.4. Mitochondrial DNA (mtDNA)
2.5. DNA Polymerase Subunit Gamma (PolG)
2.6. Mitochondrial Ribosome and Ribosomal Proteins (MRP)
2.7. Mitochondrial Proteins
3. Novel Metabolic Switch Targeted Anti-Cancer Therapeutic Approaches
3.1. Oxidative Phosphorylation (OXPHOS)
3.2. The Mitochondrial Respiratory Chain (mRC)
3.3. Mitochondrial Metabolic Regulation and Drug Resistance
4. Autophagy: A Potential Target in Cancer Treatment
4.1. Autophagy
4.2. The Role of Autophagy in Cancer Treatment
5. Cancer Chemoresistance: Mitochondrial Fusion, Fission, Biogenesis, and Mitophagy
5.1. Mitochondrial Fusion and Fission
5.2. Mitochondrial Biogenesis and Mitochondrial Selective Autophagy (Mitophagy)
5.3. Autophagy Acts as a Chemotherapy Resistance Machinery in the Mitochondrial Pathway
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| ADP | adenosine biphosphate |
| ATP | adenosine triphosphate |
| ATGs | autophagy genes |
| AMPK | AMP-activated protein kinase |
| BCL-2 | B-cell lymphoma 2 |
| Ca2+ | calcium ion |
| CaMKK | calcium/calmodulin-dependent protein kinase kinase |
| CsA | cyclosporin A |
| DNA | deoxyribonucleic acid |
| mtDNA | mitochondrial DNA |
| DNA-PKcs | DNA-dependent protein kinase catalytic subunit |
| Drp1 | dynamin-related protein 1 |
| DRAM | damage regulated autophagy modulator |
| DAPK1 | death-associated protein kinase 1 |
| DNA-PKcs | DNA-dependent protein kinase catalytic subunit |
| mtDNA | mitochondrial deoxyribonucleic acid (DNA) |
| ERK | extracellular signal regulated kinases |
| ETC | electron transport chain |
| eIF2 | eukaryotic initiation factor 2 |
| FADH2 | adenine dinucleotide |
| FIP200 | focal adhesion kinase family interacting protein of 200 kDa |
| GDP | guanosine diphosphate |
| GT | guanosine triphosphate |
| GLUT | glucose transporter |
| HK | hexokinase |
| HMGB1 | high mobility group box 1 |
| HMGN5 | high-mobility group nucleosome-binding domain 5 |
| HSP90AA1 | heat shock protein 90AA1 |
| IGF2 | insulin growth factor 2 |
| IMM | inner mitochondrial membrane |
| IRS1/2 | insulin receptor substrate 1 and 2 |
| JNK | c-Jun N-terminal kinase |
| LC3 I/II | light chain I and II |
| LDH | lactate dehydrogenase |
| LKB1 | liver kinase B1 |
| MCT | monocarboxylate transporters |
| MCU | mitochondrial calcium uniporter |
| MEK | mitogen-activated protein kinase/ERK kinase |
| MAM | mitochondrial-associated membrane |
| mtROS | mitochondria-derived reactive oxygen species |
| MOMP | mitochondrial outer membrane permeabilization |
| Mnf1/2 | mitofusin 1 and 2 |
| NADH | nicotinamide adenine dinucleotide (NAD) hydrogen |
| NDRG1 | N-myc down-stream regulated gene 1 |
| OPA1 | optic atrophy 1 |
| OXPHOS | oxidative phosphorylation |
| OMM | outer mitochondrial membrane |
| PI3K | phosphatidylinositol 3-kinase |
| PDK1 | phosphoinositide-dependent protein kinase 1 |
| PERK | protein kinase R (PKR)-like endoplasmic reticulum kinase |
| PE | phosphatidylethanolamine |
| PTEN | phosphatase and tensin |
| PIP2 | phosphatidylinositol-4,5-bisphosphate |
| PIP3 | phosphatidylinositol-3,4,5-trisphosphate |
| PKB | phosphoinositide-dependent protein kinase B |
| PGC1α | proliferator-activated receptor-gamma co-activator 1-alpha |
| PFK | phosphofructokinase |
| PGAM | phosphoglycerate mutase |
| PI3K | phosphatidylinositol 3-kinase |
| PolG | DNA polymerase gamma subunit |
| mPPT | mitochondrial permeability transition pore complex |
| RAS | rat sarcoma |
| RAF | rapidly accelerated fibrosarcoma |
| RCC | respiratory chain complex |
| Rheb | ras homolog enriched in the brain |
| ROS | reactive oxygen species |
| mRNA | messenger ribonucleic acid |
| tRNA | transfer ribonucleic acid |
| TCA | tricarboxylic acid |
| TIGAR | TP53-induced glycolysis and apoptosis regulator |
| TSC1/2 | tuberous sclerosis complex 1 and 2 |
| mTOR | mammalian target of rapamycin complex |
| ULK1 | unc-51-like kinase 1 |
| VDAC1 | voltage-dependent anion channel 1 |
| VPS34 | vacuolar protein sorting 34 |
References
- Yeldag, G.; Rice, A.; Del Rio Hernandez, A. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers 2018, 10, 471. [Google Scholar] [CrossRef] [Green Version]
- Assaraf, Y.G.; Brozovic, A.; Goncalves, A.C.; Jurkovicova, D.; Line, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updates 2019, 46, 100645. [Google Scholar] [CrossRef] [PubMed]
- Vaidya, F.U.; Sufiyan Chhipa, A.; Mishra, V.; Gupta, V.K.; Rawat, S.G.; Kumar, A.; Pathak, C. Molecular and Cellular Paradigms of Multidrug Resistance in Cancer. In Cancer Reports; Wiley Online Library: Hoboken, NJ, USA, 2020; p. e1291. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, J.Y.; Yoon, H. Chemotherapy Resistance Explained through Endoplasmic Reticulum Stress-Dependent Signaling. Cancers 2019, 11, 338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahar, E.; Kim, J.Y.; Kim, H.S.; Yoon, H. Establishment of Acquired Cisplatin Resistance in Ovarian Cancer Cell Lines Characterized by Enriched Metastatic Properties with Increased Twist Expression. Int. J. Mol. Sci. 2020, 21, 7613. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, J.Y.; Kim, D.C.; Kim, H.S.; Yoon, H. Combination of Niraparib, Cisplatin and Twist Knockdown in Cisplatin-Resistant Ovarian Cancer Cells Potentially Enhances Synthetic Lethality through ER-Stress Mediated Mitochondrial Apoptosis Pathway. Int. J. Mol. Sci. 2021, 22, 3916. [Google Scholar] [CrossRef] [PubMed]
- Bohovych, I.; Chan, S.S.; Khalimonchuk, O. Mitochondrial protein quality control: The mechanisms guarding mitochondrial health. Antioxid. Redox Signal. 2015, 22, 977–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, H.H.; Cao, Y.T.; Zhang, L.L.; Huang, F.; Yi, C. The Role of Mitochondrial Dynamics and Mitophagy in Carcinogenesis, Metastasis and Therapy. Front. Cell Dev. Biol. 2020, 8, 413. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Feng, Z.; Gu, J.; Li, X.; Dong, Q.; Liu, K.; Li, Y.; OuYang, L. microRNA-488 inhibits chemoresistance of ovarian cancer cells by targeting Six1 and mitochondrial function. Oncotarget 2017, 8, 80981–80993. [Google Scholar] [CrossRef] [Green Version]
- Osellame, L.D.; Blacker, T.S.; Duchen, M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The Mitochondrion. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK26894/ (accessed on 2 February 2022).
- Avendano-Monsalve, M.C.; Ponce-Rojas, J.C.; Funes, S. From cytosol to mitochondria: The beginning of a protein journey. Biol. Chem. 2020, 401, 645–661. [Google Scholar] [CrossRef]
- Scaglia, F.; Wong, L.J. Human mitochondrial transfer RNAs: Role of pathogenic mutation in disease. Muscle Nerve 2008, 37, 150–171. [Google Scholar] [CrossRef] [PubMed]
- Fox, T.D. Mitochondrial protein synthesis, import, and assembly. Genetics 2012, 192, 1203–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; D’Agostino, D.P. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef]
- D’Souza, G.G.; Wagle, M.A.; Saxena, V.; Shah, A. Approaches for targeting mitochondria in cancer therapy. Biochim. Biophys. Acta 2011, 1807, 689–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiendleder, S.; Schmutz, S.M.; Erhardt, G.; Green, R.D.; Plante, Y. Transmitochondrial differences and varying levels of heteroplasmy in nuclear transfer cloned cattle. Mol. Reprod. Dev. 1999, 54, 24–31. [Google Scholar] [CrossRef]
- Gulbins, E.; Dreschers, S.; Bock, J. Role of mitochondria in apoptosis. Exp. Physiol. 2003, 88, 85–90. [Google Scholar] [CrossRef]
- Waterhouse, N.J.; Goldstein, J.C.; Kluck, R.M.; Newmeyer, D.D.; Green, D.R. The (Holey) study of mitochondria in apoptosis. Methods Cell Biol. 2001, 66, 365–391. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnoczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529 Pt 1, 57–68. [Google Scholar] [CrossRef]
- Sterea, A.M.; El Hiani, Y. The Role of Mitochondrial Calcium Signaling in the Pathophysiology of Cancer Cells. Adv. Exp. Med. Biol. 2020, 1131, 747–770. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Stowe, D.F.; Camara, A.K. Mitochondrial reactive oxygen species production in excitable cells: Modulators of mitochondrial and cell function. Antioxid. Redox Signal. 2009, 11, 1373–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A. VDAC1 functions in Ca2+ homeostasis and cell life and death in health and disease. Cell Calcium 2018, 69, 81–100. [Google Scholar] [CrossRef]
- Shoshan-Barmatz, V.; Ben-Hail, D.; Admoni, L.; Krelin, Y.; Tripathi, S.S. The mitochondrial voltage-dependent anion channel 1 in tumor cells. Biochim. Biophys. Acta 2015, 1848, 2547–2575. [Google Scholar] [CrossRef] [Green Version]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A.; Arif, T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front. Oncol. 2017, 7, 154. [Google Scholar] [CrossRef]
- Camara, A.K.S.; Zhou, Y.; Wen, P.C.; Tajkhorshid, E.; Kwok, W.M. Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front. Physiol. 2017, 8, 460. [Google Scholar] [CrossRef] [Green Version]
- Guzun, R.; Gonzalez-Granillo, M.; Karu-Varikmaa, M.; Grichine, A.; Usson, Y.; Kaambre, T.; Guerrero-Roesch, K.; Kuznetsov, A.; Schlattner, U.; Saks, V. Regulation of respiration in muscle cells in vivo by VDAC through interaction with the cytoskeleton and MtCK within Mitochondrial Interactosome. Biochim. Biophys. Acta 2012, 1818, 1545–1554. [Google Scholar] [CrossRef]
- Colombini, M. Voltage gating in the mitochondrial channel, VDAC. J. Membr. Biol. 1989, 111, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Zakar, M.; Rosenthal, K.; Abu-Hamad, S. Key regions of VDAC1 functioning in apoptosis induction and regulation by hexokinase. Biochim. Biophys. Acta 2009, 1787, 421–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, T.; Colombini, M. Regulation of metabolite flux through voltage-gating of VDAC channels. J. Membr. Biol. 1997, 157, 271–279. [Google Scholar] [CrossRef]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Gomez, L.; Thiebaut, P.A.; Paillard, M.; Ducreux, S.; Abrial, M.; Crola Da Silva, C.; Durand, A.; Alam, M.R.; Van Coppenolle, F.; Sheu, S.S.; et al. The SR/ER-mitochondria calcium crosstalk is regulated by GSK3beta during reperfusion injury. Cell Death Differ. 2016, 23, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273. [Google Scholar] [CrossRef] [Green Version]
- Keinan, N.; Pahima, H.; Ben-Hail, D.; Shoshan-Barmatz, V. The role of calcium in VDAC1 oligomerization and mitochondria-mediated apoptosis. Biochim. Biophys. Acta 2013, 1833, 1745–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camara, A.K.; Aldakkak, M.; Heisner, J.S.; Rhodes, S.S.; Riess, M.L.; An, J.; Heinen, A.; Stowe, D.F. ROS scavenging before 27 degrees C ischemia protects hearts and reduces mitochondrial ROS, Ca2+ overload, and changes in redox state. Am. J. Physiol. Cell Physiol. 2007, 292, C2021–C2031. [Google Scholar] [CrossRef]
- Camara, A.K.; Bienengraeber, M.; Stowe, D.F. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front. Physiol. 2011, 2, 13. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.; Kuznetsov, A. Mitochondrial permeability transition and cell death: The role of cyclophilin d. Front. Physiol. 2013, 4, 76. [Google Scholar] [CrossRef] [Green Version]
- Javadov, S.; Karmazyn, M.; Escobales, N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J. Pharmacol. Exp. Ther. 2009, 330, 670–678. [Google Scholar] [CrossRef] [Green Version]
- Azoulay-Zohar, H.; Israelson, A.; Abu-Hamad, S.; Shoshan-Barmatz, V. In self-defence: Hexokinase promotes voltage-dependent anion channel closure and prevents mitochondria-mediated apoptotic cell death. Biochem. J. 2004, 377, 347–355. [Google Scholar] [CrossRef]
- Zaid, H.; Abu-Hamad, S.; Israelson, A.; Nathan, I.; Shoshan-Barmatz, V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005, 12, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Konishi, A.; Kodama, T.; Tsujimoto, Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitoch.hondrial changes and cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 3100–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, S.; Matsuoka, Y.; Shinohara, Y.; Yoneda, Y.; Tsujimoto, Y. Essential role of voltage-dependent anion channel in various forms of apoptosis in mammalian cells. J. Cell Biol. 2001, 152, 237–250. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Shimizu, S. VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ. 2000, 7, 1174–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Vashisht, A.A.; Tchieu, J.; Wohlschlegel, J.A.; Dreier, L. Voltage-dependent anion channels (VDACs) recruit Parkin to defective mitochondria to promote mitochondrial autophagy. J. Biol. Chem. 2012, 287, 40652–40660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol. 2015, 16, 545–553. [Google Scholar] [CrossRef]
- Chaudhuri, D.; Clapham, D.E. Outstanding questions regarding the permeation, selectivity, and regulation of the mitochondrial calcium uniporter. Biochem. Biophys. Res. Commun. 2014, 449, 367–369. [Google Scholar] [CrossRef]
- Pendin, D.; Greotti, E.; Pozzan, T. The elusive importance of being a mitochondrial Ca2+ uniporter. Cell Calcium 2014, 55, 139–145. [Google Scholar] [CrossRef]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca2+ and apoptosis. Cell Calcium 2012, 52, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Hajnoczky, G.; Csordas, G.; Das, S.; Garcia-Perez, C.; Saotome, M.; Sinha Roy, S.; Yi, M. Mitochondrial calcium signalling and cell death: Approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium 2006, 40, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Bonora, M.; Patergnani, S.; Rimessi, A.; De Marchi, E.; Suski, J.M.; Bononi, A.; Giorgi, C.; Marchi, S.; Missiroli, S.; Poletti, F.; et al. ATP synthesis and storage. Purinergic Signal. 2012, 8, 343–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallilankaraman, K.; Cardenas, C.; Doonan, P.J.; Chandramoorthy, H.C.; Irrinki, K.M.; Golenar, T.; Csordas, G.; Madireddi, P.; Yang, J.; Muller, M.; et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat. Cell Biol. 2012, 14, 1336–1343. [Google Scholar] [CrossRef] [Green Version]
- Mallilankaraman, K.; Doonan, P.; Cardenas, C.; Chandramoorthy, H.C.; Muller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgo, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [Green Version]
- Alam, M.R.; Groschner, L.N.; Parichatikanond, W.; Kuo, L.; Bondarenko, A.I.; Rost, R.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Mitochondrial Ca2+ uptake 1 (MICU1) and mitochondrial Ca2+ uniporter (MCU) contribute to metabolism-secretion coupling in clonal pancreatic beta-cells. J. Biol. Chem. 2012, 287, 34445–34454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomar, D.; Dong, Z.; Shanmughapriya, S.; Koch, D.A.; Thomas, T.; Hoffman, N.E.; Timbalia, S.A.; Goldman, S.J.; Breves, S.L.; Corbally, D.P.; et al. MCUR1 Is a Scaffold Factor for the MCU Complex Function and Promotes Mitochondrial Bioenergetics. Cell Rep. 2016, 15, 1673–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Ozono, M.; Watanabe, A.; Maeda, K.; Nara, A.; Hashida, M.; Ido, Y.; Hiroshima, Y.; Yamada, A.; Terada, H.; et al. Functional analysis of coiled-coil domains of MCU in mitochondrial calcium uptake. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 148061. [Google Scholar] [CrossRef] [PubMed]
- Garg, V.; Suzuki, J.; Paranjpe, I.; Unsulangi, T.; Boyman, L.; Milescu, L.S.; Lederer, W.J.; Kirichok, Y. The mechanism of MICU-dependent gating of the mitochondrial Ca2+uniporter. eLife 2021, 10, e69312. [Google Scholar] [CrossRef]
- Suh, D.H.; Kim, M.K.; Kim, H.S.; Chung, H.H.; Song, Y.S. Mitochondrial permeability transition pore as a selective target for anti-cancer therapy. Front. Oncol. 2013, 3, 41. [Google Scholar] [CrossRef] [Green Version]
- Brenner, C.; Grimm, S. The permeability transition pore complex in cancer cell death. Oncogene 2006, 25, 4744–4756. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blachly-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.; Brdiczka, D. The permeability transition pore in cell death. Apoptosis 2007, 12, 841–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasola, A.; Bernardi, P. The mitochondrial permeability transition pore and its adaptive responses in tumor cells. Cell Calcium 2014, 56, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Kent, A.C.; El Baradie, K.B.Y.; Hamrick, M.W. Targeting the Mitochondrial Permeability Transition Pore to Prevent Age-Associated Cell Damage and Neurodegeneration. Oxid. Med. Cell. Longev. 2021, 2021, 6626484. [Google Scholar] [CrossRef]
- Zunino, S.J.; Storms, D.H. Resveratrol-induced apoptosis is enhanced in acute lymphoblastic leukemia cells by modulation of the mitochondrial permeability transition pore. Cancer Lett. 2006, 240, 123–134. [Google Scholar] [CrossRef]
- Ling, X.; Zhou, Y.; Li, S.W.; Yan, B.; Wen, L. Modulation of mitochondrial permeability transition pore affects multidrug resistance in human hepatocellular carcinoma cells. Int. J. Biol. Sci. 2010, 6, 773–783. [Google Scholar] [CrossRef] [Green Version]
- Huttemann, M.; Lee, I.; Samavati, L.; Yu, H.; Doan, J.W. Regulation of mitochondrial oxidative phosphorylation through cell signaling. Biochim. Biophys. Acta 2007, 1773, 1701–1720. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Bermudez, J.; Cuezva, J.M. The ATPase Inhibitory Factor 1 (IF1): A master regulator of energy metabolism and of cell survival. Biochim. Biophys. Acta 2016, 1857, 1167–1182. [Google Scholar] [CrossRef]
- Seelert, H.; Dencher, N.A. ATP synthase superassemblies in animals and plants: Two or more are better. Biochim. Biophys. Acta 2011, 1807, 1185–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucharczyk, R.; Zick, M.; Bietenhader, M.; Rak, M.; Couplan, E.; Blondel, M.; Caubet, S.D.; di Rago, J.P. Mitochondrial ATP synthase disorders: Molecular mechanisms and the quest for curative therapeutic approaches. Biochim. Biophys. Acta 2009, 1793, 186–199. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.C.; Chang, H.Y.; Hsu, C.H.; Kuo, W.H.; Chang, K.J.; Juan, H.F. Targeting therapy for breast carcinoma by ATP synthase inhibitor aurovertin B. J. Proteome Res. 2008, 7, 1433–1444. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Huang, H.C.; Huang, T.C.; Yang, P.C.; Wang, Y.C.; Juan, H.F. Ectopic ATP synthase blockade suppresses lung adenocarcinoma growth by activating the unfolded protein response. Cancer Res. 2012, 72, 4696–4706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.Y.; Huang, T.C.; Chen, N.N.; Huang, H.C.; Juan, H.F. Combination therapy targeting ectopic ATP synthase and 26S proteasome induces ER stress in breast cancer cells. Cell Death Dis. 2014, 5, e1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Ramirez, V.D. Inhibition of mitochondrial proton F0F1-ATPase/ATP synthase by polyphenolic phytochemicals. Br. J. Pharmacol. 2000, 130, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Tuquet, C.; Dupont, J.; Mesneau, A.; Roussaux, J. Effects of tamoxifen on the electron transport chain of isolated rat liver mitochondria. Cell Biol. Toxicol 2000, 16, 207–219. [Google Scholar] [CrossRef]
- Kallio, A.; Zheng, A.; Dahllund, J.; Heiskanen, K.M.; Harkonen, P. Role of mitochondria in tamoxifen-induced rapid death of MCF-7 breast cancer cells. Apoptosis 2005, 10, 1395–1410. [Google Scholar] [CrossRef]
- Shchepina, L.A.; Pletjushkina, O.Y.; Avetisyan, A.V.; Bakeeva, L.E.; Fetisova, E.K.; Izyumov, D.S.; Saprunova, V.B.; Vyssokikh, M.Y.; Chernyak, B.V.; Skulachev, V.P. Oligomycin, inhibitor of the F0 part of H+-ATP-synthase, suppresses the TNF-induced apoptosis. Oncogene 2002, 21, 8149–8157. [Google Scholar] [CrossRef] [Green Version]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Shadel, G.S.; Horvath, T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell 2015, 163, 560–569. [Google Scholar] [CrossRef] [Green Version]
- Gammella, E.; Recalcati, S.; Cairo, G. Dual Role of ROS as Signal and Stress Agents: Iron Tips the Balance in favor of Toxic Effects. Oxid. Med. Cell. Longev. 2016, 2016, 8629024. [Google Scholar] [CrossRef] [Green Version]
- Diebold, L.; Chandel, N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016, 100, 86–93. [Google Scholar] [CrossRef]
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398. [Google Scholar] [CrossRef] [Green Version]
- Sipos, I.; Tretter, L.; Adam-Vizi, V. Quantitative relationship between inhibition of respiratory complexes and formation of reactive oxygen species in isolated nerve terminals. J. Neurochem. 2003, 84, 112–118. [Google Scholar] [CrossRef]
- Zhen, D.; Chen, Y.; Tang, X. Metformin reverses the deleterious effects of high glucose on osteoblast function. J. Diabetes Complicat. 2010, 24, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohen, R.; Nyska, A. Oxidation of biological systems: Oxidative stress phenomena, antioxidants, redox reactions, and methods for their quantification. Toxicol. Pathol. 2002, 30, 620–650. [Google Scholar] [CrossRef] [Green Version]
- Kruspig, B.; Valter, K.; Skender, B.; Zhivotovsky, B.; Gogvadze, V. Targeting succinate:ubiquinone reductase potentiates the efficacy of anticancer therapy. Biochim. Biophys. Acta 2016, 1863, 2065–2071. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenk, B.; Fulda, S. Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death. Oncogene 2015, 34, 5796–5806. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Tuveson, D.A. The promise and perils of antioxidants for cancer patients. N. Engl. J. Med. 2014, 371, 177–178. [Google Scholar] [CrossRef] [PubMed]
- Dall’Acqua, S.; Linardi, M.A.; Bortolozzi, R.; Clauser, M.; Marzocchini, S.; Maggi, F.; Nicoletti, M.; Innocenti, G.; Basso, G.; Viola, G. Natural daucane esters induces apoptosis in leukaemic cells through ROS production. Phytochemistry 2014, 108, 147–156. [Google Scholar] [CrossRef]
- Castaldo, S.A.; Freitas, J.R.; Conchinha, N.V.; Madureira, P.A. The Tumorigenic Roles of the Cellular REDOX Regulatory Systems. Oxid. Med. Cell. Longev. 2016, 2016, 8413032. [Google Scholar] [CrossRef] [Green Version]
- Isaia, G.C.; Limone, P.; Bodrato, L.; Avataneo, T.; Potenzoni, F.; Gremo, L.; Molinatti, G.M. Is magnetic resonance imaging more sensitive than computed tomography in the location of corticotropin secreting pituitary adenomas? Ann. Ital. Med. Int. 1989, 4, 122–124. [Google Scholar]
- Hayes, J.D.; McMahon, M. NRF2 and KEAP1 mutations: Permanent activation of an adaptive response in cancer. Trends Biochem. Sci. 2009, 34, 176–188. [Google Scholar] [CrossRef]
- Greenlee, H.; Kwan, M.L.; Kushi, L.H.; Song, J.; Castillo, A.; Weltzien, E.; Quesenberry, C.P., Jr.; Caan, B.J. Antioxidant supplement use after breast cancer diagnosis and mortality in the Life After Cancer Epidemiology (LACE) cohort. Cancer 2012, 118, 2048–2058. [Google Scholar] [CrossRef] [Green Version]
- Harris, H.R.; Orsini, N.; Wolk, A. Vitamin C and survival among women with breast cancer: A meta-analysis. Eur. J. Cancer 2014, 50, 1223–1231. [Google Scholar] [CrossRef]
- Bober, P.; Alexovic, M.; Talian, I.; Tomkova, Z.; Viscorova, Z.; Benckova, M.; Andrasina, I.; Ciccocioppo, R.; Petrovic, D.; Adamek, M.; et al. Proteomic analysis of the vitamin C effect on the doxorubicin cytotoxicity in the MCF-7 breast cancer cell line. J. Cancer Res. Clin. Oncol. 2017, 143, 35–42. [Google Scholar] [CrossRef]
- Printz, C. High vitamin D levels increase survival rates in patients with metastatic colorectal cancer. Cancer 2015, 121, 2105. [Google Scholar] [CrossRef]
- DiMauro, S. Mitochondrial diseases. Biochim. Biophys. Acta 2004, 1658, 80–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisnovsky, S.P.; Wilson, J.J.; Radford, R.J.; Pereira, M.P.; Chan, M.R.; Laposa, R.R.; Lippard, S.J.; Kelley, S.O. Targeting mitochondrial DNA with a platinum-based anticancer agent. Chem. Biol. 2013, 20, 1323–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, K.J.; Yang, Z.; Schumaker, L.; Guo, Z. Mitochondria as a critical target of the chemotheraputic agent cisplatin in head and neck cancer. J. Bioenerg. Biomembr. 2007, 39, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Hibasami, H.; Maekawa, S.; Tagawa, T.; Nakashima, K. Preferential binding of cisplatin to mitochondrial DNA and suppression of ATP generation in human malignant melanoma cells. Biochem. Int. 1990, 20, 949–955. [Google Scholar]
- Yang, Z.; Schumaker, L.M.; Egorin, M.J.; Zuhowski, E.G.; Guo, Z.; Cullen, K.J. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: Possible role in apoptosis. Clin. Cancer Res. 2006, 12, 5817–5825. [Google Scholar] [CrossRef] [Green Version]
- Lomeli, N.; Di, K.; Czerniawski, J.; Guzowski, J.F.; Bota, D.A. Cisplatin-induced mitochondrial dysfunction is associated with impaired cognitive function in rats. Free Radic. Biol. Med. 2017, 102, 274–286. [Google Scholar] [CrossRef] [Green Version]
- Podratz, J.L.; Knight, A.M.; Ta, L.E.; Staff, N.P.; Gass, J.M.; Genelin, K.; Schlattau, A.; Lathroum, L.; Windebank, A.J. Cisplatin induced mitochondrial DNA damage in dorsal root ganglion neurons. Neurobiol. Dis. 2011, 41, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Montopoli, M.; Bellanda, M.; Lonardoni, F.; Ragazzi, E.; Dorigo, P.; Froldi, G.; Mammi, S.; Caparrotta, L. “Metabolic reprogramming” in ovarian cancer cells resistant to cisplatin. Curr. Cancer Drug Targets 2011, 11, 226–235. [Google Scholar] [CrossRef]
- Copeland, W.C. Inherited mitochondrial diseases of DNA replication. Annu. Rev. Med. 2008, 59, 131–146. [Google Scholar] [CrossRef] [Green Version]
- Hance, N.; Ekstrand, M.I.; Trifunovic, A. Mitochondrial DNA polymerase gamma is essential for mammalian embryogenesis. Hum. Mol. Genet. 2005, 14, 1775–1783. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.H.; Naviaux, R.K. The clinical diagnosis of POLG disease and other mitochondrial DNA depletion disorders. Methods 2010, 51, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Ayyasamy, V.; Owens, K.M.; Koul, M.S.; Vujcic, M. Mutations in mitochondrial DNA polymerase-gamma promote breast tumorigenesis. J. Hum. Genet. 2009, 54, 516–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, J.S. Mitochondrial medicine: Pharmacological targeting of mitochondria in disease. Br. J. Pharmacol. 2007, 151, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Owens, K.M.; Bajpai, P.; Desouki, M.M.; Srinivasasainagendra, V.; Tiwari, H.K.; Singh, K.K. Mitochondrial DNA Polymerase POLG1 Disease Mutations and Germline Variants Promote Tumorigenic Properties. PLoS ONE 2015, 10, e0139846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copeland, W.C.; Longley, M.J. DNA polymerase gamma in mitochondrial DNA replication and repair. Sci. World J. 2003, 3, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinz, K.G.; Bogenhagen, D.F. The influence of the DNA polymerase gamma accessory subunit on base excision repair by the catalytic subunit. DNA Repair. 2006, 5, 121–128. [Google Scholar] [CrossRef]
- Yehudai, D.; Liyanage, S.U.; Hurren, R.; Rizoska, B.; Albertella, M.; Gronda, M.; Jeyaraju, D.V.; Wang, X.; Barghout, S.H.; MacLean, N.; et al. The thymidine dideoxynucleoside analog, alovudine, inhibits the mitochondrial DNA polymerase gamma, impairs oxidative phosphorylation and promotes monocytic differentiation in acute myeloid leukemia. Haematologica 2019, 104, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.S.; Copeland, W.C. DNA polymerase gamma and mitochondrial disease: Understanding the consequence of POLG mutations. Biochim. Biophys. Acta 2009, 1787, 312–319. [Google Scholar] [CrossRef] [Green Version]
- Attardi, G. Animal mitochondrial DNA: An extreme example of genetic economy. Int. Rev. Cytol. 1985, 93, 93–145. [Google Scholar] [CrossRef]
- Chomyn, A.; Cleeter, M.W.; Ragan, C.I.; Riley, M.; Doolittle, R.F.; Attardi, G. URF6, last unidentified reading frame of human mtDNA, codes for an NADH dehydrogenase subunit. Science 1986, 234, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, A.; Pagano, M.; Russo, G.; Russo, A. Ribosome Biogenesis and Cancer: Overview on Ribosomal Proteins. Int. J. Mol. Sci. 2021, 22, 5496. [Google Scholar] [CrossRef] [PubMed]
- Cavdar Koc, E.; Ranasinghe, A.; Burkhart, W.; Blackburn, K.; Koc, H.; Moseley, A.; Spremulli, L.L. A new face on apoptosis: Death-associated protein 3 and PDCD9 are mitochondrial ribosomal proteins. FEBS Lett. 2001, 492, 166–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.L.; Koc, H.; Koc, E.C. Identification of phosphorylation sites in mammalian mitochondrial ribosomal protein DAP3. Protein Sci. 2008, 17, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Yoo, Y.A.; Kim, M.J.; Park, J.K.; Chung, Y.M.; Lee, J.H.; Chi, S.G.; Kim, J.S.; Yoo, Y.D. Mitochondrial ribosomal protein L41 suppresses cell growth in association with p53 and p27Kip1. Mol. Cell. Biol. 2005, 25, 6603–6616. [Google Scholar] [CrossRef] [Green Version]
- Saini, N.; Balhara, J.; Adlakha, Y.K. Singh N: S29 Ribosomal protein induces mitochondria mediated apoptosis of Hep2 cells via the activation of p38 MAPK and JNK signaling. Int. J. Integr. Biol. 2009, 5, 49–57. [Google Scholar]
- Garand, C.; Guay, D.; Sereduk, C.; Chow, D.; Tsofack, S.P.; Langlois, M.; Perreault, E.; Yin, H.H.; Lebel, M. An integrative approach to identify YB-1-interacting proteins required for cisplatin resistance in MCF7 and MDA-MB-231 breast cancer cells. Cancer Sci. 2011, 102, 1410–1417. [Google Scholar] [CrossRef]
- Tsofack, S.P.; Meunier, L.; Sanchez, L.; Madore, J.; Provencher, D.; Mes-Masson, A.M.; Lebel, M. Low expression of the X-linked ribosomal protein S4 in human serous epithelial ovarian cancer is associated with a poor prognosis. BMC Cancer 2013, 13, 303. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Liu, Y.; Fremont, M.; Schwarz, S.; Siegmann, M.; Matthies, R.; Jost, J.P. A novel 52 kDa protein induces apoptosis and concurrently activates c-Jun N-terminal kinase 1 (JNK1) in mouse C3H10T1/2 fibroblasts. Gene 1998, 208, 157–166. [Google Scholar] [CrossRef]
- Gilkerson, R.W.; Margineantu, D.H.; Capaldi, R.A.; Selker, J.M. Mitochondrial DNA depletion causes morphological changes in the mitochondrial reticulum of cultured human cells. FEBS Lett. 2000, 474, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lomonosova, E.; Chinnadurai, G. BH3-only proteins in apoptosis and beyond: An overview. Oncogene 2008, 27 (Suppl. S1), S2–S19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell 2010, 37, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Lin, Y.; Wu, X. TRAIL-induced apoptosis requires Bax-dependent mitochondrial release of Smac/DIABLO. Genes Dev. 2002, 16, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Renault, T.T.; Floros, K.V.; Chipuk, J.E. BAK/BAX activation and cytochrome c release assays using isolated mitochondria. Methods 2013, 61, 146–155. [Google Scholar] [CrossRef] [Green Version]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a026716. [Google Scholar] [CrossRef] [Green Version]
- Kesavardhana, S.; Malireddi, R.K.S.; Kanneganti, T.D. Caspases in cell death, inflammation, and pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef] [Green Version]
- Imai, K.; Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef]
- Bender, A.; Opel, D.; Naumann, I.; Kappler, R.; Friedman, L.; von Schweinitz, D.; Debatin, K.M.; Fulda, S. PI3K inhibitors prime neuroblastoma cells for chemotherapy by shifting the balance towards pro-apoptotic Bcl-2 proteins and enhanced mitochondrial apoptosis. Oncogene 2011, 30, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Deng, J.; Shimamura, T.; Perera, S.; Carlson, N.E.; Cai, D.; Shapiro, G.I.; Wong, K.K.; Letai, A. Proapoptotic BH3-only BCL-2 family protein BIM connects death signaling from epidermal growth factor receptor inhibition to the mitochondrion. Cancer Res. 2007, 67, 11867–11875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gojo, I.; Zhang, B.; Fenton, R.G. The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1. Clin. Cancer Res. 2002, 8, 3527–3538. [Google Scholar] [PubMed]
- Sotgia, F.; Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.C.; Witkiewicz, A.K.; Howell, A.; Philp, N.J.; Pestell, R.G.; Lisanti, M.P. Mitochondrial metabolism in cancer metastasis: Visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle 2012, 11, 1445–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, E.L.; Pearce, E.J. Metabolic pathways in immune cell activation and quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Pavlides, S.; Sotgia, F.; Lisanti, M.P. Mitochondrial biogenesis drives tumor cell proliferation. Am. J. Pathol. 2011, 178, 1949–1952. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, C.; Puente-Moncada, N.; Reiter, R.J.; Sanchez-Sanchez, A.M.; Herrera, F.; Rodriguez-Blanco, J.; Duarte-Olivenza, C.; Turos-Cabal, M.; Antolin, I.; Martin, V. Regulation of cancer cell glucose metabolism is determinant for cancer cell fate after melatonin administration. J. Cell. Physiol. 2021, 236, 27–40. [Google Scholar] [CrossRef]
- Callao, V.; Montoya, E. Toxohormone-like factor from microorganisms with impaired respiration. Science 1961, 134, 2041–2042. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 749. [Google Scholar] [CrossRef]
- Akram, M. Mini-review on glycolysis and cancer. J. Cancer Educ. 2013, 28, 454–457. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.H.; Pelicano, H.; Zhou, Y.; Carew, J.S.; Feng, L.; Bhalla, K.N.; Keating, M.J.; Huang, P. Inhibition of glycolysis in cancer cells: A novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005, 65, 613–621. [Google Scholar] [PubMed]
- Wood, T.E.; Dalili, S.; Simpson, C.D.; Hurren, R.; Mao, X.; Saiz, F.S.; Gronda, M.; Eberhard, Y.; Minden, M.D.; Bilan, P.J.; et al. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol. Cancer Ther. 2008, 7, 3546–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraus, D.; Reckenbeil, J.; Veit, N.; Kuerpig, S.; Meisenheimer, M.; Beier, I.; Stark, H.; Winter, J.; Probstmeier, R. Targeting glucose transport and the NAD pathway in tumor cells with STF-31: A re-evaluation. Cell. Oncol. 2018, 41, 485–494. [Google Scholar] [CrossRef]
- Chen, X.S.; Li, L.Y.; Guan, Y.D.; Yang, J.M.; Cheng, Y. Anticancer strategies based on the metabolic profile of tumor cells: Therapeutic targeting of the Warburg effect. Acta Pharmacol. Sin. 2016, 37, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Walcher, W.; Petru, E. The obstetric risk in adolescent primiparous females. Geburtshilfe Frauenheilkd. 1989, 49, 491–493. [Google Scholar] [CrossRef]
- Hadjiolov, D. Glucose-6-phosphatase activity in primary rat liver tumors induced by high doses of 4-dimethylaminoazo-benzene. Z. Krebsforsch. 1969, 72, 43–46. [Google Scholar] [CrossRef]
- Weber, G.; Cantero, A. Glucose-6-phosphatase activity in normal, pre-cancerous, and neoplastic tissues. Cancer Res. 1955, 15, 105–108. [Google Scholar]
- Sancho, P.; Barneda, D.; Heeschen, C. Hallmarks of cancer stem cell metabolism. Br. J. Cancer 2016, 114, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Amadori, D.; Frassineti, G.L.; De Matteis, A.; Mustacchi, G.; Santoro, A.; Cariello, S.; Ferrari, M.; Nascimben, O.; Nanni, O.; Lombardi, A.; et al. Modulating effect of lonidamine on response to doxorubicin in metastatic breast cancer patients: Results from a multicenter prospective randomized trial. Breast Cancer Res. Treat. 1998, 49, 209–217. [Google Scholar] [CrossRef]
- Nath, K.; Nelson, D.S.; Heitjan, D.F.; Leeper, D.B.; Zhou, R.; Glickson, J.D. Lonidamine induces intracellular tumor acidification and ATP depletion in breast, prostate and ovarian cancer xenografts and potentiates response to doxorubicin. NMR Biomed. 2015, 28, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer’s Achilles’ heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, S.E.; Chandel, N.S. Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 2015, 11, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Griguer, C.E.; Oliva, C.R.; Gillespie, G.Y. Glucose metabolism heterogeneity in human and mouse malignant glioma cell lines. J. Neurooncol. 2005, 74, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.A.; Richardson, A.D.; Filipp, F.V.; Knutzen, C.A.; Chiang, G.G.; Ronai, Z.A.; Osterman, A.L.; Smith, J.W. Comparative metabolic flux profiling of melanoma cell lines: Beyond the Warburg effect. J. Biol. Chem. 2011, 286, 42626–42634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef]
- Christofk, H.R.; Vander Heiden, M.G.; Wu, N.; Asara, J.M.; Cantley, L.C. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 2008, 452, 181–186. [Google Scholar] [CrossRef]
- Koritzinsky, M. Metformin: A Novel Biological Modifier of Tumor Response to Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2015, 93, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitiello, G.A.; Medina, B.D.; Zeng, S.; Bowler, T.G.; Zhang, J.Q.; Loo, J.K.; Param, N.J.; Liu, M.; Moral, A.J.; Zhao, J.N.; et al. Mitochondrial Inhibition Augments the Efficacy of Imatinib by Resetting the Metabolic Phenotype of Gastrointestinal Stromal Tumor. Clin. Cancer Res. 2018, 24, 972–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinnaird, A.; Dromparis, P.; Saleme, B.; Gurtu, V.; Watson, K.; Paulin, R.; Zervopoulos, S.; Stenson, T.; Sutendra, G.; Pink, D.B.; et al. Metabolic Modulation of Clear-cell Renal Cell Carcinoma with Dichloroacetate, an Inhibitor of Pyruvate Dehydrogenase Kinase. Eur. Urol. 2016, 69, 734–744. [Google Scholar] [CrossRef]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109, djx071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutendra, G.; Michelakis, E.D. Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology. Front. Oncol. 2013, 3, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velpula, K.K.; Bhasin, A.; Asuthkar, S.; Tsung, A.J. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013, 73, 7277–7289. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wu, N.; Ma, L.; Liu, M.; Liu, G.; Zhang, Y.; Lin, X. Oleanolic acid suppresses aerobic glycolysis in cancer cells by switching pyruvate kinase type M isoforms. PLoS ONE 2014, 9, e91606. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.; Bhagat, S.D.; Gupta, A.; Samaiya, A.; Srivastava, A.; Shukla, S. Dietary-phytochemical mediated reversion of cancer-specific splicing inhibits Warburg effect in head and neck cancer. BMC Cancer 2019, 19, 1031. [Google Scholar] [CrossRef]
- Zu, X.L.; Guppy, M. Cancer metabolism: Facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 2004, 313, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Mayer, A. Availability, not respiratory capacity governs oxygen consumption of solid tumors. Int. J. Biochem. Cell Biol. 2012, 44, 1477–1481. [Google Scholar] [CrossRef] [PubMed]
- Yu, M. Generation, function and diagnostic value of mitochondrial DNA copy number alterations in human cancers. Life Sci. 2011, 89, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Reznik, E.; Miller, M.L.; Senbabaoglu, Y.; Riaz, N.; Sarungbam, J.; Tickoo, S.K.; Al-Ahmadie, H.A.; Lee, W.; Seshan, V.E.; Hakimi, A.A.; et al. Mitochondrial DNA copy number variation across human cancers. eLife 2016, 5, e10769. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; Garone, C.; Naini, A. Metabolic myopathies. Curr. Rheumatol. Rep. 2010, 12, 386–393. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S. Pathogenesis and treatment of mitochondrial myopathies: Recent advances. Acta Myol. 2010, 29, 333–338. [Google Scholar]
- Dimauro, S.; Rustin, P. A critical approach to the therapy of mitochondrial respiratory chain and oxidative phosphorylation diseases. Biochim. Biophys. Acta 2009, 1792, 1159–1167. [Google Scholar] [CrossRef] [Green Version]
- Bao, B.; Azmi, A.S.; Ali, S.; Zaiem, F.; Sarkar, F.H. Metformin may function as anti-cancer agent via targeting cancer stem cells: The potential biological significance of tumor-associated miRNAs in breast and pancreatic cancers. Ann. Transl. Med. 2014, 2, 59. [Google Scholar] [CrossRef]
- Brunmair, B.; Staniek, K.; Gras, F.; Scharf, N.; Althaym, A.; Clara, R.; Roden, M.; Gnaiger, E.; Nohl, H.; Waldhausl, W.; et al. Thiazolidinediones, like metformin, inhibit respiratory complex I: A common mechanism contributing to their antidiabetic actions? Diabetes 2004, 53, 1052–1059. [Google Scholar] [CrossRef] [Green Version]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef]
- Taddia, L.; D’Arca, D.; Ferrari, S.; Marraccini, C.; Severi, L.; Ponterini, G.; Assaraf, Y.G.; Marverti, G.; Costi, M.P. Inside the biochemical pathways of thymidylate synthase perturbed by anticancer drugs: Novel strategies to overcome cancer chemoresistance. Drug Resist. Updates 2015, 23, 20–54. [Google Scholar] [CrossRef]
- Apicella, M.; Giannoni, E.; Fiore, S.; Ferrari, K.J.; Fernandez-Perez, D.; Isella, C.; Granchi, C.; Minutolo, F.; Sottile, A.; Comoglio, P.M.; et al. Increased Lactate Secretion by Cancer Cells Sustains Non-cell-autonomous Adaptive Resistance to MET and EGFR Targeted Therapies. Cell Metab. 2018, 28, 848–865.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.M.; Alifano, M.; Lincet, H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist. Updates 2018, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Chen, S.; Yang, W.; Cheng, X.; Ye, Y.; Mao, J.; Wu, X.; Huang, L.; Ji, J. FGFR4 Links Glucose Metabolism and Chemotherapy Resistance in Breast Cancer. Cell. Physiol. Biochem. 2018, 47, 151–160. [Google Scholar] [CrossRef] [Green Version]
- Pelicano, H.; Zhang, W.; Liu, J.; Hammoudi, N.; Dai, J.; Xu, R.H.; Pusztai, L.; Huang, P. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: Role of mTOR pathway and therapeutic potential. Breast Cancer Res. 2014, 16, 434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varghese, E.; Samuel, S.M.; Liskova, A.; Samec, M.; Kubatka, P.; Busselberg, D. Targeting Glucose Metabolism to Overcome Resistance to Anticancer Chemotherapy in Breast Cancer. Cancers 2020, 12, 2252. [Google Scholar] [CrossRef]
- Samuel, S.M.; Varghese, E.; Koklesova, L.; Liskova, A.; Kubatka, P.; Busselberg, D. Counteracting Chemoresistance with Metformin in Breast Cancers: Targeting Cancer Stem Cells. Cancers 2020, 12, 2482. [Google Scholar] [CrossRef]
- Peng, J.; Cui, Y.; Xu, S.; Wu, X.; Huang, Y.; Zhou, W.; Wang, S.; Fu, Z.; Xie, H. Altered glycolysis results in drug-resistant in clinical tumor therapy. Oncol. Lett. 2021, 21, 369. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Miao, W.; Huang, M.; Li, L.; Dai, X.; Wang, Y. Elevated Hexokinase II Expression Confers Acquired Resistance to 4-Hydroxytamoxifen in Breast Cancer Cells. Mol. Cell. Proteom. 2019, 18, 2273–2284. [Google Scholar] [CrossRef] [PubMed]
- Krasnov, G.S.; Dmitriev, A.A.; Lakunina, V.A.; Kirpiy, A.A.; Kudryavtseva, A.V. Targeting VDAC-bound hexokinase II: A promising approach for concomitant anti-cancer therapy. Expert Opin. Ther. Targets 2013, 17, 1221–1233. [Google Scholar] [CrossRef]
- Catanzaro, D.; Gabbia, D.; Cocetta, V.; Biagi, M.; Ragazzi, E.; Montopoli, M.; Carrara, M. Silybin counteracts doxorubicin resistance by inhibiting GLUT1 expression. Fitoterapia 2018, 124, 42–48. [Google Scholar] [CrossRef]
- Scambia, G.; De Vincenzo, R.; Ranelletti, F.O.; Panici, P.B.; Ferrandina, G.; D’Agostino, G.; Fattorossi, A.; Bombardelli, E.; Mancuso, S. Antiproliferative effect of silybin on gynaecological malignancies: Synergism with cisplatin and doxorubicin. Eur. J. Cancer 1996, 32, 877–882. [Google Scholar] [CrossRef]
- Colombo, V.; Lupi, M.; Falcetta, F.; Forestieri, D.; D’Incalci, M.; Ubezio, P. Chemotherapeutic activity of silymarin combined with doxorubicin or paclitaxel in sensitive and multidrug-resistant colon cancer cells. Cancer Chemother. Pharmacol. 2011, 67, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Chuang, J.H.; Chou, M.H.; Tai, M.H.; Lin, T.K.; Liou, C.W.; Chen, T.; Hsu, W.M.; Wang, P.W. 2-Deoxyglucose treatment complements the cisplatin- or BH3-only mimetic-induced suppression of neuroblastoma cell growth. Int. J. Biochem. Cell Biol. 2013, 45, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Maschek, G.; Savaraj, N.; Priebe, W.; Braunschweiger, P.; Hamilton, K.; Tidmarsh, G.F.; De Young, L.R.; Lampidis, T.J. 2-deoxy-D-glucose increases the efficacy of adriamycin and paclitaxel in human osteosarcoma and non-small cell lung cancers in vivo. Cancer Res. 2004, 64, 31–34. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Mathur, R.; Dwarakanath, B.S. The glycolytic inhibitor 2-deoxy-D-glucose enhances the efficacy of etoposide in ehrlich ascites tumor-bearing mice. Cancer Biol. Ther. 2005, 4, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lena, M.; Lorusso, V.; Latorre, A.; Fanizza, G.; Gargano, G.; Caporusso, L.; Guida, M.; Catino, A.; Crucitta, E.; Sambiasi, D.; et al. Paclitaxel, cisplatin and lonidamine in advanced ovarian cancer. A phase II study. Eur. J. Cancer 2001, 37, 364–368. [Google Scholar] [CrossRef]
- Angioli, R.; Janicek, M.; Sevin, B.; Estape, R.; Averette, H.; Koechli, O.; Untch, M.; Penalver, M. Use of lonidamine to potentiate the effect of cisplatin and carboplatin on platinum resistant human ovarian cancer cells. Int. J. Oncol. 1997, 11, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Biroccio, A.; Del Bufalo, D.; Fanciulli, M.; Bruno, T.; Zupi, G.; Floridi, A. bcl-2 inhibits mitochondrial metabolism and lonidamine-induced apoptosis in adriamycin-resistant MCF7 cells. Int. J. Cancer 1999, 82, 125–130. [Google Scholar] [CrossRef]
- Li, Y.C.; Fung, K.P.; Kwok, T.T.; Lee, C.Y.; Suen, Y.K.; Kong, S.K. Mitochondrial targeting drug lonidamine triggered apoptosis in doxorubicin-resistant HepG2 cells. Life Sci. 2002, 71, 2729–2740. [Google Scholar] [CrossRef]
- Calvino, E.; Estan, M.C.; Simon, G.P.; Sancho, P.; Boyano-Adanez Mdel, C.; de Blas, E.; Breard, J.; Aller, P. Increased apoptotic efficacy of lonidamine plus arsenic trioxide combination in human leukemia cells. Reactive oxygen species generation and defensive protein kinase (MEK/ERK, Akt/mTOR) modulation. Biochem. Pharmacol. 2011, 82, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Chong, D.; Ma, L.; Liu, F.; Zhang, Z.; Zhao, S.; Huo, Q.; Zhang, P.; Zheng, H.; Liu, H. Synergistic antitumor effect of 3-bromopyruvate and 5-fluorouracil against human colorectal cancer through cell cycle arrest and induction of apoptosis. Anticancer Drugs 2017, 28, 831–840. [Google Scholar] [CrossRef]
- Attia, Y.M.; El-Abhar, H.S.; Al Marzabani, M.M.; Shouman, S.A. Targeting glycolysis by 3-bromopyruvate improves tamoxifen cytotoxicity of breast cancer cell lines. BMC Cancer 2015, 15, 838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, Y.; Yadav, P.; Kumari, N.; Kalra, N.; Bhatt, A.N. Hexokinase II inhibition by 3-bromopyruvate sensitizes myeloid leukemic cells K-562 to anti-leukemic drug, daunorubicin. Biosci. Rep. 2019, 39, BSR20190880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, S.K. Inhibition of liver hexokinase by dehydroascorbic acid and alloxan. Nature 1959, 184 (Suppl. S21), 1638–1640. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Flescher, E. Methyl jasmonate: A plant stress hormone as an anti-cancer drug. Phytochemistry 2009, 70, 1600–1609. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Liang, Q.; Zhou, Y.; Jiang, L.L.; Gu, W.M.; Luo, M.Y.; Tang, Y.B.; Wang, Y.; Lu, W.; Huang, M.; et al. A Novel Allosteric Inhibitor of Phosphoglycerate Mutase 1 Suppresses Growth and Metastasis of Non-Small-Cell Lung Cancer. Cell Metab. 2019, 30, 1107–1119.e1108. [Google Scholar] [CrossRef]
- Qian, Y.; Bi, L.; Yang, Y.; Wang, D. Effect of pyruvate kinase M2-regulating aerobic glycolysis on chemotherapy resistance of estrogen receptor-positive breast cancer. Anticancer. Drugs 2018, 29, 616–627. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, F.; Wu, X.R. Inhibition of Pyruvate Kinase M2 Markedly Reduces Chemoresistance of Advanced Bladder Cancer to Cisplatin. Sci. Rep. 2017, 7, 45983. [Google Scholar] [CrossRef] [Green Version]
- Shang, D.; Wu, J.; Guo, L.; Xu, Y.; Liu, L.; Lu, J. Metformin increases sensitivity of osteosarcoma stem cells to cisplatin by inhibiting expression of PKM2. Int. J. Oncol. 2017, 50, 1848–1856. [Google Scholar] [CrossRef]
- Gatti, M.; Solari, A.; Pattarozzi, A.; Campanella, C.; Thellung, S.; Maniscalco, L.; De Maria, R.; Wurth, R.; Corsaro, A.; Bajetto, A.; et al. In vitro and in vivo characterization of stem-like cells from canine osteosarcoma and assessment of drug sensitivity. Exp. Cell Res. 2018, 363, 48–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, S.H.; Seo, S.K.; Park, Y.; Kim, E.K.; Seong, M.K.; Kim, H.A.; Song, J.Y.; Hwang, S.G.; Lee, J.K.; Noh, W.C.; et al. Dichloroacetate potentiates tamoxifen-induced cell death in breast cancer cells via downregulation of the epidermal growth factor receptor. Oncotarget 2016, 7, 59809–59819. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Lu, Y.; Xie, Y.; Qiu, S.; Li, X.; Fan, Z. Rational combination with PDK1 inhibition overcomes cetuximab resistance in head and neck squamous cell carcinoma. JCI Insight 2019, 4, e131106. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhao, Y.; Ding, Y.; Liu, H.; Liu, Z.; Fodstad, O.; Riker, A.I.; Kamarajugadda, S.; Lu, J.; Owen, L.B.; et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol. Cancer 2010, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farabegoli, F.; Vettraino, M.; Manerba, M.; Fiume, L.; Roberti, M.; Di Stefano, G. Galloflavin, a new lactate dehydrogenase inhibitor, induces the death of human breast cancer cells with different glycolytic attitude by affecting distinct signaling pathways. Eur. J. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef]
- Lee, J.S.; Lee, H.; Jang, H.; Woo, S.M.; Park, J.B.; Lee, S.H.; Kang, J.H.; Kim, H.Y.; Song, J.; Kim, S.Y. Targeting Oxidative Phosphorylation Reverses Drug Resistance in Cancer Cells by Blocking Autophagy Recycling. Cells 2020, 9, 2013. [Google Scholar] [CrossRef]
- Lee, S.; Lee, J.S.; Seo, J.; Lee, S.H.; Kang, J.H.; Song, J.; Kim, S.Y. Targeting Mitochondrial Oxidative Phosphorylation Abrogated Irinotecan Resistance in NSCLC. Sci. Rep. 2018, 8, 15707. [Google Scholar] [CrossRef]
- Masoud, R.; Reyes-Castellanos, G.; Lac, S.; Garcia, J.; Dou, S.; Shintu, L.; Abdel Hadi, N.; Gicquel, T.; El Kaoutari, A.; Dieme, B.; et al. Targeting Mitochondrial Complex I Overcomes Chemoresistance in High OXPHOS Pancreatic Cancer. Cell Rep. Med. 2020, 1, 100143. [Google Scholar] [CrossRef]
- Odier, F.; Vago, P.; Quiot, J.M.; Devauchelle, G.; Bureau, J.P. Cytometric study of the effects of destruxin E on leukemic cells in mice. Comptes Rendus Acad. Sci. III 1987, 305, 575–578. [Google Scholar]
- Giddings, E.L.; Champagne, D.P.; Wu, M.H.; Laffin, J.M.; Thornton, T.M.; Valenca-Pereira, F.; Culp-Hill, R.; Fortner, K.A.; Romero, N.; East, J.; et al. Mitochondrial ATP fuels ABC transporter-mediated drug efflux in cancer chemoresistance. Nat. Commun. 2021, 12, 2804. [Google Scholar] [CrossRef]
- Oliva, C.R.; Zhang, W.; Langford, C.; Suto, M.J.; Griguer, C.E. Repositioning chlorpromazine for treating chemoresistant glioma through the inhibition of cytochrome c oxidase bearing the COX4-1 regulatory subunit. Oncotarget 2017, 8, 37568–37583. [Google Scholar] [CrossRef]
- Li, Y.C.; Fung, K.P.; Kwok, T.T.; Lee, C.Y.; Suen, Y.K.; Kong, S.K. Mitochondria-targeting drug oligomycin blocked P-glycoprotein activity and triggered apoptosis in doxorubicin-resistant HepG2 cells. Chemotherapy 2004, 50, 55–62. [Google Scholar] [CrossRef]
- Shi, Y.; Lim, S.K.; Liang, Q.; Iyer, S.V.; Wang, H.Y.; Wang, Z.; Xie, X.; Sun, D.; Chen, Y.J.; Tabar, V.; et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 2019, 567, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Dartier, J.; Lemaitre, E.; Chourpa, I.; Goupille, C.; Servais, S.; Chevalier, S.; Maheo, K.; Dumas, J.F. ATP-dependent activity and mitochondrial localization of drug efflux pumps in doxorubicin-resistant breast cancer cells. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1075–1084. [Google Scholar] [CrossRef]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Mazure, N.M.; Pouyssegur, J. Hypoxia-induced autophagy: Cell death or cell survival? Curr. Opin. Cell Biol. 2010, 22, 177–180. [Google Scholar] [CrossRef]
- Ureshino, R.P.; Rocha, K.K.; Lopes, G.S.; Bincoletto, C.; Smaili, S.S. Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid. Redox Signal. 2014, 21, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.Y.; Juhasz, G.; Goraksha-Hicks, P.; Arsham, A.M.; Mallin, D.R.; Muller, L.K.; Neufeld, T.P. Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem. Soc. Trans. 2009, 37, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy-unanswered questions. J. Cell Sci. 2011, 124, 161–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyo, J.O.; Nah, J.; Jung, Y.K. Molecules and their functions in autophagy. Exp. Mol. Med. 2012, 44, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraft, C.; Martens, S. Mechanisms and regulation of autophagosome formation. Curr. Opin. Cell Biol. 2012, 24, 496–501. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Yoshimori, T. A current perspective of autophagosome biogenesis. Cell Res. 2014, 24, 58–68. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Hanada, T.; Noda, N.N.; Satomi, Y.; Ichimura, Y.; Fujioka, Y.; Takao, T.; Inagaki, F.; Ohsumi, Y. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J. Biol. Chem. 2007, 282, 37298–37302. [Google Scholar] [CrossRef] [Green Version]
- Noda, N.N.; Fujioka, Y.; Hanada, T.; Ohsumi, Y.; Inagaki, F. Structure of the Atg12-Atg5 conjugate reveals a platform for stimulating Atg8-PE conjugation. EMBO Rep. 2013, 14, 206–211. [Google Scholar] [CrossRef] [Green Version]
- Sakoh-Nakatogawa, M.; Matoba, K.; Asai, E.; Kirisako, H.; Ishii, J.; Noda, N.N.; Inagaki, F.; Nakatogawa, H.; Ohsumi, Y. Atg12-Atg5 conjugate enhances E2 activity of Atg3 by rearranging its catalytic site. Nat. Struct. Mol. Biol. 2013, 20, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: Beyond the usual suspects’ review series. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. Role of the Apg12 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2003, 35, 553–561. [Google Scholar] [CrossRef]
- Mizushima, N.; Sugita, H.; Yoshimori, T.; Ohsumi, Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 1998, 273, 33889–33892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef]
- Matsushita, M.; Suzuki, N.N.; Obara, K.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. Structure of Atg5.Atg16, a complex essential for autophagy. J. Biol. Chem. 2007, 282, 6763–6772. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Q.; Li, W.; Li, P.; Yang, M.; Wu, C.; Eichinger, L. The Role of ATG16 in Autophagy and The Ubiquitin Proteasome System. Cells 2018, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Romanov, J.; Walczak, M.; Ibiricu, I.; Schuchner, S.; Ogris, E.; Kraft, C.; Martens, S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J 2012, 31, 4304–4317. [Google Scholar] [CrossRef]
- Fujioka, Y.; Noda, N.N.; Nakatogawa, H.; Ohsumi, Y.; Inagaki, F. Dimeric coiled-coil structure of Saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J. Biol. Chem. 2010, 285, 1508–1515. [Google Scholar] [CrossRef] [Green Version]
- Kuma, A.; Mizushima, N.; Ishihara, N.; Ohsumi, Y. Formation of the approximately 350-kDa Apg12-Apg5.Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J. Biol. Chem. 2002, 277, 18619–18625. [Google Scholar] [CrossRef] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J. Biol. Chem. 2004, 279, 47704–47710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirisako, T.; Ichimura, Y.; Okada, H.; Kabeya, Y.; Mizushima, N.; Yoshimori, T.; Ohsumi, M.; Takao, T.; Noda, T.; Ohsumi, Y. The reversible modification regulates the membrane-binding state of Apg8/Aut7 essential for autophagy and the cytoplasm to vacuole targeting pathway. J. Cell Biol. 2000, 151, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Hanada, T.; Satomi, Y.; Takao, T.; Ohsumi, Y. The amino-terminal region of Atg3 is essential for association with phosphatidylethanolamine in Atg8 lipidation. FEBS Lett. 2009, 583, 1078–1083. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, P.; Juhasz, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef]
- Wang, X.; Proud, C.G. mTORC2 is a tyrosine kinase. Cell Res. 2016, 26, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M.S. The Role of Mammalian Target of Rapamycin (mTOR) in Insulin Signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Chen, L.; Zhao, L.; Xu, Y.; Peng, X.; Wang, X.; Ding, L.; Jin, J.; Teng, H.; Wang, Y.; et al. Ubiquitination of Rheb governs growth factor-induced mTORC1 activation. Cell Res. 2019, 29, 136–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angarola, B.; Ferguson, S.M. Coordination of Rheb lysosomal membrane interactions with mTORC1 activation. F1000Research 2020, 9, 450. [Google Scholar] [CrossRef] [PubMed]
- Ganley, I.G.; Lam, D.H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Peng, W.; Du, T.; Zhang, Z.; Du, F.; Jin, J.; Gong, A. Knockdown of autophagy-related gene LC3 enhances the sensitivity of HepG2 cells to epirubicin. Exp. Ther. Med. 2015, 9, 1271–1276. [Google Scholar] [CrossRef]
- Sun, W.; Li, J.; Zhou, L.; Han, J.; Liu, R.; Zhang, H.; Ning, T.; Gao, Z.; Liu, B.; Chen, X.; et al. The c-Myc/miR-27b-3p/ATG10 regulatory axis regulates chemoresistance in colorectal cancer. Theranostics 2020, 10, 1981–1996. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [Green Version]
- Mancias, J.D.; Kimmelman, A.C. Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J. Mol. Biol. 2016, 428, 1659–1680. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and current strategies for targeting autophagy for cancer treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garber, K. Inducing indigestion: Companies embrace autophagy inhibitors. J. Natl. Cancer Inst. 2011, 103, 708–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Altman, B.J.; Jacobs, S.R.; Mason, E.F.; Michalek, R.D.; MacIntyre, A.N.; Coloff, J.L.; Ilkayeva, O.; Jia, W.; He, Y.W.; Rathmell, J.C. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 2011, 30, 1855–1867. [Google Scholar] [CrossRef] [Green Version]
- Feldman, M.E.; Shokat, K.M. New inhibitors of the PI3K-Akt-mTOR pathway: Insights into mTOR signaling from a new generation of Tor Kinase Domain Inhibitors (TORKinibs). Curr. Top. Microbiol. Immunol. 2010, 347, 241–262. [Google Scholar] [CrossRef]
- Sheen, J.H.; Zoncu, R.; Kim, D.; Sabatini, D.M. Defective regulation of autophagy upon leucine deprivation reveals a targetable liability of human melanoma cells in vitro and in vivo. Cancer Cell 2011, 19, 613–628. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Meco, M.T. Targeting leucine addiction and autophagy in melanoma. Pigment Cell Melanoma Res. 2011, 24, 588–589. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Bursch, W.; Karwan, A.; Mayer, M.; Dornetshuber, J.; Frohwein, U.; Schulte-Hermann, R.; Fazi, B.; Di Sano, F.; Piredda, L.; Piacentini, M.; et al. Cell death and autophagy: Cytokines, drugs, and nutritional factors. Toxicology 2008, 254, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Dasari, S.K.; Bialik, S.; Levin-Zaidman, S.; Levin-Salomon, V.; Merrill, A.H., Jr.; Futerman, A.H.; Kimchi, A. Signalome-wide RNAi screen identifies GBA1 as a positive mediator of autophagic cell death. Cell Death Differ. 2017, 24, 1288–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, S.K.; Schejter, E.; Bialik, S.; Shkedy, A.; Levin-Salomon, V.; Levin-Zaidman, S.; Kimchi, A. Death by over-eating: The Gaucher disease associated gene GBA1, identified in a screen for mediators of autophagic cell death, is necessary for developmental cell death in Drosophila midgut. Cell Cycle 2017, 16, 2003–2010. [Google Scholar] [CrossRef] [Green Version]
- Cecconi, F.; Levine, B. The role of autophagy in mammalian development: Cell makeover rather than cell death. Dev. Cell 2008, 15, 344–357. [Google Scholar] [CrossRef] [Green Version]
- Le Bot, N. Autophagy: A new regulator of development. Nat. Cell Biol. 2007, 9, 741. [Google Scholar] [CrossRef] [PubMed]
- Kelekar, A. Autophagy. Ann. N. Y. Acad. Sci. 2005, 1066, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Musiwaro, P.; Smith, M.; Manifava, M.; Walker, S.A.; Ktistakis, N.T. Characteristics and requirements of basal autophagy in HEK 293 cells. Autophagy 2013, 9, 1407–1417. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [Green Version]
- Nagaria, T.S.; Williams, J.L.; Leduc, C.; Squire, J.A.; Greer, P.A.; Sangrar, W. Flavopiridol synergizes with sorafenib to induce cytotoxicity and potentiate antitumorigenic activity in EGFR/HER-2 and mutant RAS/RAF breast cancer model systems. Neoplasia 2013, 15, 939–951. [Google Scholar] [CrossRef] [Green Version]
- Nagaria, T.S.; Shi, C.; Leduc, C.; Hoskin, V.; Sikdar, S.; Sangrar, W.; Greer, P.A. Combined targeting of Raf and Mek synergistically inhibits tumorigenesis in triple negative breast cancer model systems. Oncotarget 2017, 8, 80804–80819. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Wang, H.; Li, W.; Zhong, J.; Yu, R.; Huang, X.; Wang, H.; Tan, Z.; Wang, J.; Zhang, Y. Pazopanib, a novel multi-kinase inhibitor, shows potent antitumor activity in colon cancer through PUMA-mediated apoptosis. Oncotarget 2017, 8, 3289–3303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codogno, P.; Morel, E. FOXO3a Provides a Quickstep from Autophagy Inhibition to Apoptosis in Cancer Therapy. Dev. Cell 2018, 44, 537–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzwalter, B.E.; Thorburn, A. FOXO3 links autophagy to apoptosis. Autophagy 2018, 14, 1467–1468. [Google Scholar] [CrossRef] [PubMed]
- Fitzwalter, B.E.; Towers, C.G.; Sullivan, K.D.; Andrysik, Z.; Hoh, M.; Ludwig, M.; O’Prey, J.; Ryan, K.M.; Espinosa, J.M.; Morgan, M.J.; et al. Autophagy Inhibition Mediates Apoptosis Sensitization in Cancer Therapy by Relieving FOXO3a Turnover. Dev. Cell 2018, 44, 555–565.e553. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Huang, Y.; Liu, Y.; Zhao, Y.; He, X.; Zhang, L.; Wang, F.; Zhang, Y. Ipatasertib, a novel Akt inhibitor, induces transcription factor FoxO3a and NF-kappaB directly regulates PUMA-dependent apoptosis. Cell Death Dis. 2018, 9, 911. [Google Scholar] [CrossRef] [Green Version]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [Green Version]
- Din, F.V.; Valanciute, A.; Houde, V.P.; Zibrova, D.; Green, K.A.; Sakamoto, K.; Alessi, D.R.; Dunlop, M.G. Aspirin inhibits mTOR signaling, activates AMP-activated protein kinase, and induces autophagy in colorectal cancer cells. Gastroenterology 2012, 142, 1504–1515.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, B.; Querfurth, H.W. Palmitate activates mTOR/p70S6K through AMPK inhibition and hypophosphorylation of raptor in skeletal muscle cells: Reversal by oleate is similar to metformin. Biochimie 2015, 118, 141–150. [Google Scholar] [CrossRef]
- Iksen; Pothongsrisit, S.; Pongrakhananon, V. Targeting the PI3K/AKT/mTOR Signaling Pathway in Lung Cancer: An Update Regarding Potential Drugs and Natural Products. Molecules 2021, 26, 4100. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, S.; Wang, J.; Jiang, Y. Blockade of AMPK-Mediated cAMP-PKA-CREB/ATF1 Signaling Synergizes with Aspirin to Inhibit Hepatocellular Carcinoma. Cancers 2021, 13, 1738. [Google Scholar] [CrossRef]
- Fattahi, S.; Amjadi-Moheb, F.; Tabaripour, R.; Ashrafi, G.H.; Akhavan-Niaki, H. PI3K/AKT/mTOR signaling in gastric cancer: Epigenetics and beyond. Life Sci. 2020, 262, 118513. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin. Cancer Biol. 2019, 59, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/AKT/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Abudu, Y.P.; Claude-Taupin, A.; Gu, Y.; Kumar, S.; Choi, S.W.; Peters, R.; Mudd, M.H.; Allers, L.; Salemi, M.; et al. Galectins control MTOR and AMPK in response to lysosomal damage to induce autophagy. Autophagy 2019, 15, 169–171. [Google Scholar] [CrossRef] [Green Version]
- Shaw, R.J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009, 196, 65–80. [Google Scholar] [CrossRef]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [Green Version]
- Anerillas, C.; Abdelmohsen, K.; Gorospe, M. Regulation of senescence traits by MAPKs. Geroscience 2020, 42, 397–408. [Google Scholar] [CrossRef]
- Maruyama, J.; Naguro, I.; Takeda, K.; Ichijo, H. Stress-activated MAP kinase cascades in cellular senescence. Curr. Med. Chem. 2009, 16, 1229–1235. [Google Scholar] [CrossRef]
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576. [Google Scholar] [CrossRef] [Green Version]
- Tee, A.R.; Manning, B.D.; Roux, P.P.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003, 13, 1259–1268. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMP-activated protein kinase: A cellular energy sensor with a key role in metabolic disorders and in cancer. Biochem. Soc. Trans. 2011, 39, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carling, D. AMPK signalling in health and disease. Curr. Opin. Cell Biol. 2017, 45, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Carling, D.; Mayer, F.V.; Sanders, M.J.; Gamblin, S.J. AMP-activated protein kinase: Nature’s energy sensor. Nat. Chem. Biol. 2011, 7, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ke, R.; Xu, Q.; Li, C.; Luo, L.; Huang, D. Mechanisms of AMPK in the maintenance of ATP balance during energy metabolism. Cell Biol. Int. 2018, 42, 384–392. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Xin, F.J.; Wang, J.; Hu, J.; Zhang, Y.Y.; Wan, S.; Cao, L.S.; Lu, C.; Li, P.; Yan, S.F.; et al. Conserved regulatory elements in AMPK. Nature 2013, 498, E8–E10. [Google Scholar] [CrossRef]
- Viollet, B.; Mounier, R.; Leclerc, J.; Yazigi, A.; Foretz, M.; Andreelli, F. Targeting AMP-activated protein kinase as a novel therapeutic approach for the treatment of metabolic disorders. Diabetes Metab. 2007, 33, 395–402. [Google Scholar] [CrossRef]
- Stahmann, N.; Woods, A.; Carling, D.; Heller, R. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase beta. Mol. Cell. Biol. 2006, 26, 5933–5945. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willows, R.; Sanders, M.J.; Xiao, B.; Patel, B.R.; Martin, S.R.; Read, J.; Wilson, J.R.; Hubbard, J.; Gamblin, S.J.; Carling, D. Phosphorylation of AMPK by upstream kinases is required for activity in mammalian cells. Biochem. J. 2017, 474, 3059–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, S.A.; Boudeau, J.; Reid, J.L.; Mustard, K.J.; Udd, L.; Makela, T.P.; Alessi, D.R.; Hardie, D.G. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2003, 2, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellingson, W.J.; Chesser, D.G.; Winder, W.W. Effects of 3-phosphoglycerate and other metabolites on the activation of AMP-activated protein kinase by LKB1-STRAD-MO25. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E400–E407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessi, D.R.; Sakamoto, K.; Bayascas, J.R. LKB1-dependent signaling pathways. Annu. Rev. Biochem. 2006, 75, 137–163. [Google Scholar] [CrossRef] [PubMed]
- Forcet, C.; Billaud, M. Dialogue between LKB1 and AMPK: A hot topic at the cellular pole. Sci. STKE 2007, 2007, pe51. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Corradetti, M.N.; Inoki, K.; Bardeesy, N.; DePinho, R.A.; Guan, K.L. Regulation of the TSC pathway by LKB1: Evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004, 18, 1533–1538. [Google Scholar] [CrossRef] [Green Version]
- Nobukini, T.; Thomas, G. The mTOR/S6K signalling pathway: The role of the TSC1/2 tumour suppressor complex and the proto-oncogene Rheb. Novartis Found. Symp. 2004, 262, discussion 154–149, 265–148. [Google Scholar]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Tasdemir, E.; Chiara Maiuri, M.; Morselli, E.; Criollo, A.; D’Amelio, M.; Djavaheri-Mergny, M.; Cecconi, F.; Tavernarakis, N.; Kroemer, G. A dual role of p53 in the control of autophagy. Autophagy 2008, 4, 810–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denisenko, T.V.; Pivnyuk, A.D.; Zhivotovsky, B. p53-Autophagy-Metastasis Link. Cancers 2018, 10, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef]
- Kenzelmann Broz, D.; Spano Mello, S.; Bieging, K.T.; Jiang, D.; Dusek, R.L.; Brady, C.A.; Sidow, A.; Attardi, L.D. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 2013, 27, 1016–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, M.; Flores, E.R. The family that eats together stays together: New p53 family transcriptional targets in autophagy. Genes Dev. 2013, 27, 971–974. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Tavana, O.; Gu, W. p53 modifications: Exquisite decorations of the powerful guardian. J. Mol. Cell Biol. 2019, 11, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Hu, W.; Chen, S.; Thorne, R.F.; Wu, M. TP53, TP53 Target Genes (DRAM, TIGAR), and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 127–149. [Google Scholar] [CrossRef]
- Chollat-Namy, M.; Ben Safta-Saadoun, T.; Haferssas, D.; Meurice, G.; Chouaib, S.; Thiery, J. The pharmalogical reactivation of p53 function improves breast tumor cell lysis by granzyme B and NK cells through induction of autophagy. Cell Death Dis. 2019, 10, 695. [Google Scholar] [CrossRef]
- Budanov, A.V.; Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008, 134, 451–460. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Song, Z.; Liang, B.; Jia, L.; Ma, S.; Liu, X. Radiation induces autophagic cell death via the p53/DRAM signaling pathway in breast cancer cells. Oncol. Rep. 2016, 35, 3639–3647. [Google Scholar] [CrossRef] [Green Version]
- Garufi, A.; Pistritto, G.; Baldari, S.; Toietta, G.; Cirone, M.; D’Orazi, G. p53-Dependent PUMA to DRAM antagonistic interplay as a key molecular switch in cell-fate decision in normal/high glucose conditions. J. Exp. Clin. Cancer Res. 2017, 36, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [Green Version]
- Pei, L.; Shang, Y.; Jin, H.; Wang, S.; Wei, N.; Yan, H.; Wu, Y.; Yao, C.; Wang, X.; Zhu, L.Q.; et al. DAPK1-p53 interaction converges necrotic and apoptotic pathways of ischemic neuronal death. J. Neurosci. 2014, 34, 6546–6556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, N.; Chen, D.; Zhou, X.Z.; Lee, T.H. Death-Associated Protein Kinase 1 Phosphorylation in Neuronal Cell Death and Neurodegenerative Disease. Int. J. Mol. Sci. 2019, 20, 3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, B.; Kraus, M.; Burch, L.; Stevens, C.; Craig, A.; Gordon-Weeks, P.; Hupp, T.R. DAPK-1 binding to a linear peptide motif in MAP1B stimulates autophagy and membrane blebbing. J. Biol. Chem. 2008, 283, 9999–10014. [Google Scholar] [CrossRef] [Green Version]
- Steinmann, S.; Scheibe, K.; Erlenbach-Wuensch, K.; Neufert, C.; Schneider-Stock, R. Death-associated protein kinase: A molecule with functional antagonistic duality and a potential role in inflammatory bowel disease (Review). Int. J. Oncol. 2015, 47, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Ravanan, P.; Talwar, P. Death Associated Protein Kinase 1 (DAPK1): A Regulator of Apoptosis and Autophagy. Front. Mol. Neurosci. 2016, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Mrakovcic, M.; Frohlich, L.F. p53-Mediated Molecular Control of Autophagy in Tumor Cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Tasdemir, E.; Maiuri, M.C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Jin, L.; Huang, X.; Geng, S.; He, C.; Hu, X. p53 signaling and autophagy in cancer: A revolutionary strategy could be developed for cancer treatment. Autophagy 2011, 7, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, Z.; Xie, J.M.; Wang, G.; Qian, L.Q.; Guan, X.M.; Shen, X.P.; Qin, Z.H.; Shen, G.H.; Li, X.Q.; et al. TIGAR knockdown enhanced the anticancer effect of aescin via regulating autophagy and apoptosis in colorectal cancer cells. Acta Pharmacol. Sin. 2019, 40, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Zhao, X.; Lu, J.; Qian, G.; Zheng, J.C.; Ge, S. Knockdown of TIGAR by RNA interference induces apoptosis and autophagy in HepG2 hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2013, 437, 300–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spoerri, P.E.; Srivastava, N.; Vernadakis, A. Role of GABAA receptors in the GABA attenuation of ethanol neurotoxicity. J. Neurosci. Res. 1996, 44, 499–506. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Katz, M.; Amit, I.; Yarden, Y. Regulation of MAPKs by growth factors and receptor tyrosine kinases. Biochim. Biophys. Acta 2007, 1773, 1161–1176. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Song, K.; Yen, I.; Brandhuber, B.J.; Anderson, D.J.; Alvarado, R.; Ludlam, M.J.; Stokoe, D.; Gloor, S.L.; Vigers, G.; et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 2010, 464, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: A new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.; Kim, Y.M.; Chin, Y.W.; Kang, K.S. Schisandrol A Exhibits Estrogenic Activity via Estrogen Receptor alpha-Dependent Signaling Pathway in Estrogen Receptor-Positive Breast Cancer Cells. Pharmaceutics 2021, 13, 1082. [Google Scholar] [CrossRef]
- Komatsu, N.; Fujita, Y.; Matsuda, M.; Aoki, K. mTORC1 upregulation via ERK-dependent gene expression change confers intrinsic resistance to MEK inhibitors in oncogenic KRas-mutant cancer cells. Oncogene 2015, 34, 5607–5616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogier-Denis, E.; Pattingre, S.; El Benna, J.; Codogno, P. Erk1/2-dependent phosphorylation of Galpha-interacting protein stimulates its GTPase accelerating activity and autophagy in human colon cancer cells. J. Biol. Chem. 2000, 275, 39090–39095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattingre, S.; Bauvy, C.; Codogno, P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. J. Biol. Chem. 2003, 278, 16667–16674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranek, M.J.; Kokkonen-Simon, K.M.; Chen, A.; Dunkerly-Eyring, B.L.; Vera, M.P.; Oeing, C.U.; Patel, C.H.; Nakamura, T.; Zhu, G.; Bedja, D.; et al. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature 2019, 566, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Mahoney, E.; Lucas, D.M.; Gupta, S.V.; Wagner, A.J.; Herman, S.E.; Smith, L.L.; Yeh, Y.Y.; Andritsos, L.; Jones, J.A.; Flynn, J.M.; et al. ER stress and autophagy: New discoveries in the mechanism of action and drug resistance of the cyclin-dependent kinase inhibitor flavopiridol. Blood 2012, 120, 1262–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef]
- Lv, S.; Sun, E.C.; Xu, Q.Y.; Zhang, J.K.; Wu, D.L. Endoplasmic reticulum stress-mediated autophagy contributes to bluetongue virus infection via the PERK-eIF2alpha pathway. Biochem. Biophys. Res. Commun. 2015, 466, 406–412. [Google Scholar] [CrossRef]
- Avivar-Valderas, A.; Salas, E.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Nagi, C.; Debnath, J.; Aguirre-Ghiso, J.A. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol. Cell. Biol. 2011, 31, 3616–3629. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Yan, D.Y.; Wang, C.; Ma, Z.; Deng, Y.; Liu, W.; Xu, B. IRE1 signaling pathway mediates protective autophagic response against manganese-induced neuronal apoptosis in vivo and in vitro. Sci. Total Environ. 2020, 712, 136480. [Google Scholar] [CrossRef]
- Zheng, Y.; Tu, J.; Wang, X.; Yu, Y.; Li, J.; Jin, Y.; Wu, J. The Therapeutic Effect of Melatonin on GC by Inducing Cell Apoptosis and Autophagy Induced by Endoplasmic Reticulum Stress. OncoTargets Ther. 2019, 12, 10187–10198. [Google Scholar] [CrossRef] [Green Version]
- Borsello, T.; Croquelois, K.; Hornung, J.P.; Clarke, P.G. N-methyl-d-aspartate-triggered neuronal death in organotypic hippocampal cultures is endocytic, autophagic and mediated by the c-Jun N-terminal kinase pathway. Eur. J. Neurosci. 2003, 18, 473–485. [Google Scholar] [CrossRef] [PubMed]
- Walton, M.; MacGibbon, G.; Young, D.; Sirimanne, E.; Williams, C.; Gluckman, P.; Dragunow, M. Do c-Jun, c-Fos, and amyloid precursor protein play a role in neuronal death or survival? J. Neurosci. Res. 1998, 53, 330–342. [Google Scholar] [CrossRef]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef] [Green Version]
- Daitoku, H.; Fukamizu, A. FOXO transcription factors in the regulatory networks of longevity. J. Biochem. 2007, 141, 769–774. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Pietrocola, F.; Bravo-San Pedro, J.M.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, 856–880. [Google Scholar] [CrossRef]
- Kimmelman, A.C. The dynamic nature of autophagy in cancer. Genes Dev. 2011, 25, 1999–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiuri, M.C.; Tasdemir, E.; Criollo, A.; Morselli, E.; Vicencio, J.M.; Carnuccio, R.; Kroemer, G. Control of autophagy by oncogenes and tumor suppressor genes. Cell Death Differ. 2009, 16, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Yu, J.; Brown, K.; Levine, B. Beclin 1 contains a leucine-rich nuclear export signal that is required for its autophagy and tumor suppressor function. Cancer Res. 2001, 61, 3443–3449. [Google Scholar]
- Lenardo, M.J.; McPhee, C.K.; Yu, L. Autophagic cell death. Methods Enzymol. 2009, 453, 17–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, D.; Nicolson, S.; Kumar, S. Cell death by autophagy: Facts and apparent artefacts. Cell Death Differ. 2012, 19, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, S.; Yoshida, T.; Tsujioka, M.; Arakawa, S. Autophagic cell death and cancer. Int. J. Mol. Sci. 2014, 15, 3145–3153. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.S.; Blanco, E.; Kim, Y.S.; Rodriguez, A.A.; Zhao, H.; Huang, T.H.; Chen, C.L.; Jin, G.; Landis, M.D.; Burey, L.A.; et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells 2014, 32, 2309–2323. [Google Scholar] [CrossRef] [Green Version]
- Balic, A.; Sorensen, M.D.; Trabulo, S.M.; Sainz, B., Jr.; Cioffi, M.; Vieira, C.R.; Miranda-Lorenzo, I.; Hidalgo, M.; Kleeff, J.; Erkan, M.; et al. Chloroquine targets pancreatic cancer stem cells via inhibition of CXCR4 and hedgehog signaling. Mol. Cancer Ther. 2014, 13, 1758–1771. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, S.; Nakata, K.; Sagara, A.; Guan, W.; Ikenaga, N.; Ohuchida, K.; Nakamura, M. Efficient pre-treatment for pancreatic cancer using chloroquine-loaded nanoparticles targeting pancreatic stellate cells. Oncol. Lett. 2021, 22, 633. [Google Scholar] [CrossRef]
- Jiang, H.; Gomez-Manzano, C.; Aoki, H.; Alonso, M.M.; Kondo, S.; McCormick, F.; Xu, J.; Kondo, Y.; Bekele, B.N.; Colman, H.; et al. Examination of the therapeutic potential of Delta-24-RGD in brain tumor stem cells: Role of autophagic cell death. J. Natl. Cancer Inst. 2007, 99, 1410–1414. [Google Scholar] [CrossRef] [Green Version]
- Camuzard, O.; Trojani, M.C.; Santucci-Darmanin, S.; Pagnotta, S.; Breuil, V.; Carle, G.F.; Pierrefite-Carle, V. Autophagy in Osteosarcoma Cancer Stem Cells Is Critical Process which Can Be Targeted by the Antipsychotic Drug Thioridazine. Cancers 2020, 12, 3675. [Google Scholar] [CrossRef]
- Choudhury, S.; Kolukula, V.K.; Preet, A.; Albanese, C.; Avantaggiati, M.L. Dissecting the pathways that destabilize mutant p53: The proteasome or autophagy? Cell Cycle 2013, 12, 1022–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscat, J.; Diaz-Meco, M.T. p62: A versatile multitasker takes on cancer. Trends Biochem. Sci. 2012, 37, 230–236. [Google Scholar] [CrossRef] [Green Version]
- Duran, A.; Linares, J.F.; Galvez, A.S.; Wikenheiser, K.; Flores, J.M.; Diaz-Meco, M.T.; Moscat, J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008, 13, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Isakson, P.; Bjoras, M.; Boe, S.O.; Simonsen, A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010, 116, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Goussetis, D.J.; Gounaris, E.; Wu, E.J.; Vakana, E.; Sharma, B.; Bogyo, M.; Altman, J.K.; Platanias, L.C. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood 2012, 120, 3555–3562. [Google Scholar] [CrossRef] [Green Version]
- Goussetis, D.J.; Gounaris, E.; Platanias, L.C. BCR-ABL1-induced leukemogenesis and autophagic targeting by arsenic trioxide. Autophagy 2013, 9, 93–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auberger, P. BCR-ABL/p62/SQSTM1: A cannibal embrace. Blood 2012, 120, 3389–3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, C.P.; Budina, A.; Balaburski, G.; Bergenstock, M.K.; Murphy, M. Autophagy in tumor suppression and cancer therapy. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 71–100. [Google Scholar] [CrossRef]
- Avalos, Y.; Canales, J.; Bravo-Sagua, R.; Criollo, A.; Lavandero, S.; Quest, A.F. Tumor suppression and promotion by autophagy. Biomed. Res. Int. 2014, 2014, 603980. [Google Scholar] [CrossRef]
- Perez-Mancera, P.A.; Young, A.R.; Narita, M. Inside and out: The activities of senescence in cancer. Nat. Rev. Cancer 2014, 14, 547–558. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscat, J.; Diaz-Meco, M.T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscat, J.; Diaz-Meco, M.T.; Wooten, M.W. Signal integration and diversification through the p62 scaffold protein. Trends Biochem. Sci. 2007, 32, 95–100. [Google Scholar] [CrossRef]
- Liu, E.Y.; Ryan, K.M. Autophagy and cancer--issues we need to digest. J. Cell Sci. 2012, 125, 2349–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a molecular target for cancer treatment. Eur. J. Pharm. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Chen, Y.; Zhang, H.; Min, S.; Yu, B.; He, B.; Jin, A. Comparison of mesenchymal stromal cells from human bone marrow and adipose tissue for the treatment of spinal cord injury. Cytotherapy 2013, 15, 434–448. [Google Scholar] [CrossRef]
- Nagelkerke, A.; Bussink, J.; Sweep, F.C.; Span, P.N. The unfolded protein response as a target for cancer therapy. Biochim. Biophys. Acta 2014, 1846, 277–284. [Google Scholar] [CrossRef]
- Chan, D.C. Mitochondrial fusion and fission in mammals. Annu. Rev. Cell Dev. Biol. 2006, 22, 79–99. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Zhang, J.; Yu, M.; Xie, Y.; Huang, Y.; Wolff, D.W.; Abel, P.W.; Tu, Y. Mitochondrial dynamics regulates migration and invasion of breast cancer cells. Oncogene 2013, 32, 4814–4824. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Metabolic regulation of mitochondrial dynamics. J. Cell Biol. 2016, 212, 379–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wai, T.; Langer, T. Mitochondrial Dynamics and Metabolic Regulation. Trends Endocrinol. Metab. 2016, 27, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Scorrano, L. Keeping mitochondria in shape: A matter of life and death. Eur. J. Clin. Investig. 2013, 43, 886–893. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Nomura, M.; Jofuku, A.; Kato, H.; Suzuki, S.O.; Masuda, K.; Otera, H.; Nakanishi, Y.; Nonaka, I.; Goto, Y.; et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009, 11, 958–966. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Delerue, T.; Millet, A.M.; Moulis, M.F.; David, C.; Daloyau, M.; Arnaune-Pelloquin, L.; Davezac, N.; Mils, V.; Miquel, M.C.; et al. Mitochondrial fusion/fission dynamics in neurodegeneration and neuronal plasticity. Neurobiol. Dis. 2016, 90, 3–19. [Google Scholar] [CrossRef]
- van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Quintana-Cabrera, R.; Quirin, C.; Glytsou, C.; Corrado, M.; Urbani, A.; Pellattiero, A.; Calvo, E.; Vazquez, J.; Enriquez, J.A.; Gerle, C.; et al. The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat. Commun. 2018, 9, 3399. [Google Scholar] [CrossRef]
- Skulachev, V.P. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem. Sci. 2001, 26, 23–29. [Google Scholar] [CrossRef]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Jeong, S.Y.; Karbowski, M.; Smith, C.L.; Youle, R.J. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell 2004, 15, 5001–5011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [Green Version]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira-da-Silva, A.; Valacca, C.; Rios, E.; Populo, H.; Soares, P.; Sobrinho-Simoes, M.; Scorrano, L.; Maximo, V.; Campello, S. Mitochondrial dynamics protein Drp1 is overexpressed in oncocytic thyroid tumors and regulates cancer cell migration. PLoS ONE 2015, 10, e0122308. [Google Scholar] [CrossRef] [Green Version]
- Westrate, L.M.; Sayfie, A.D.; Burgenske, D.M.; MacKeigan, J.P. Persistent mitochondrial hyperfusion promotes G2/M accumulation and caspase-dependent cell death. PLoS ONE 2014, 9, e91911. [Google Scholar] [CrossRef]
- Qian, W.; Choi, S.; Gibson, G.A.; Watkins, S.C.; Bakkenist, C.J.; Van Houten, B. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J. Cell Sci. 2012, 125, 5745–5757. [Google Scholar] [CrossRef] [Green Version]
- Santel, A.; Frank, S.; Gaume, B.; Herrler, M.; Youle, R.J.; Fuller, M.T. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J. Cell Sci. 2003, 116, 2763–2774. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.L.; Meng, S.; Chen, Y.; Feng, J.X.; Gu, D.D.; Yu, B.; Li, Y.J.; Yang, J.Y.; Liao, S.; Chan, D.C.; et al. MFN1 structures reveal nucleotide-triggered dimerization critical for mitochondrial fusion. Nature 2017, 542, 372–376. [Google Scholar] [CrossRef]
- Hoppins, S.; Nunnari, J. The molecular mechanism of mitochondrial fusion. Biochim. Biophys. Acta 2009, 1793, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihara, N.; Eura, Y.; Mihara, K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 2004, 117, 6535–6546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipolat, S.; Martins de Brito, O.; Dal Zilio, B.; Scorrano, L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 15927–15932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santel, A.; Fuller, M.T. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 2001, 114, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Fujita, Y.; Oka, T.; Mihara, K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006, 25, 2966–2977. [Google Scholar] [CrossRef] [PubMed]
- Duvezin-Caubet, S.; Jagasia, R.; Wagener, J.; Hofmann, S.; Trifunovic, A.; Hansson, A.; Chomyn, A.; Bauer, M.F.; Attardi, G.; Larsson, N.G.; et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J. Biol. Chem. 2006, 281, 37972–37979. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Xiao, W.; Shan, S.; Jiang, C.; Chen, M.; Zhang, Y.; Lu, S.; Chen, J.; Zhang, C.; Chen, Q.; et al. Multi-patterned dynamics of mitochondrial fission and fusion in a living cell. PLoS ONE 2012, 7, e19879. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [Green Version]
- Kong, B.; Tsuyoshi, H.; Orisaka, M.; Shieh, D.B.; Yoshida, Y.; Tsang, B.K. Mitochondrial dynamics regulating chemoresistance in gynecological cancers. Ann. N. Y. Acad. Sci. 2015, 1350, 1–16. [Google Scholar] [CrossRef]
- Hou, L.; Wang, R.; Wei, H.; Li, S.; Liu, L.; Lu, X.; Yu, H.; Liu, Z. ABT737 enhances ovarian cancer cells sensitivity to cisplatin through regulation of mitochondrial fission via Sirt3 activation. Life Sci. 2019, 232, 116561. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Wang, Q.; Fung, E.; Xue, K.; Tsang, B.K. p53 is required for cisplatin-induced processing of the mitochondrial fusion protein L-Opa1 that is mediated by the mitochondrial metallopeptidase Oma1 in gynecologic cancers. J. Biol. Chem. 2014, 289, 27134–27145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingnate, C.; Charoenkwan, K.; Kumfu, S.; Chattipakorn, N.; Chattipakorn, S.C. Possible Roles of Mitochondrial Dynamics and the Effects of Pharmacological Interventions in Chemoresistant Ovarian Cancer. EBioMedicine 2018, 34, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer 2017, 3, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Glytsou, C.; Zhou, H.; Narang, S.; Reyna, D.E.; Lopez, A.; Sakellaropoulos, T.; Gong, Y.; Kloetgen, A.; Yap, Y.S.; et al. Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov. 2019, 9, 890–909. [Google Scholar] [CrossRef]
- Ramsey, H.E.; Fischer, M.A.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensintaffar, J.; Hogdal, L.J.; et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef] [Green Version]
- Decker, C.W.; Garcia, J.; Gatchalian, K.; Arceneaux, D.; Choi, C.; Han, D.; Hernandez, J.B. Mitofusin-2 mediates doxorubicin sensitivity and acute resistance in Jurkat leukemia cells. Biochem. Biophys. Rep. 2020, 24, 100824. [Google Scholar] [CrossRef]
- Otasevic, V.; Surlan, L.; Vucetic, M.; Tulic, I.; Buzadzic, B.; Stancic, A.; Jankovic, A.; Velickovic, K.; Golic, I.; Markelic, M.; et al. Expression patterns of mitochondrial OXPHOS components, mitofusin 1 and dynamin-related protein 1 are associated with human embryo fragmentation. Reprod. Fertil. Dev. 2016, 28, 319–327. [Google Scholar] [CrossRef]
- Casinelli, G.; LaRosa, J.; Sharma, M.; Cherok, E.; Banerjee, S.; Branca, M.; Edmunds, L.; Wang, Y.; Sims-Lucas, S.; Churley, L.; et al. N-Myc overexpression increases cisplatin resistance in neuroblastoma via deregulation of mitochondrial dynamics. Cell Death Discov. 2016, 2, 16082. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, T.; Ferraz, L.S. Therapeutic potential of targeting mitochondrial dynamics in cancer. Biochem. Pharmacol. 2020, 182, 114282. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Shi, F.; Li, Y.; Li, W.; Yu, X.; Zhao, L.; Zhou, M.; Hu, J.; Luo, X.; Tang, M.; et al. Drp1-dependent remodeling of mitochondrial morphology triggered by EBV-LMP1 increases cisplatin resistance. Signal Transduct. Target. Ther. 2020, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.D.; Huang, T.J.; Peng, L.X.; Yang, C.F.; Liu, R.Y.; Huang, H.B.; Chu, Q.Q.; Yang, H.J.; Huang, J.L.; Zhu, Z.Y.; et al. Epstein-Barr Virus_Encoded LMP1 upregulates microRNA-21 to promote the resistance of nasopharyngeal carcinoma cells to cisplatin-induced Apoptosis by suppressing PDCD4 and Fas-L. PLoS ONE 2013, 8, e78355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Kim, B.; Cho, U.; Park, I.S.; Kim, S.I.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Mitochondrial fission causes cisplatin resistance under hypoxic conditions via ROS in ovarian cancer cells. Oncogene 2019, 38, 7089–7105. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Wang, J.; Huang, Y.; Wu, H.; Xia, T.; Xiao, J.; Chen, X.; Li, H.; Qiu, Y.; Wang, Y.; et al. ERK/Drp1-dependent mitochondrial fission is involved in the MSC-induced drug resistance of T-cell acute lymphoblastic leukemia cells. Cell Death Dis. 2016, 7, e2459. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Luan, T.; Chen, Y.; Bao, X.; Huang, Y.; Fu, S.; Wang, H.; Wang, J. LASS2 regulates invasion and chemoresistance via ERK/Drp1 modulated mitochondrial dynamics in bladder cancer cells. J. Cancer 2018, 9, 1017–1024. [Google Scholar] [CrossRef]
- Luan, T.; Fu, S.; Huang, L.; Zuo, Y.; Ding, M.; Li, N.; Chen, J.; Wang, H.; Wang, J. MicroRNA-98 promotes drug resistance and regulates mitochondrial dynamics by targeting LASS2 in bladder cancer cells. Exp. Cell Res. 2018, 373, 188–197. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chiang, S.F.; Chen, W.T.; Ke, T.W.; Chen, T.W.; You, Y.S.; Lin, C.Y.; Chao, K.S.C.; Huang, C.Y. HMGB1 promotes ERK-mediated mitochondrial Drp1 phosphorylation for chemoresistance through RAGE in colorectal cancer. Cell Death Dis. 2018, 9, 1004. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Xu, L.; Qi, L.; Wang, C.; Xu, N.; Liu, S.; Li, S.; Tian, H.; Liu, W.; Xu, Y.; et al. ABT737 induces mitochondrial pathway apoptosis and mitophagy by regulating DRP1-dependent mitochondrial fission in human ovarian cancer cells. Biomed. Pharmacother. 2017, 96, 22–29. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015, 22, 1399–1401. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Wang, K.Z.; Chu, C.T. After the banquet: Mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 2013, 9, 1663–1676. [Google Scholar] [CrossRef] [Green Version]
- Dominy, J.E.; Puigserver, P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a015008. [Google Scholar] [CrossRef] [PubMed]
- Viscomi, C.; Bottani, E.; Civiletto, G.; Cerutti, R.; Moggio, M.; Fagiolari, G.; Schon, E.A.; Lamperti, C.; Zeviani, M. In vivo correction of COX deficiency by activation of the AMPK/PGC-1alpha axis. Cell Metab. 2011, 14, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Haq, R.; Shoag, J.; Andreu-Perez, P.; Yokoyama, S.; Edelman, H.; Rowe, G.C.; Frederick, D.T.; Hurley, A.D.; Nellore, A.; Kung, A.L.; et al. Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 2013, 23, 302–315. [Google Scholar] [CrossRef] [Green Version]
- McGuirk, S.; Gravel, S.P.; Deblois, G.; Papadopoli, D.J.; Faubert, B.; Wegner, A.; Hiller, K.; Avizonis, D.; Akavia, U.D.; Jones, R.G.; et al. PGC-1alpha supports glutamine metabolism in breast cancer. Cancer Metab. 2013, 1, 22. [Google Scholar] [CrossRef] [Green Version]
- Vazquez, F.; Lim, J.H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.H.; Luo, C.; Vazquez, F.; Puigserver, P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res. 2014, 74, 3535–3545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaGory, E.L.; Wu, C.; Taniguchi, C.M.; Ding, C.C.; Chi, J.T.; von Eyben, R.; Scott, D.A.; Richardson, A.D.; Giaccia, A.J. Suppression of PGC-1alpha Is Critical for Reprogramming Oxidative Metabolism in Renal Cell Carcinoma. Cell Rep. 2015, 12, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Feilchenfeldt, J.; Brundler, M.A.; Soravia, C.; Totsch, M.; Meier, C.A. Peroxisome proliferator-activated receptors (PPARs) and associated transcription factors in colon cancer: Reduced expression of PPARgamma-coactivator 1 (PGC-1). Cancer Lett. 2004, 203, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ba, Y.; Liu, C.; Sun, G.; Ding, L.; Gao, S.; Hao, J.; Yu, Z.; Zhang, J.; Zen, K.; et al. PGC-1alpha induces apoptosis in human epithelial ovarian cancer cells through a PPARgamma-dependent pathway. Cell Res. 2007, 17, 363–373. [Google Scholar] [CrossRef]
- Neill, T.; Torres, A.; Buraschi, S.; Owens, R.T.; Hoek, J.B.; Baffa, R.; Iozzo, R.V. Decorin induces mitophagy in breast carcinoma cells via peroxisome proliferator-activated receptor gamma coactivator-1alpha (PGC-1alpha) and mitostatin. J. Biol. Chem. 2014, 289, 4952–4968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ba, Y.; Zhang, Y.; Zhang, C.Y. Peroxisome proliferator activated receptor gamma coactivator-1 induces apoptosis in human epithelial ovarian cancer cells. Zhonghua Yi Xue Za Zhi 2007, 87, 1430–1433. [Google Scholar] [PubMed]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [Green Version]
- Lemasters, J.J. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. 2005, 8, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kondo-Okamoto, N.; Ohsumi, Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev. Cell 2009, 17, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Methods for detecting autophagy and determining autophagy-induced cell death. Can. J. Physiol. Pharmacol. 2010, 88, 285–295. [Google Scholar] [CrossRef]
- Levine, B.; Klionsky, D.J. Development by self-digestion: Molecular mechanisms and biological functions of autophagy. Dev. Cell 2004, 6, 463–477. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Yuan, J. Autophagy in cell death: An innocent convict? J. Clin. Investig. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Criollo, A.; Kroemer, G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010, 29, 515–516. [Google Scholar] [CrossRef] [Green Version]
- Tan, E.Y.; Campo, L.; Han, C.; Turley, H.; Pezzella, F.; Gatter, K.C.; Harris, A.L.; Fox, S.B. BNIP3 as a progression marker in primary human breast cancer; opposing functions in in situ versus invasive cancer. Clin. Cancer Res. 2007, 13, 467–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, A.G.; Maugeri, G.; Magro, G.; Salvatorelli, L.; Drago, F.; D’Agata, V. Expression pattern of parkin isoforms in lung adenocarcinomas. Tumour Biol. 2015, 36, 5133–5141. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zuo, Z.; Lu, S.; Wang, L.; Liu, A.; Liu, X. Silencing of PINK1 represses cell growth, migration and induces apoptosis of lung cancer cells. Biomed. Pharmacother. 2018, 106, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Wu, H.; Wang, T.W.; Yang, N.; Guo, X.; Jang, X.J. Hydrogen peroxide-induced mitophagy contributes to laryngeal cancer cells survival via the upregulation of FUNDC1. Clin. Transl. Oncol. 2019, 21, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Miyata, H.; Makino, T.; Masuike, Y.; Furukawa, H.; Tanaka, K.; Miyazaki, Y.; Takahashi, T.; Kurokawa, Y.; Yamasaki, M.; et al. High Expression of the Mitophagy-Related Protein Pink1 is Associated with a Poor Response to Chemotherapy and a Poor Prognosis for Patients Treated with Neoadjuvant Chemotherapy for Esophageal Squamous Cell Carcinoma. Ann. Surg. Oncol. 2017, 24, 4025–4032. [Google Scholar] [CrossRef]
- Li, G.B.; Fu, R.Q.; Shen, H.M.; Zhou, J.; Hu, X.Y.; Liu, Y.X.; Li, Y.N.; Zhang, H.W.; Liu, X.; Zhang, Y.H.; et al. Polyphyllin I induces mitophagic and apoptotic cell death in human breast cancer cells by increasing mitochondrial PINK1 levels. Oncotarget 2017, 8, 10359–10374. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.H.; Lee, A.Y.; Yu, K.N.; Park, J.; Kim, K.P.; Cho, M.H. Dihydroergotamine Tartrate Induces Lung Cancer Cell Death through Apoptosis and Mitophagy. Chemotherapy 2016, 61, 304–312. [Google Scholar] [CrossRef]
- Regula, K.M.; Ens, K.; Kirshenbaum, L.A. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ. Res. 2002, 91, 226–231. [Google Scholar] [CrossRef] [Green Version]
- Kanzawa, T.; Zhang, L.; Xiao, L.; Germano, I.M.; Kondo, Y.; Kondo, S. Arsenic trioxide induces autophagic cell death in malignant glioma cells by upregulation of mitochondrial cell death protein BNIP3. Oncogene 2005, 24, 980–991. [Google Scholar] [CrossRef] [Green Version]
- Daido, S.; Kanzawa, T.; Yamamoto, A.; Takeuchi, H.; Kondo, Y.; Kondo, S. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004, 64, 4286–4293. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.P.; Yang, M.C.; Liu, H.S.; Lin, Y.S.; Lei, H.Y. Concanavalin A induces autophagy in hepatoma cells and has a therapeutic effect in a murine in situ hepatoma model. Hepatology 2007, 45, 286–296. [Google Scholar] [CrossRef]
- Panigrahi, D.P.; Praharaj, P.P.; Bhol, C.S.; Mahapatra, K.K.; Patra, S.; Behera, B.P.; Mishra, S.R.; Bhutia, S.K. The emerging, multifaceted role of mitophagy in cancer and cancer therapeutics. Semin. Cancer Biol. 2020, 66, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Okatsu, K.; Oka, T.; Iguchi, M.; Imamura, K.; Kosako, H.; Tani, N.; Kimura, M.; Go, E.; Koyano, F.; Funayama, M.; et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat. Commun. 2012, 3, 1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazlauskaite, A.; Kondapalli, C.; Gourlay, R.; Campbell, D.G.; Ritorto, M.S.; Hofmann, K.; Alessi, D.R.; Knebel, A.; Trost, M.; Muqit, M.M. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem. J. 2014, 460, 127–139. [Google Scholar] [CrossRef] [Green Version]
- Kazlauskaite, A.; Martinez-Torres, R.J.; Wilkie, S.; Kumar, A.; Peltier, J.; Gonzalez, A.; Johnson, C.; Zhang, J.; Hope, A.G.; Peggie, M.; et al. Binding to serine 65-phosphorylated ubiquitin primes Parkin for optimal PINK1-dependent phosphorylation and activation. EMBO Rep. 2015, 16, 939–954. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Yao, N.; Wang, C.; Hu, N.; Li, Y.; Liu, M.; Lei, Y.; Chen, M.; Chen, L.; Chen, C.; Lan, P.; et al. Inhibition of PINK1/Parkin-dependent mitophagy sensitizes multidrug-resistant cancer cells to B5G1, a new betulinic acid analog. Cell Death Dis. 2019, 10, 232. [Google Scholar] [CrossRef]
- Zhu, Y.; Massen, S.; Terenzio, M.; Lang, V.; Chen-Lindner, S.; Eils, R.; Novak, I.; Dikic, I.; Hamacher-Brady, A.; Brady, N.R. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J. Biol. Chem. 2013, 288, 1099–1113. [Google Scholar] [CrossRef] [Green Version]
- Novak, I.; Kirkin, V.; McEwan, D.G.; Zhang, J.; Wild, P.; Rozenknop, A.; Rogov, V.; Lohr, F.; Popovic, D.; Occhipinti, A.; et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010, 11, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhujabal, Z.; Birgisdottir, A.B.; Sjottem, E.; Brenne, H.B.; Overvatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Novak, I.; Dikic, I. Autophagy receptors in developmental clearance of mitochondria. Autophagy 2011, 7, 301–303. [Google Scholar] [CrossRef]
- Yan, C.; Luo, L.; Guo, C.Y.; Goto, S.; Urata, Y.; Shao, J.H.; Li, T.S. Doxorubicin-induced mitophagy contributes to drug resistance in cancer stem cells from HCT8 human colorectal cancer cells. Cancer Lett. 2017, 388, 34–42. [Google Scholar] [CrossRef]
- Hou, H.; Er, P.; Cheng, J.; Chen, X.; Ding, X.; Wang, Y.; Chen, X.; Yuan, Z.; Pang, Q.; Wang, P.; et al. High expression of FUNDC1 predicts poor prognostic outcomes and is a promising target to improve chemoradiotherapy effects in patients with cervical cancer. Cancer Med. 2017, 6, 1871–1881. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Radoshevich, L.; Murrow, L.; Chen, N.; Fernandez, E.; Roy, S.; Fung, C.; Debnath, J. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell 2010, 142, 590–600. [Google Scholar] [CrossRef] [Green Version]
- Hua, L.; Zhu, G.; Wei, J. MicroRNA-1 overexpression increases chemosensitivity of non-small cell lung cancer cells by inhibiting autophagy related 3-mediated autophagy. Cell Biol. Int. 2018, 42, 1240–1249. [Google Scholar] [CrossRef]
- Zhang, W.; Dong, Y.Z.; Du, X.; Peng, X.N.; Shen, Q.M. MiRNA-153-3p promotes gefitinib-sensitivity in non-small cell lung cancer by inhibiting ATG5 expression and autophagy. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 2444–2452. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.H.; Yeom, H.; Lee, M. ATG5 knockout promotes paclitaxel sensitivity in drug-resistant cells via induction of necrotic cell death. Korean J. Physiol. Pharmacol. 2020, 24, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Eom, S.Y.; Hwang, S.H.; Yeom, H.; Lee, M. An ATG5 knockout promotes paclitaxel resistance in v-Ha-ras-transformed NIH 3T3 cells. Biochem. Biophys. Res. Commun. 2019, 513, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Chen, T.; Li, G.; Xu, C.; Xu, Z.; Zhang, J.; He, K.; Zheng, L.; Guan, Z.; Su, X.; et al. Lower Beclin 1 downregulates HER2 expression to enhance tamoxifen sensitivity and predicts a favorable outcome for ER positive breast cancer. Oncotarget 2017, 8, 52156–52177. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Liu, S.; Chen, H.; Lao, L. MicroRNA-30a downregulation contributes to chemoresistance of osteosarcoma cells through activating Beclin-1-mediated autophagy. Oncol. Rep. 2016, 35, 1757–1763. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Wu, L.; Ding, H.; Wang, Y.; Zhang, Y.; Chen, X.; Chen, X.; Zhang, C.Y.; Zhang, Q.; Zen, K. MicroRNA-30a sensitizes tumor cells to cis-platinum via suppressing beclin 1-mediated autophagy. J. Biol. Chem. 2012, 287, 4148–4156. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.; Hu, C.; Gu, F.; Xia, Q.; Yao, C.; Zhang, L.; Qiang, L.; Gao, S.; Gao, Y. Co-delivery of autophagy inhibitor ATG7 siRNA and docetaxel for breast cancer treatment. J. Control. Release 2017, 266, 272–286. [Google Scholar] [CrossRef]
- An, Y.; Zhang, Z.; Shang, Y.; Jiang, X.; Dong, J.; Yu, P.; Nie, Y.; Zhao, Q. miR-23b-3p regulates the chemoresistance of gastric cancer cells by targeting ATG12 and HMGB2. Cell Death Dis. 2015, 6, e1766. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef]
- Nguyen, E.V.; Huhtinen, K.; Goo, Y.A.; Kaipio, K.; Andersson, N.; Rantanen, V.; Hynninen, J.; Lahesmaa, R.; Carpen, O.; Goodlett, D.R. Hyper-phosphorylation of Sequestosome-1 Distinguishes Resistance to Cisplatin in Patient Derived High Grade Serous Ovarian Cancer Cells. Mol. Cell. Proteom. 2017, 16, 1377–1392. [Google Scholar] [CrossRef] [Green Version]
- Daido, S.; Yamamoto, A.; Fujiwara, K.; Sawaya, R.; Kondo, S.; Kondo, Y. Inhibition of the DNA-dependent protein kinase catalytic subunit radiosensitizes malignant glioma cells by inducing autophagy. Cancer Res. 2005, 65, 4368–4375. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Wang, W.; Li, Y.; Yang, D.; Li, X.; Shen, C.; Liu, Y.; Ke, X.; Guo, S.; Guo, Z. HSP90AA1-mediated autophagy promotes drug resistance in osteosarcoma. J. Exp. Clin. Cancer Res. 2018, 37, 201. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Gao, R.; Wang, J.; Yuan, W.; Wang, C.; Zhou, X. High-mobility group nucleosome-binding domain 5 increases drug resistance in osteosarcoma through upregulating autophagy. Tumour Biol. 2014, 35, 6357–6363. [Google Scholar] [CrossRef] [PubMed]
- Luk, F.; Yu, Y.; Walsh, W.R.; Yang, J.L. IGF1R-targeted therapy and its enhancement of doxorubicin chemosensitivity in human osteosarcoma cell lines. Cancer Investig. 2011, 29, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Bae, D.H.; Jansson, P.J.; Huang, M.L.; Kovacevic, Z.; Kalinowski, D.; Lee, C.S.; Sahni, S.; Richardson, D.R. The role of NDRG1 in the pathology and potential treatment of human cancers. J. Clin. Pathol. 2013, 66, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant, H.E.; Helleday, T. Inhibition of poly (ADP-ribose) polymerase activates ATM which is required for subsequent homologous recombination repair. Nucleic Acids Res. 2006, 34, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Golla, R.M.; Li, M.; Shen, Y.; Ji, M.; Yan, Y.; Fu, K.; Greiner, T.C.; McKeithan, T.W.; Chan, W.C. Inhibition of poly(ADP-ribose) polymerase (PARP) and ataxia telangiectasia mutated (ATM) on the chemosensitivity of mantle cell lymphoma to agents that induce DNA strand breaks. Hematol. Oncol. 2012, 30, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J., III; et al. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Zeh, H.J., III; Lotze, M.T. High mobility group box 1 (HMGB1) activates an autophagic response to oxidative stress. Antioxid. Redox Signal. 2011, 15, 2185–2195. [Google Scholar] [CrossRef]
- Cicco, S.; Cicco, G.; Racanelli, V.; Vacca, A. Neutrophil Extracellular Traps (NETs) and Damage-Associated Molecular Patterns (DAMPs): Two Potential Targets for COVID-19 Treatment. Mediat. Inflamm. 2020, 2020, 7527953. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, X.; Gilvary, D.L.; Tejera, M.M.; Eksioglu, E.A.; Wei, S.; Djeu, J.Y. HMGB1 induction of clusterin creates a chemoresistant niche in human prostate tumor cells. Sci. Rep. 2015, 5, 15085. [Google Scholar] [CrossRef] [PubMed]
- Azizian-Farsani, F.; Abedpoor, N.; Hasan Sheikhha, M.; Gure, A.O.; Nasr-Esfahani, M.H.; Ghaedi, K. Receptor for Advanced Glycation End Products Acts as a Fuel to Colorectal Cancer Development. Front. Oncol. 2020, 10, 552283. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Sugihara, E.; Yamaguchi-Iwai, S.; Tamaki, S.; Koyama, Y.; Kamel, W.; Ueki, A.; Ishikawa, T.; Chiyoda, T.; Osuka, S.; et al. IGF2 preserves osteosarcoma cell survival by creating an autophagic state of dormancy that protects cells against chemotherapeutic stress. Cancer Res. 2014, 74, 6531–6541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahni, S.; Gillson, J.; Park, K.C.; Chiang, S.; Leck, L.Y.W.; Jansson, P.J.; Richardson, D.R. NDRG1 suppresses basal and hypoxia-induced autophagy at both the initiation and degradation stages and sensitizes pancreatic cancer cells to lysosomal membrane permeabilization. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129625. [Google Scholar] [CrossRef]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Zhivotovsky, B.; Orrenius, S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium 2011, 50, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Bong, A.H.L.; Monteith, G.R. Calcium signaling and the therapeutic targeting of cancer cells. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1786–1794. [Google Scholar] [CrossRef]
- Dubois, C.; Kondratskyi, A.; Bidaux, G.; Noyer, L.; Vancauwenberghe, E.; Farfariello, V.; Toillon, R.A.; Roudbaraki, M.; Tierny, D.; Bonnal, J.L.; et al. Co-targeting Mitochondrial Ca2+ Homeostasis and Autophagy Enhances Cancer Cells’ Chemosensitivity. iScience 2020, 23, 101263. [Google Scholar] [CrossRef]
- Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafa, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca2+ Signaling in Health, Disease and Therapy. Cells 2021, 10, 1317. [Google Scholar] [CrossRef]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Kondratskyi, A.; Kondratska, K.; Skryma, R.; Klionsky, D.J.; Prevarskaya, N. Ion channels in the regulation of autophagy. Autophagy 2018, 14, 3–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmohsen, K.; Srikantan, S.; Tominaga, K.; Kang, M.J.; Yaniv, Y.; Martindale, J.L.; Yang, X.; Park, S.S.; Becker, K.G.; Subramanian, M.; et al. Growth inhibition by miR-519 via multiple p21-inducing pathways. Mol. Cell. Biol. 2012, 32, 2530–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.D.; Xia, X.; Lv, X.F.; Yu, B.X.; Yuan, J.N.; Mai, X.Y.; Shang, J.Y.; Zhou, J.G.; Liang, S.J.; Pang, R.P. Inhibition of Orai1-mediated Ca2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J. Cell Mol. Med. 2017, 21, 904–915. [Google Scholar] [CrossRef]
- Hoyer-Hansen, M.; Bastholm, L.; Szyniarowski, P.; Campanella, M.; Szabadkai, G.; Farkas, T.; Bianchi, K.; Fehrenbacher, N.; Elling, F.; Rizzuto, R.; et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 2007, 25, 193–205. [Google Scholar] [CrossRef]
- Mathiasen, I.S.; Sergeev, I.N.; Bastholm, L.; Elling, F.; Norman, A.W.; Jaattela, M. Calcium and calpain as key mediators of apoptosis-like death induced by vitamin D compounds in breast cancer cells. J. Biol. Chem. 2002, 277, 30738–30745. [Google Scholar] [CrossRef] [Green Version]
- Decuypere, J.P.; Welkenhuyzen, K.; Luyten, T.; Ponsaerts, R.; Dewaele, M.; Molgo, J.; Agostinis, P.; Missiaen, L.; De Smedt, H.; Parys, J.B.; et al. Ins(1,4,5)P3 receptor-mediated Ca2+ signaling and autophagy induction are interrelated. Autophagy 2011, 7, 1472–1489. [Google Scholar] [CrossRef] [Green Version]
- Bednarczyk, P.; Kicinska, A.; Laskowski, M.; Kulawiak, B.; Kampa, R.; Walewska, A.; Krajewska, M.; Jarmuszkiewicz, W.; Szewczyk, A. Evidence for a mitochondrial ATP-regulated potassium channel in human dermal fibroblasts. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 309–318. [Google Scholar] [CrossRef]
- Laskowski, M.; Kulawiak, B. Topology of the mitochondrial potassium ion channels. Postepy Biochem. 2016, 62, 199–205. [Google Scholar]
- Nakaya, H. Role of ATP-sensitive K+ channels in cardiac arrhythmias. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 237–243. [Google Scholar] [CrossRef]
- Galecka, S.; Kulawiak, B.; Bednarczyk, P.; Singh, H.; Szewczyk, A. Single channel properties of mitochondrial large conductance potassium channel formed by BK-VEDEC splice variant. Sci. Rep. 2021, 11, 10925. [Google Scholar] [CrossRef] [PubMed]
- Bednarczyk, P.; Kowalczyk, J.E.; Beresewicz, M.; Dolowy, K.; Szewczyk, A.; Zablocka, B. Identification of a voltage-gated potassium channel in gerbil hippocampal mitochondria. Biochem. Biophys. Res. Commun. 2010, 397, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Toczylowska-Maminska, R.; Olszewska, A.; Laskowski, M.; Bednarczyk, P.; Skowronek, K.; Szewczyk, A. Potassium channel in the mitochondria of human keratinocytes. J. Investig. Dermatol. 2014, 134, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.; Kim, S.H.; Hwang, E.M.; Kwon, O.S.; Yang, H.Y.; Kim, E.S.; Choi, T.H.; Park, J.Y.; Hong, S.G.; Han, J. Expression of thermosensitive two-pore domain K+ channels in human keratinocytes cell line HaCaT cells. Exp. Dermatol. 2007, 16, 1016–1022. [Google Scholar] [CrossRef]
- Rotko, D.; Kunz, W.S.; Szewczyk, A.; Kulawiak, B. Signaling pathways targeting mitochondrial potassium channels. Int. J. Biochem. Cell Biol. 2020, 125, 105792. [Google Scholar] [CrossRef] [PubMed]
- Krabbendam, I.E.; Honrath, B.; Culmsee, C.; Dolga, A.M. Mitochondrial Ca2+-activated K(+) channels and their role in cell life and death pathways. Cell Calcium 2018, 69, 101–111. [Google Scholar] [CrossRef]
- Testai, L.; Rapposelli, S.; Martelli, A.; Breschi, M.C.; Calderone, V. Mitochondrial potassium channels as pharmacological target for cardioprotective drugs. Med. Res. Rev. 2015, 35, 520–553. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.B.; Ho, A.S.; Rucker, J.; Garlid, A.O.; Chen, L.; Sidor, A.; Garlid, K.D.; O’Rourke, B. Mitochondrial ROMK channel is a molecular component of mitoK(ATP). Circ. Res. 2012, 111, 446–454. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, M.; Augustynek, B.; Bednarczyk, P.; Zochowska, M.; Kalisz, J.; O’Rourke, B.; Szewczyk, A.; Kulawiak, B. Single-Channel Properties of the ROMK-Pore-Forming Subunit of the Mitochondrial ATP-Sensitive Potassium Channel. Int. J. Mol. Sci. 2019, 20, 5323. [Google Scholar] [CrossRef] [Green Version]
- Papanicolaou, K.N.; Ashok, D.; Liu, T.; Bauer, T.M.; Sun, J.; Li, Z.; da Costa, E.; D’Orleans, C.C.; Nathan, S.; Lefer, D.J.; et al. Global knockout of ROMK potassium channel worsens cardiac ischemia-reperfusion injury but cardiomyocyte-specific knockout does not: Implications for the identity of mitoKATP. J. Mol. Cell. Cardiol. 2020, 139, 176–189. [Google Scholar] [CrossRef]
- Kulawiak, B.; Bednarczyk, P.; Szewczyk, A. Multidimensional Regulation of Cardiac Mitochondrial Potassium Channels. Cells 2021, 10, 1554. [Google Scholar] [CrossRef]
- Wrzosek, A.; Augustynek, B.; Zochowska, M.; Szewczyk, A. Mitochondrial Potassium Channels as Druggable Targets. Biomolecules 2020, 10, 1200. [Google Scholar] [CrossRef] [PubMed]
- Checchetto, V.; Prosdocimi, E.; Leanza, L. Mitochondrial Kv1.3: A New Target in Cancer Biology? Cell. Physiol. Biochem. 2019, 53, 52–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leanza, L.; Henry, B.; Sassi, N.; Zoratti, M.; Chandy, K.G.; Gulbins, E.; Szabo, I. Inhibitors of mitochondrial Kv1.3 channels induce Bax/Bak-independent death of cancer cells. EMBO Mol. Med. 2012, 4, 577–593. [Google Scholar] [CrossRef] [PubMed]
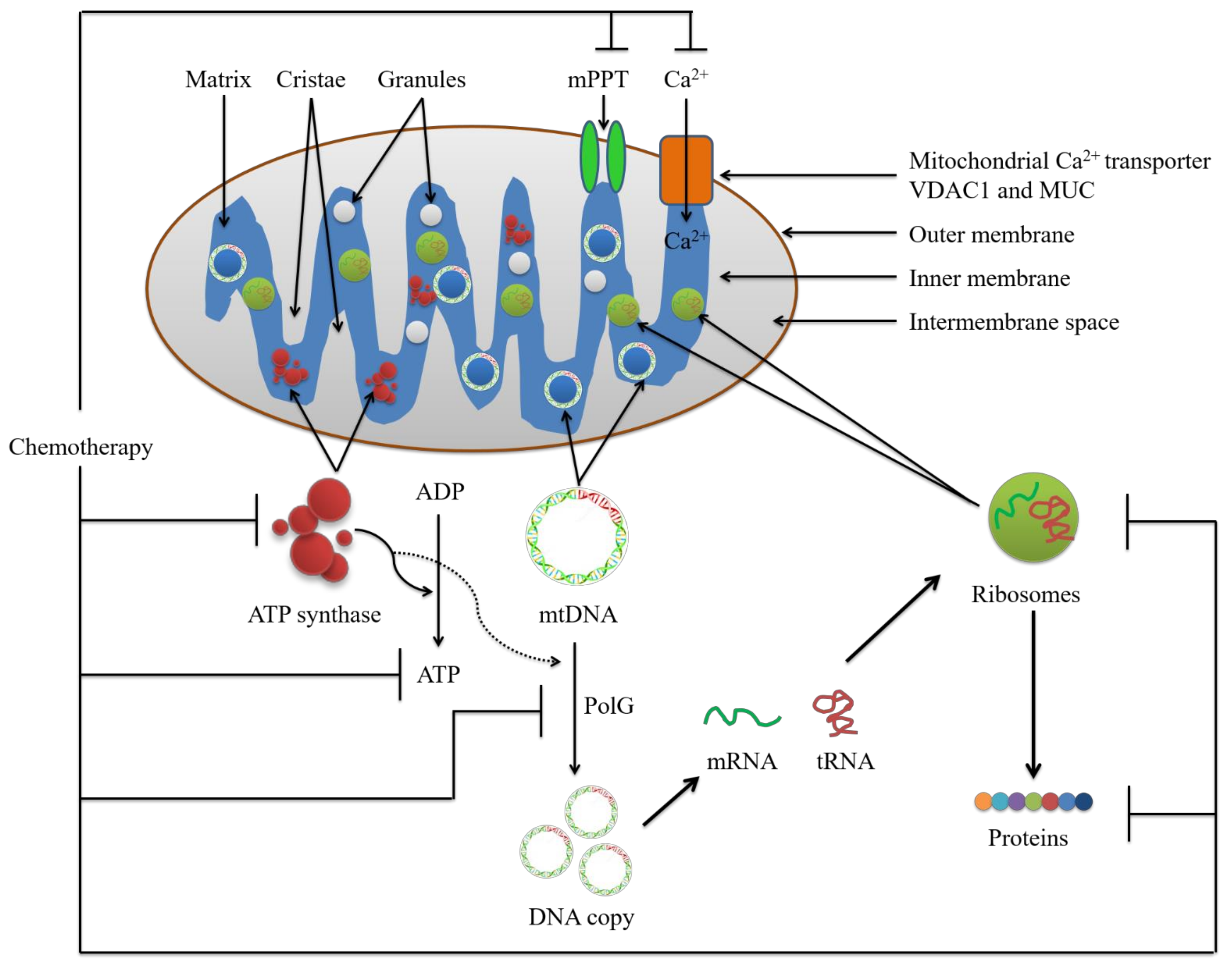


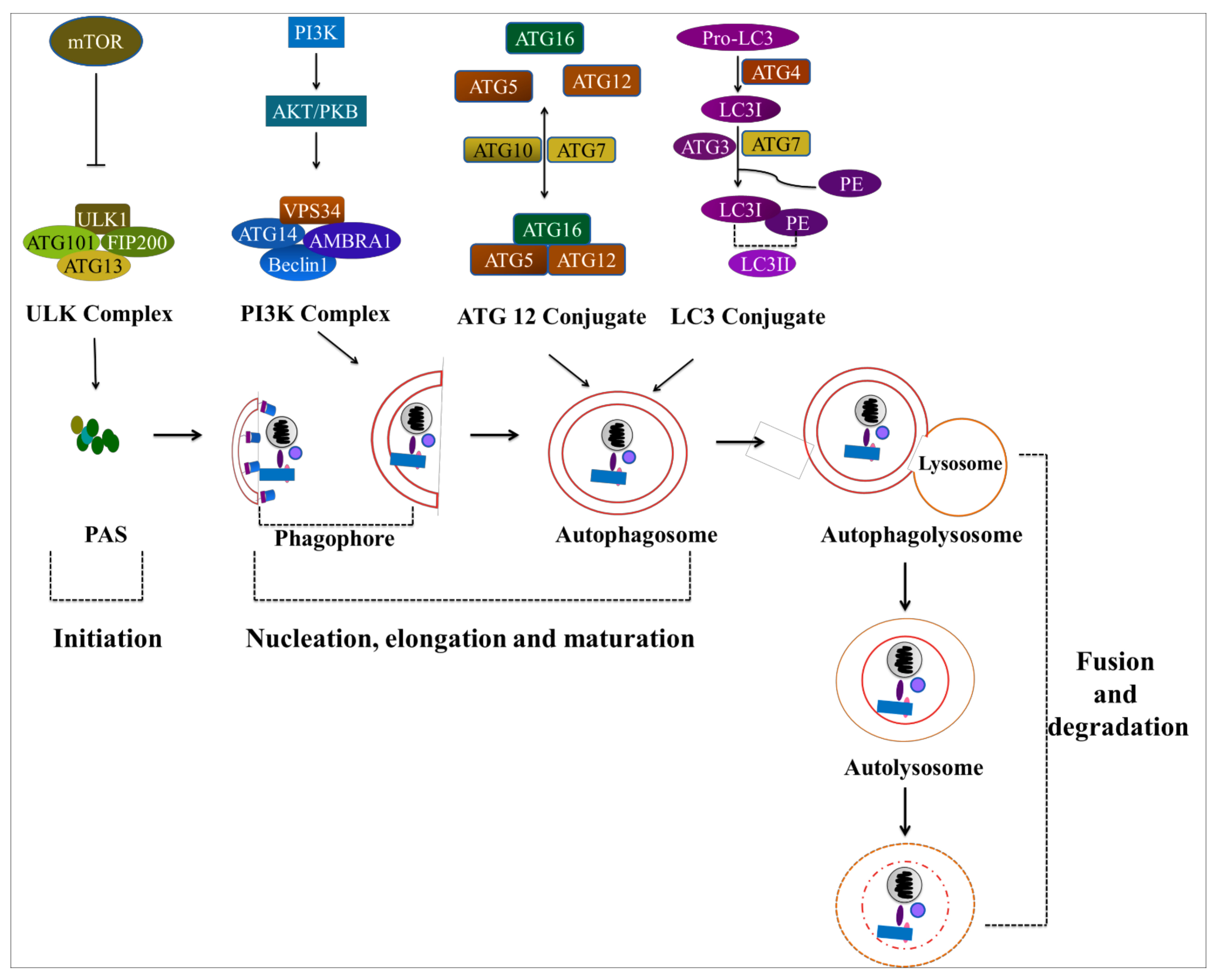

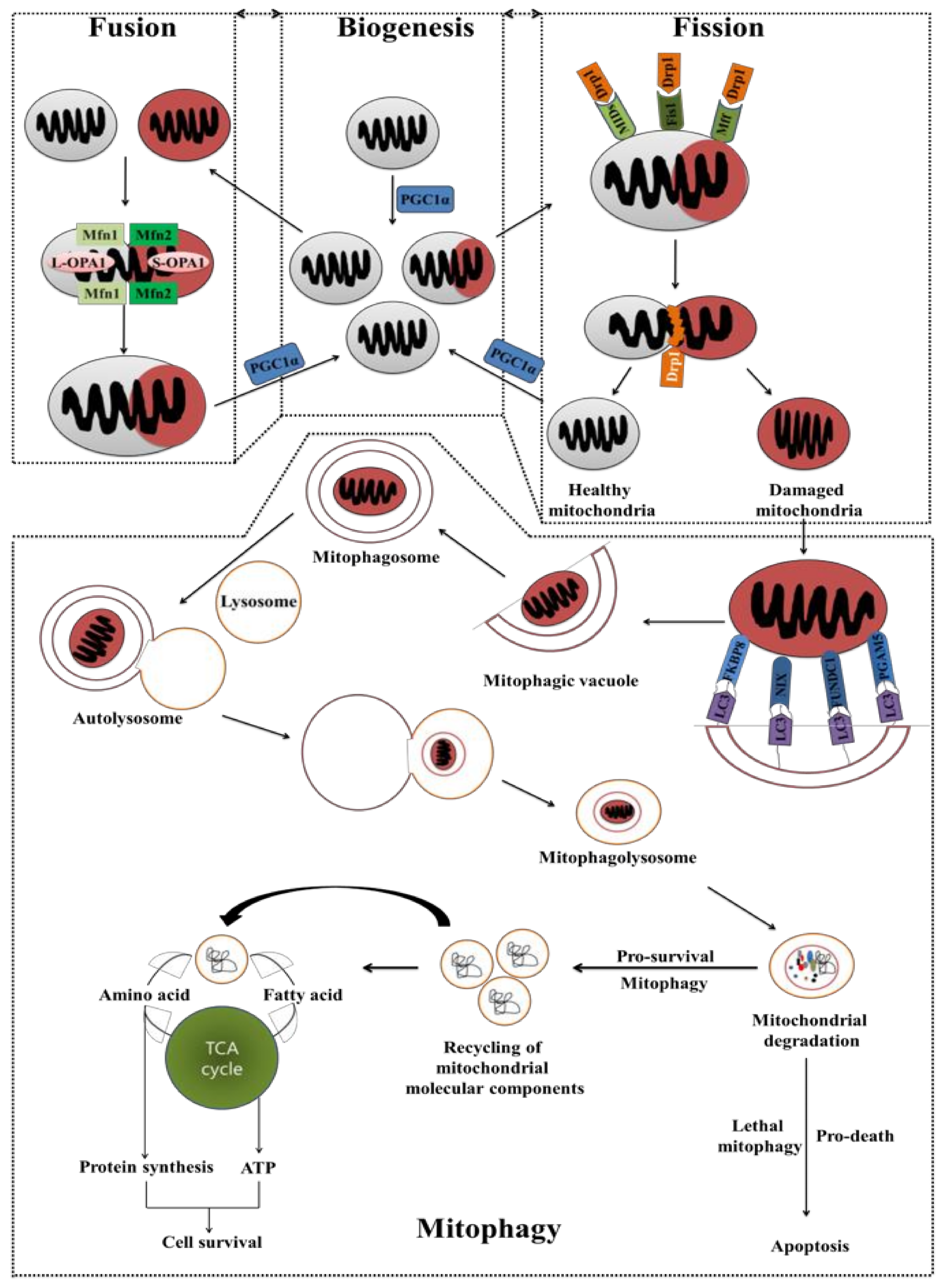


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets | Drugs | Augment the Effects of Chemotherapy | Refs. |
|---|---|---|---|
| GLUT1 | Silybin | Doxorubicin, cisplatin and paclitaxel | [203,204,205] |
| STF-31 | Gemcitabine, paclitaxel and 5-fluorouracil | [156] | |
| Hexokinase (HK) | 2-deoxyglucose (2-DG) | Cisplatin | [206] |
| Doxorubicin | [207] | ||
| Etoposide | [208] | ||
| Lonidamine | Cisplatin and paclitaxel | [209] | |
| Carboplatin | [210] | ||
| Doxorubicin | [150,151,211,212,213] | ||
| 3-bromopyruvate (3-BP) | 5-fluorouracil | [214] | |
| Tamoxifen | [215] | ||
| Daunorubicin | [216] | ||
| Dehydroascorbic acid (DHA) | Cisplatin or sorafenib | [202,217] | |
| HK2—VDAC complexes | Methyl jasmonate (MJ) | Cisplatin and doxorubicin | [218] |
| Phosphoglycerate mutase 1 (PGAM1) | HKB99 | Erlotinib | [219] |
| Pyruvate kinase M2 (PKM2) | 2-DG | Doxorubicin | [220] |
| Shikonin | Tamoxifen | [221] | |
| Metformin | Cisplatin, doxorubicin and 5-fluorouracil | [222,223] | |
| Pyruvate dehydrogenase (PDH) complex | Dichloroacetate (DCA) | Tamoxifen | [224,225] |
| Cetuximab | [226] | ||
| Lactate dehydrogenase (LDH) | Oxalate | Taxol | [227] |
| Galloflavin | 4-hydroxy-tamoxifen | [167,228] | |
| Aldehyde dehydrogenase (ALDH) | Gossypol | Irinotecan | [229,230] |
| Complex I | Phenformin | Gemcitabine and irinotecan | [229,230,231] |
| Deguelin | Vemurafenib | [232] | |
| Rotenone | Doxorubicin | [233] | |
| Complex IV | Chlorpromazine | Temozolomide | [234] |
| ATP synthase | Oligomycin | Doxorubicin | [233,235] |
| C-Gboxin | Gboxin | [236] | |
| Mitochondrial uncoupler | FCCP | Doxorubicin | [237] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahar, E.; Han, S.-Y.; Kim, J.-Y.; Yoon, H. Chemotherapy Resistance: Role of Mitochondrial and Autophagic Components. Cancers 2022, 14, 1462. https://doi.org/10.3390/cancers14061462
Bahar E, Han S-Y, Kim J-Y, Yoon H. Chemotherapy Resistance: Role of Mitochondrial and Autophagic Components. Cancers. 2022; 14(6):1462. https://doi.org/10.3390/cancers14061462
Chicago/Turabian StyleBahar, Entaz, Sun-Young Han, Ji-Ye Kim, and Hyonok Yoon. 2022. "Chemotherapy Resistance: Role of Mitochondrial and Autophagic Components" Cancers 14, no. 6: 1462. https://doi.org/10.3390/cancers14061462