
Divergent Metabolic Effects of Metformin Merge to Enhance Eicosapentaenoic Acid Metabolism and Inhibit Ovarian Cancer In Vivo


 ,
,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Media
2.2. Seahorse Metabolic Analysis
2.3. Cell Proliferation Assays
2.4. Metabolomics
2.4.1. Metabolite Assessment
2.4.2. Data Analysis
2.4.3. Pathway Analysis
2.5. Mouse Studies
2.5.1. Ethics Statement
2.5.2. Tumor Inoculation and Treatment
2.6. Total RNA Isolation and Real-Time PCR
2.7. Western Blot Analysis
2.8. Enzyme-Linked Immunosorbent Assay
2.9. Cell Cycle Analysis
2.10. Hoechst Staining
2.11. Immunohistochemistry
2.12. TUNEL Assay
2.13. Statistical Methods
3. Results
3.1. Metformin Induces Sustained Effect on Cellular Bioenergetics
3.2. Metformin Induces Differential Cell Line Specific Metabolomic Changes
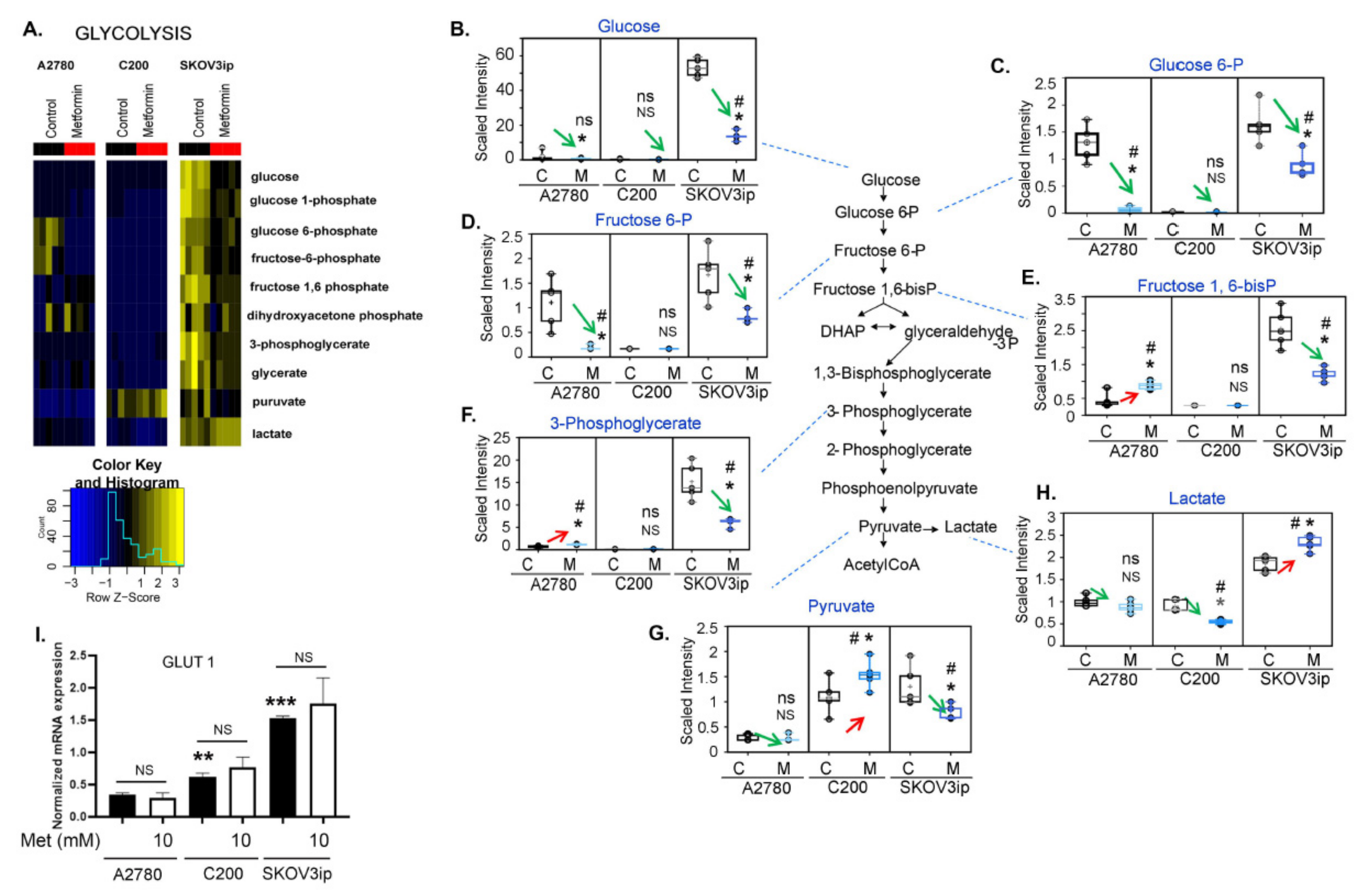
3.3. Changes in Glycolysis Metabolites following Metformin Treatment Were Cell Line Specific
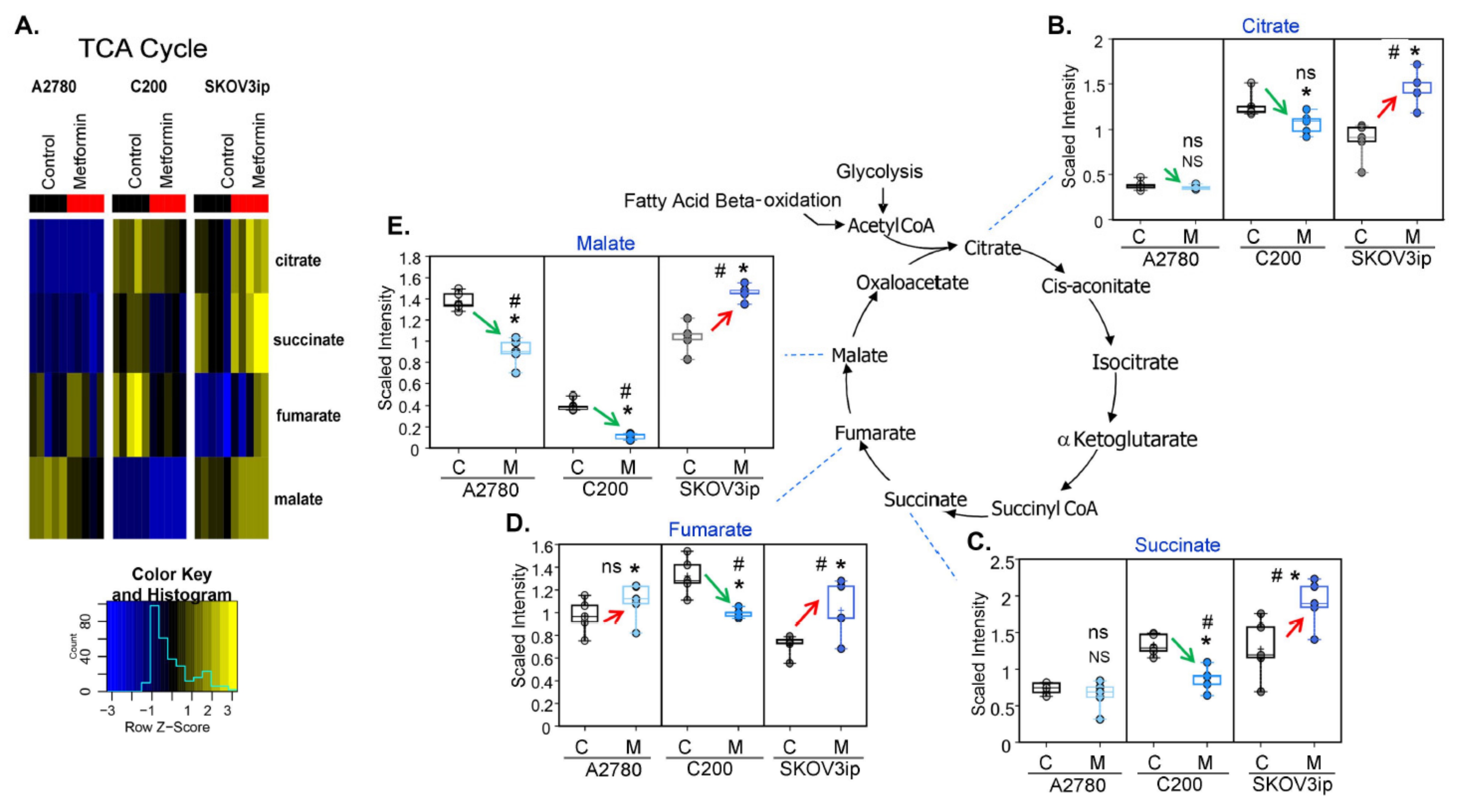
3.4. Changes in TCA Metabolites following Metformin Treatment Were Cell-Line Specific
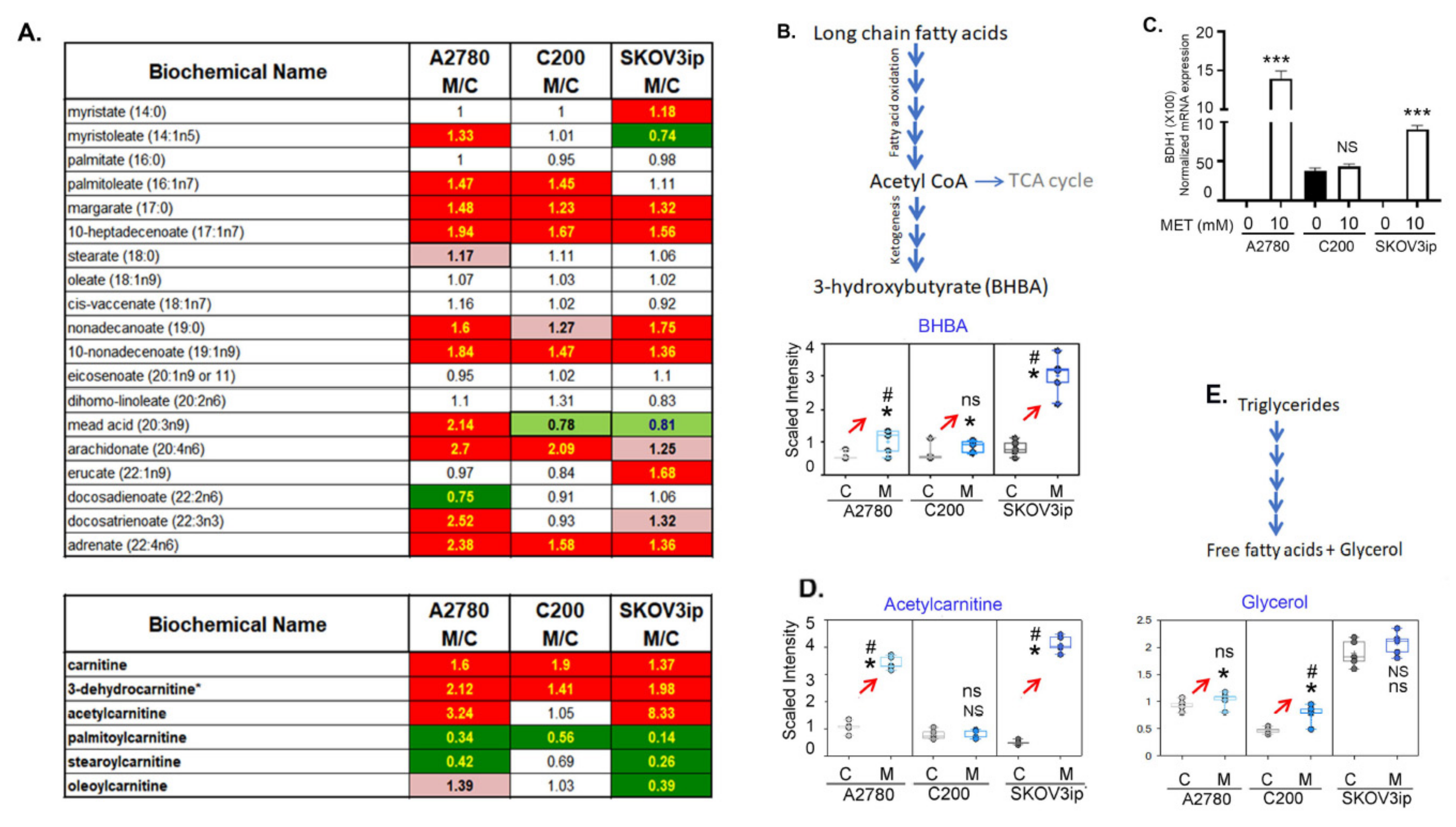
3.5. Fatty Acid β-Oxidation Was Increased by Metformin
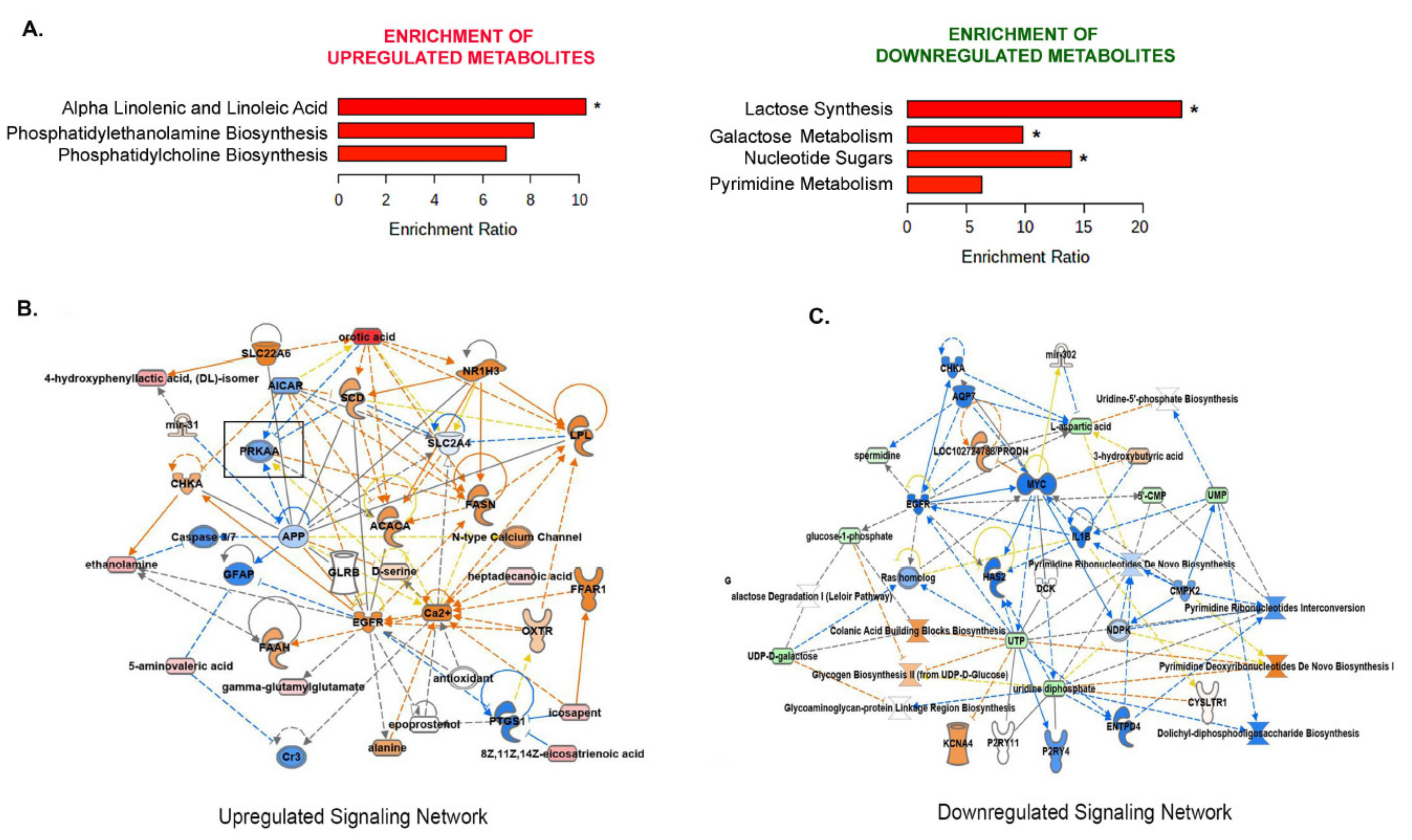
3.6. Alteration of Other Major Metabolic Pathways by Metformin
3.6.1. Metformin Inhibits the Pyrimidine Metabolic Pathway
3.6.2. Metformin Decreases the Polyamine Biosynthesis
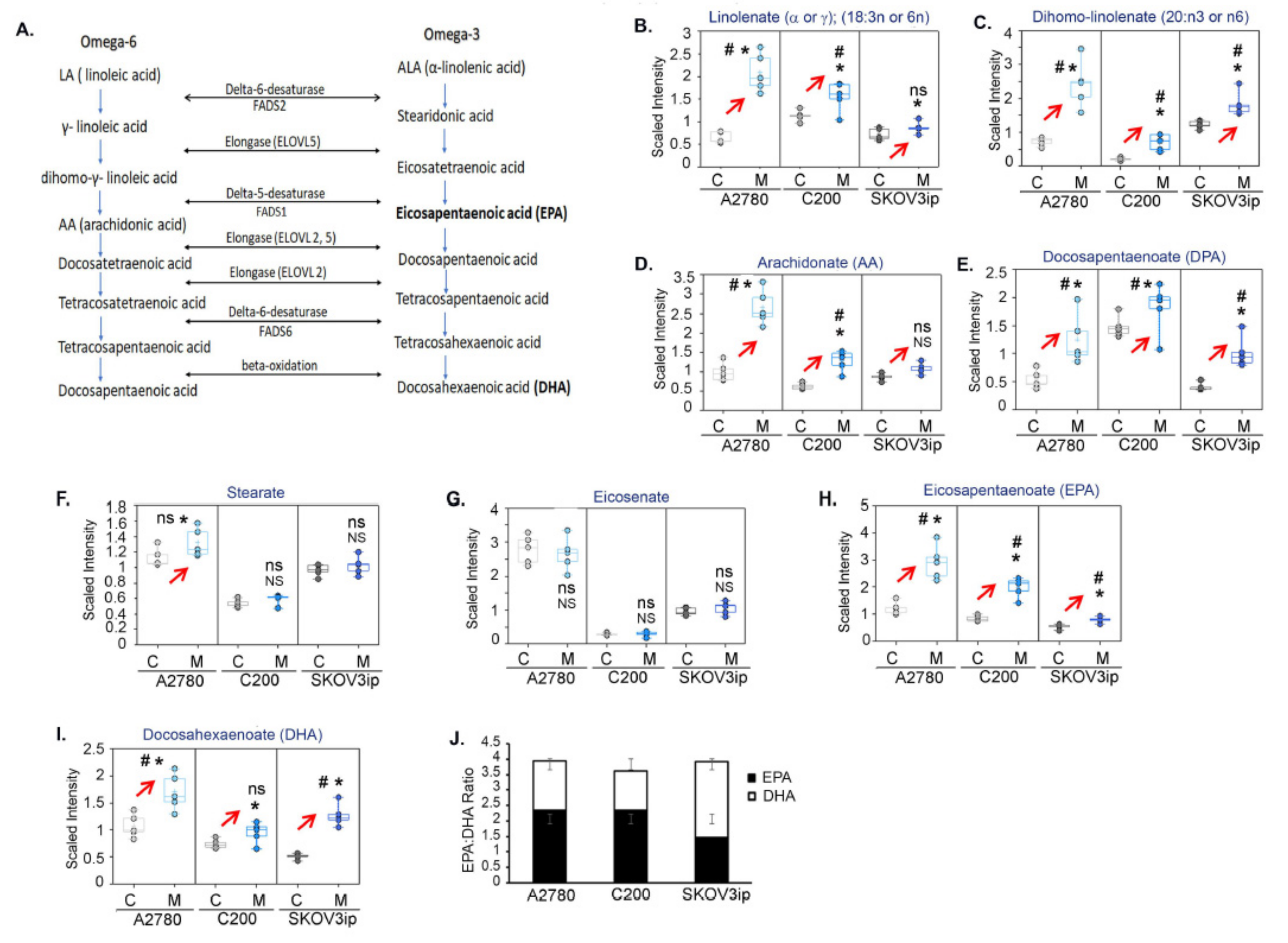
3.6.3. Metformin Increases Essential Fatty Acids
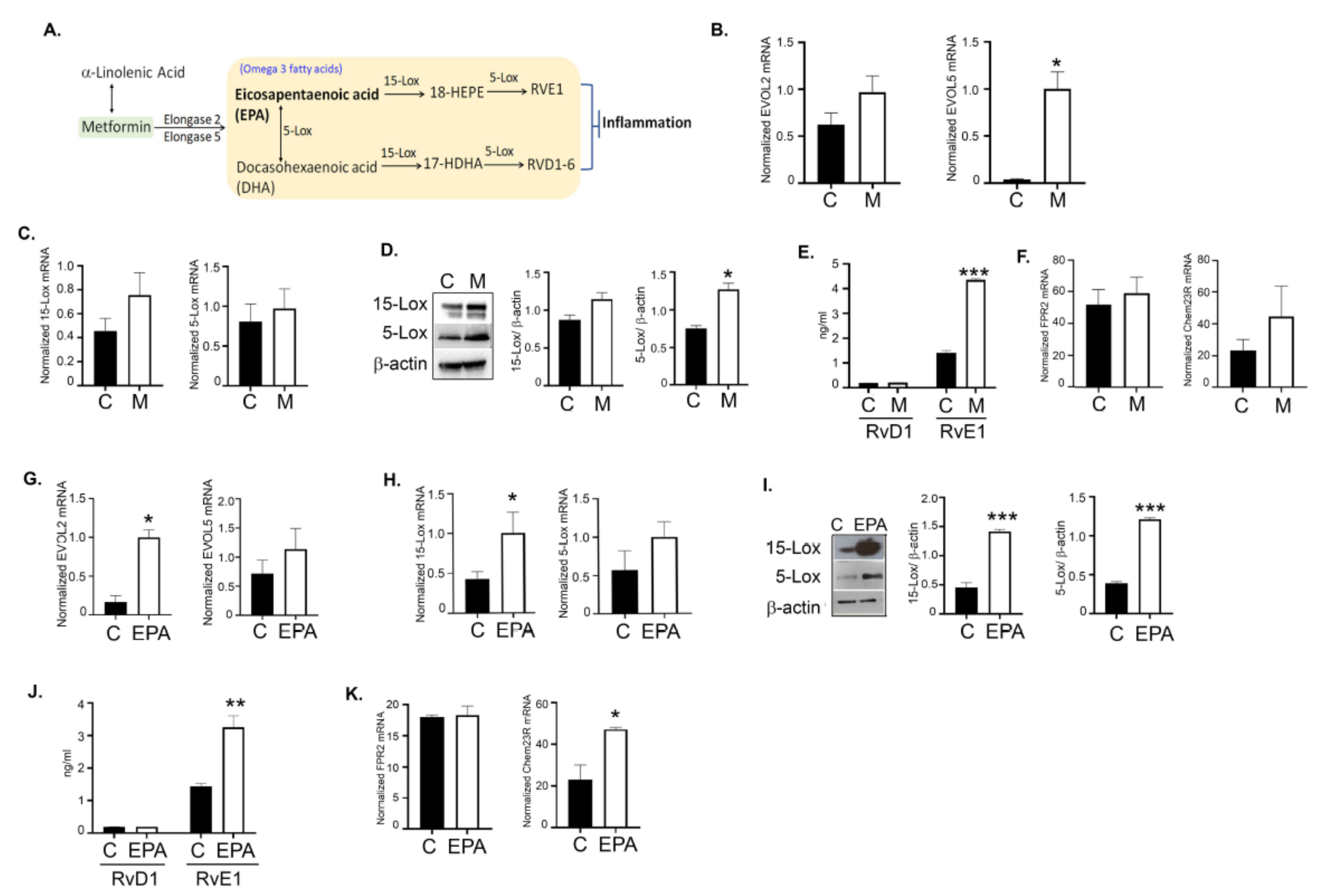
3.7. Metformin Upregulates the Alpha-Linolenic and Linoleic Acid Metabolic Pathway
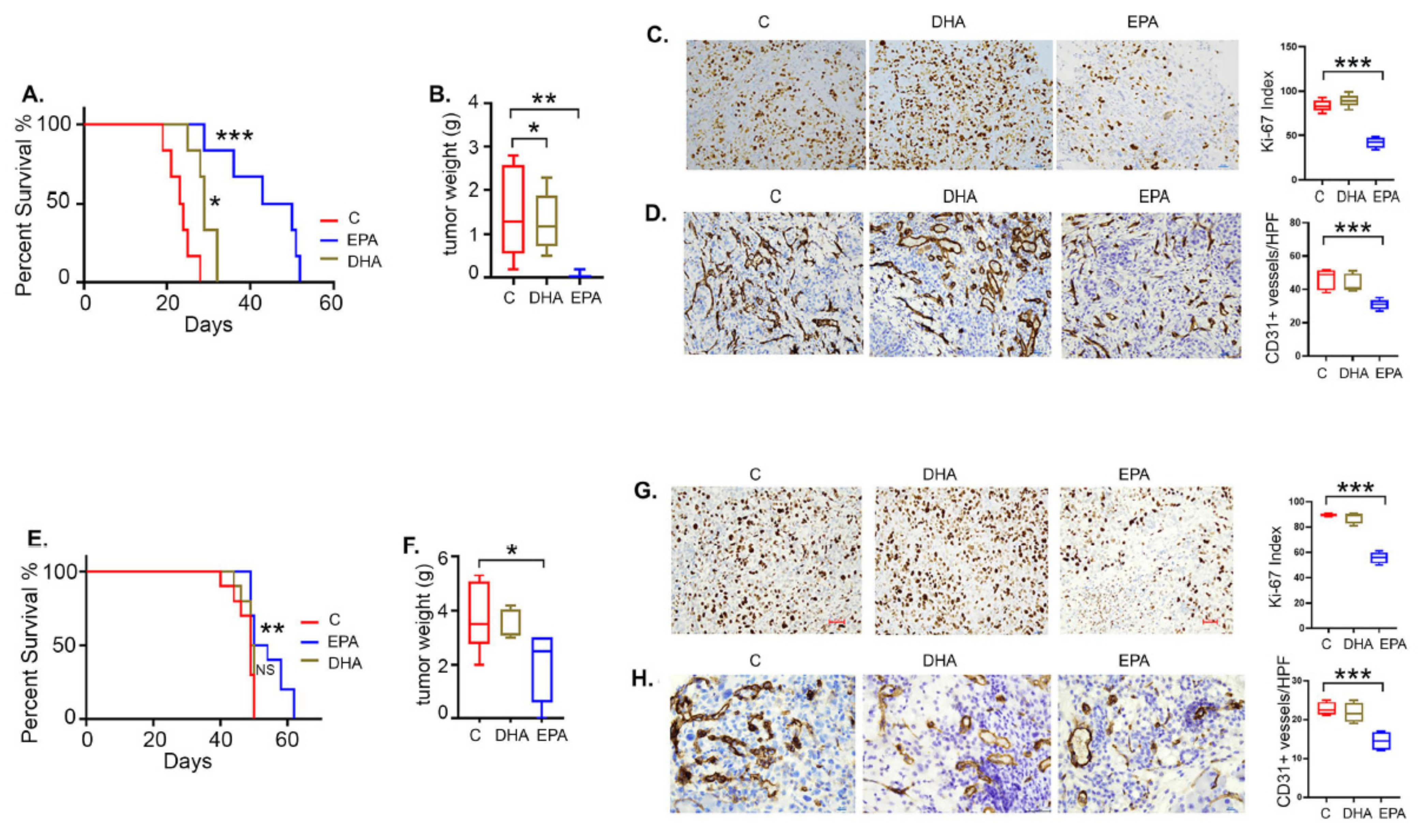
3.8. EPA Is More Effective than DHA in Restricting Ovarian Tumor Growth and Improving Survival
3.9. EPA Induces Apoptotic Cell Death in Ovarian Tumors
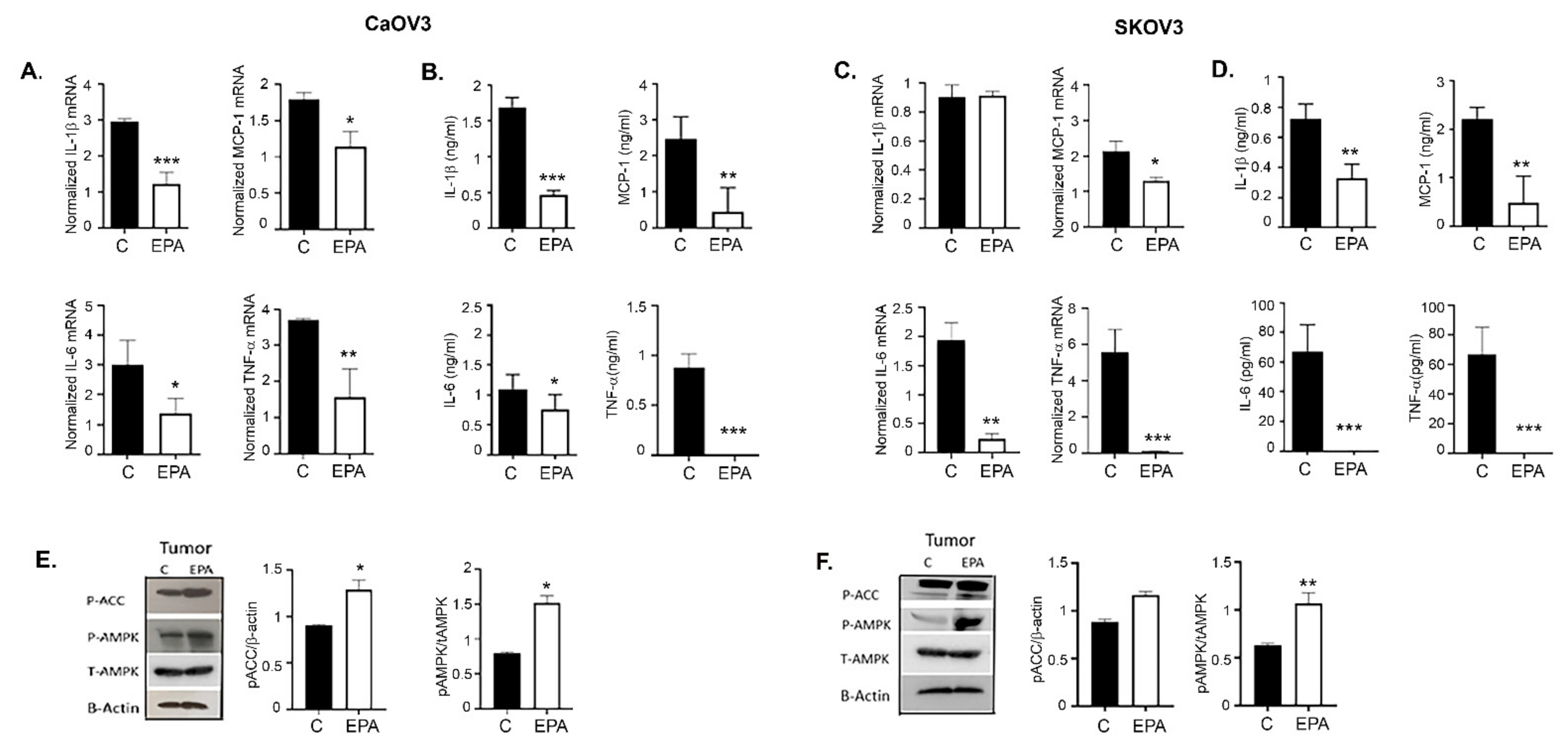
3.10. EPA Treatments Lower the Inflammatory Milieu in Ovarian Tumors and Its Microenvironment
3.11. Metformin and EPA Induced the Downstream Anti-Inflammatory Pathway Mediators
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Gotlieb, W.H.; Saumet, J.; Beauchamp, M.C.; Gu, J.; Lau, S.; Pollak, M.N.; Bruchim, I. In vitro metformin anti-neoplastic activity in epithelial ovarian cancer. Gynecol. Oncol. 2008, 110, 246–250. [Google Scholar] [CrossRef]
- Lengyel, E.; Litchfield, L.M.; Mitra, A.K.; Nieman, K.M.; Mukherjee, A.; Zhang, Y.; Johnson, A.; Bradaric, M.; Lee, W.; Romero, I.L. Metformin inhibits ovarian cancer growth and increases sensitivity to paclitaxel in mouse models. Am. J. Obstet. Gynecol. 2015, 212, 479.e1–479.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattan, R.; Giri, S.; Hartmann, L.C.; Shridhar, V. Metformin attenuates ovarian cancer cell growth in an AMP-kinase dispensable manner. J. Cell Mol. Med. 2011, 15, 166–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rattan, R.; Graham, R.P.; Maguire, J.L.; Giri, S.; Shridhar, V. Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia 2011, 13, 483–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shank, J.J.; Yang, K.; Ghannam, J.; Cabrera, L.; Johnston, C.J.; Reynolds, R.K.; Buckanovich, R.J. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecol. Oncol. 2012, 127, 390–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, I.L.; McCormick, A.; McEwen, K.A.; Park, S.; Karrison, T.; Yamada, S.D.; Pannain, S.; Lengyel, E. Relationship of type II diabetes and metformin use to ovarian cancer progression, survival, and chemosensitivity. Obstet. Gynecol. 2012, 119, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Meuter, A.; Thapa, P.; Langstraat, C.; Giri, S.; Chien, J.; Rattan, R.; Cliby, W.; Shridhar, V. Metformin intake is associated with better survival in ovarian cancer: A case-control study. Cancer 2013, 119, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.B.; Lei, K.J.; Liu, J.P.; Jia, Y.M. Continuous use of metformin can improve survival in type 2 diabetic patients with ovarian cancer: A retrospective study. Medicine 2017, 96, e7605. [Google Scholar] [CrossRef]
- Shi, J.; Liu, B.; Wang, H.; Zhang, T.; Yang, L. Association of metformin use with ovarian cancer incidence and prognosis: A systematic review and meta-analysis. Int. J. Gynecol. Cancer 2019, 29, 140–146. [Google Scholar] [CrossRef]
- Chae, Y.K.; Arya, A.; Malecek, M.K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget 2016, 7, 40767–40780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothermundt, C.; Hayoz, S.; Templeton, A.J.; Winterhalder, R.; Strebel, R.T.; Bartschi, D.; Pollak, M.; Lui, L.; Endt, K.; Schiess, R.; et al. Metformin in chemotherapy-naive castration-resistant prostate cancer: A multicenter phase 2 trial (SAKK 08/09). Eur. Urol. 2014, 66, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Morales, D.R.; Morris, A.D. Metformin in cancer treatment and prevention. Ann. Rev. Med. 2015, 66, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Swanson, K.D.; Zheng, B. Therapeutic Repurposing of Biguanides in Cancer. Trends Cancer 2021, 7, 714–730. [Google Scholar] [CrossRef]
- Rattan, R.; Ali Fehmi, R.; Munkarah, A. Metformin: An emerging new therapeutic option for targeting cancer stem cells and metastasis. J. Oncol. 2012, 2012, 928127. [Google Scholar] [CrossRef] [Green Version]
- Towler, M.C.; Hardie, D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341. [Google Scholar] [CrossRef]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Wahdan-Alaswad, R.S.; Cochrane, D.R.; Spoelstra, N.S.; Howe, E.N.; Edgerton, S.M.; Anderson, S.M.; Thor, A.D.; Richer, J.K. Metformin-induced killing of triple-negative breast cancer cells is mediated by reduction in fatty acid synthase via miRNA-193b. Horm. Cancer 2014, 5, 374–389. [Google Scholar] [CrossRef] [Green Version]
- Conza, D.; Mirra, P.; Cali, G.; Insabato, L.; Fiory, F.; Beguinot, F.; Ulianich, L. Metformin dysregulates the unfolded protein response and the WNT/beta-catenin pathway in endometrial cancer cells through an AMPK-independent mechanism. Cells 2021, 10, 1067. [Google Scholar] [CrossRef]
- Zhang, L.; He, H.; Balschi, J.A. Metformin and phenformin activate AMP-activated protein kinase in the heart by increasing cytosolic AMP concentration. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H457–H466. [Google Scholar] [CrossRef] [Green Version]
- LeBrasseur, N.K.; Kelly, M.; Tsao, T.S.; Farmer, S.R.; Saha, A.K.; Ruderman, N.B.; Tomas, E. Thiazolidinediones can rapidly activate AMP-activated protein kinase in mammalian tissues. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E175–E181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.; Zhang, H.; Ye, J. Traditional chinese medicine in treatment of metabolic syndrome. Endocr. Metab. Immune Disord. Drug Targets 2008, 8, 99–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Averet, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, T.; Cai, S.; Lu, X.; Sha, Y.; Yu, M.; Li, F. Metabonomic study of biochemical changes in the serum of type 2 diabetes mellitus patients after the treatment of metformin hydrochloride. J. Pharm. Biomed. Anal. 2009, 49, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Schuler, K.M.; Rambally, B.S.; DiFurio, M.J.; Sampey, B.P.; Gehrig, P.A.; Makowski, L.; Bae-Jump, V.L. Antiproliferative and metabolic effects of metformin in a preoperative window clinical trial for endometrial cancer. Cancer Med. 2015, 4, 161–173. [Google Scholar] [CrossRef]
- Cuyas, E.; Fernandez-Arroyo, S.; Buxo, M.; Pernas, S.; Dorca, J.; Alvarez, I.; Martinez, S.; Perez-Garcia, J.M.; Batista-Lopez, N.; Rodriguez-Sanchez, C.A.; et al. Metformin induces a fasting- and antifolate-mimicking modification of systemic host metabolism in breast cancer patients. Aging 2019, 11, 2874–2888. [Google Scholar] [CrossRef] [PubMed]
- Hsu, I.R.; Kim, S.P.; Kabir, M.; Bergman, R.N. Metabolic syndrome, hyperinsulinemia, and cancer. Am. J. Clin. Nutr. 2007, 86, s867–s871. [Google Scholar] [CrossRef]
- Xue, F.; Michels, K.B. Diabetes, metabolic syndrome, and breast cancer: A review of the current evidence. Am. J. Clin. Nutr. 2007, 86, s823–s835. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Romero, I.L.; Litchfield, L.M.; Lengyel, E.; Locasale, J.W. Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab. 2016, 24, 728–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godwin, A.K.; Meister, A.; O’Dwyer, P.J.; Huang, C.S.; Hamilton, T.C.; Anderson, M.E. High resistance to cisplatin in human ovarian cancer cell lines is associated with marked increase of glutathione synthesis. Proc. Natl. Acad. Sci. USA 1992, 89, 3070–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Wolf, J.K.; Scanlon, M.; Price, J.E.; Hung, M.C. Enhanced c-erbB-2/neu expression in human ovarian cancer cells correlates with more severe malignancy that can be suppressed by E1A. Cancer Res. 1993, 53, 891–898. [Google Scholar] [PubMed]
- Dar, S.; Chhina, J.; Mert, I.; Chitale, D.; Buekers, T.; Kaur, H.; Giri, S.; Munkarah, A.; Rattan, R. Bioenergetic Adaptations in Chemoresistant Ovarian Cancer Cells. Sci. Rep. 2017, 7, 8760. [Google Scholar] [CrossRef] [PubMed]
- Udumula, M.P.; Sakr, S.; Dar, S.; Alvero, A.B.; Ali-Fehmi, R.; Abdulfatah, E.; Li, J.; Jiang, J.; Tang, A.; Buekers, T.; et al. Ovarian cancer modulates the immunosuppressive function of CD11b(+)Gr1(+) myeloid cells via glutamine metabolism. Mol. Metab. 2021, 53, 101272. [Google Scholar] [CrossRef]
- Crowley, L.C.; Marfell, B.J.; Scott, A.P.; Waterhouse, N.J. Quantitation of Apoptosis and Necrosis by Annexin V Binding, Propidium Iodide Uptake, and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087288. [Google Scholar] [CrossRef]
- Hamaguchi, K.; Godwin, A.K.; Yakushiji, M.; O’Dwyer, P.J.; Ozols, R.F.; Hamilton, T.C. Cross-resistance to diverse drugs is associated with primary cisplatin resistance in ovarian cancer cell lines. Cancer Res. 1993, 53, 5225–5232. [Google Scholar]
- Su, M.; Feng, Y.J.; Yao, L.Q.; Cheng, M.J.; Xu, C.J.; Huang, Y.; Zhao, Y.Q.; Jiang, H. Plasticity of ovarian cancer cell SKOV3ip and vasculogenic mimicry in vivo. Int. J. Gynecol. Cancer 2008, 18, 476–486. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348 Pt 3, 607–614. [Google Scholar] [CrossRef]
- Cai, L.; Jin, X.; Zhang, J.; Li, L.; Zhao, J. Metformin suppresses Nrf2-mediated chemoresistance in hepatocellular carcinoma cells by increasing glycolysis. Aging 2020, 12, 17582–17600. [Google Scholar] [CrossRef] [PubMed]
- Fulgencio, J.P.; Kohl, C.; Girard, J.; Pegorier, J.P. Effect of metformin on fatty acid and glucose metabolism in freshly isolated hepatocytes and on specific gene expression in cultured hepatocytes. Biochem. Pharmacol. 2001, 62, 439–446. [Google Scholar] [CrossRef]
- Tokubuchi, I.; Tajiri, Y.; Iwata, S.; Hara, K.; Wada, N.; Hashinaga, T.; Nakayama, H.; Mifune, H.; Yamada, K. Beneficial effects of metformin on energy metabolism and visceral fat volume through a possible mechanism of fatty acid oxidation in human subjects and rats. PLoS ONE 2017, 12, e0171293. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A., Jr.; Murray Stewart, T.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Bai, B.; Chen, H. Metformin: A Novel Weapon Against Inflammation. Front. Pharmacol. 2021, 12, 622262. [Google Scholar] [CrossRef]
- Al-Wahab, Z.; Mert, I.; Tebbe, C.; Chhina, J.; Hijaz, M.; Morris, R.T.; Ali-Fehmi, R.; Giri, S.; Munkarah, A.R.; Rattan, R. Metformin prevents aggressive ovarian cancer growth driven by high-energy diet: Similarity with calorie restriction. Oncotarget 2015, 6, 10908–10923. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.S.; Xia, J. Using MetaboAnalyst 4.0 for Comprehensive and Integrative Metabolomics Data Analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Bilotto, S.; Russo, G.L.; Orhan, I.E.; Habtemariam, S.; Daglia, M.; Devi, K.P.; Loizzo, M.R.; Tundis, R.; Nabavi, S.M. Omega-3 polyunsaturated fatty acids and cancer: Lessons learned from clinical trials. Cancer Metastasis Rev. 2015, 34, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Udumula, M.P.; Poisson, L.; Dutta, I.; Tiwari, N.; Kim, S.; Chinna-Shankar, J.; Allo, G.; Sakr, S.; Hijaz, M.; Munkarah, A.R.; et al. Divergent Metabolic Effects of Metformin Merge to Enhance Eicosapentaenoic Acid Metabolism and Inhibit Ovarian Cancer In Vivo. 2022; Unpublished work. [Google Scholar]
- Corominas-Faja, B.; Quirantes-Pine, R.; Oliveras-Ferraros, C.; Vazquez-Martin, A.; Cufi, S.; Martin-Castillo, B.; Micol, V.; Joven, J.; Segura-Carretero, A.; Menendez, J.A. Metabolomic fingerprint reveals that metformin impairs one-carbon metabolism in a manner similar to the antifolate class of chemotherapy drugs. Aging 2012, 4, 480–498. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Belna, J.; Logan, J.; Espat, J.; Hurteau, J.A. The effects of Omega-3 fatty acids on growth regulation of epithelial ovarian cancer cell lines. Gynecol. Oncol. 2005, 99, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Yamamoto, A.; Murota, K.; Tsujiuchi, T.; Iwamori, M.; Fukushima, N. Polyunsaturated fatty acids induce ovarian cancer cell death through ROS-dependent MAP kinase activation. Biochem. Biophys. Res. Commun. 2017, 493, 468–473. [Google Scholar] [CrossRef] [PubMed]
- So, W.W.; Liu, W.N.; Leung, K.N. Omega-3 Polyunsaturated Fatty Acids Trigger Cell Cycle Arrest and Induce Apoptosis in Human Neuroblastoma LA-N-1 Cells. Nutrients 2015, 7, 6956–6973. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Kang, K.S.; Okada, K.; Zhu, B.T. EPA, an omega-3 fatty acid, induces apoptosis in human pancreatic cancer cells: Role of ROS accumulation, caspase-8 activation, and autophagy induction. J. Cell Biochem. 2013, 114, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Coscia, F.; Watters, K.M.; Curtis, M.; Eckert, M.A.; Chiang, C.Y.; Tyanova, S.; Montag, A.; Lastra, R.R.; Lengyel, E.; Mann, M. Integrative proteomic profiling of ovarian cancer cell lines reveals precursor cell associated proteins and functional status. Nat. Commun. 2016, 7, 12645. [Google Scholar] [CrossRef] [PubMed]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Edwards, I.J.; O’Flaherty, J.T. Omega-3 Fatty Acids and PPARgamma in Cancer. PPAR Res. 2008, 2008, 358052. [Google Scholar] [CrossRef] [Green Version]
- Negoescu, A.; Guillermet, C.; Lorimier, P.; Brambilla, E.; Labat-Moleur, F. Importance of DNA fragmentation in apoptosis with regard to TUNEL specificity. Biomed. Pharmacother. 1998, 52, 252–258. [Google Scholar] [CrossRef]
- Maccio, A.; Madeddu, C. Inflammation and ovarian cancer. Cytokine 2012, 58, 133–147. [Google Scholar] [CrossRef] [Green Version]
- Calder, P.C. Omega-3 fatty acids and inflammatory processes: From molecules to man. Biochem. Soc. Trans. 2017, 45, 1105–1115. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, M.; Abbasalipourkabir, R.; Ziamajidi, N. Fish oil and chicoric acid combination protects better against palmitate-induced lipid accumulation via regulating AMPK-mediated SREBP-1/FAS and PPARalpha/UCP2 pathways. Arch. Physiol. Biochem. 2020; 1–9, online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Jeong, S.; Jing, K.; Shin, S.; Kim, S.; Heo, J.Y.; Kweon, G.R.; Park, S.K.; Wu, T.; Park, J.I.; et al. Docosahexaenoic Acid Induces Cell Death in Human Non-Small Cell Lung Cancer Cells by Repressing mTOR via AMPK Activation and PI3K/Akt Inhibition. Biomed. Res. Int. 2015, 2015, 239764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Chiang, N.; Dalli, J. The resolution code of acute inflammation: Novel pro-resolving lipid mediators in resolution. Semin. Immunol. 2015, 27, 200–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panigrahy, D.; Gilligan, M.M.; Serhan, C.N.; Kashfi, K. Resolution of inflammation: An organizing principle in biology and medicine. Pharmacol. Ther. 2021, 227, 107879. [Google Scholar] [CrossRef]
- Janakiram, N.B.; Mohammed, A.; Rao, C.V. Role of lipoxins, resolvins, and other bioactive lipids in colon and pancreatic cancer. Cancer Metastasis Rev. 2011, 30, 507–523. [Google Scholar] [CrossRef]
- Krishnamoorthy, S.; Recchiuti, A.; Chiang, N.; Fredman, G.; Serhan, C.N. Resolvin D1 receptor stereoselectivity and regulation of inflammation and proresolving microRNAs. Am. J. Pathol. 2012, 180, 2018–2027. [Google Scholar] [CrossRef] [Green Version]
- Arita, M.; Ohira, T.; Sun, Y.P.; Elangovan, S.; Chiang, N.; Serhan, C.N. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 2007, 178, 3912–3917. [Google Scholar] [CrossRef] [Green Version]
- American Diabetes Association. 2. Classification and diagnosis of diabetes: Standards of medical care in diabetes-2020. Diabetes Care 2020, 43, S14–S31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Febbraro, T.; Lengyel, E.; Romero, I.L. Old drug, new trick: Repurposing metformin for gynecologic cancers? Gynecol. Oncol. 2014, 135, 614–621. [Google Scholar] [CrossRef] [Green Version]
- Bodmer, M.; Becker, C.; Meier, C.; Jick, S.S.; Meier, C.R. Use of metformin and the risk of ovarian cancer: A case-control analysis. Gynecol. Oncol. 2011, 123, 200–204. [Google Scholar] [CrossRef]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II clinical trial of metformin as a cancer stem cell-targeting agent in ovarian cancer. JCI Insight 2020, 5, e133247. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhu, J.; Zhang, H.; Liu, Y.; Sun, H. Metformin plus first-line chemotherapy versus chemotherapy alone in the treatment of epithelial ovarian cancer: A prospective open-label pilot trial. Cancer Chemother. Pharmacol. 2019, 84, 1349–1357. [Google Scholar] [CrossRef]
- Miller, R.A.; Birnbaum, M.J. An energetic tale of AMPK-independent effects of metformin. J. Clin. Investig. 2010, 120, 2267–2270. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, D.Y.; Sullivan, L.B.; Luengo, A.; Hosios, A.M.; Bush, L.N.; Gitego, N.; Davidson, S.M.; Freinkman, E.; Thomas, C.J.; Vander Heiden, M.G. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab. 2016, 24, 716–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, J.; Parakhia, R.A.; Ochs, R.S. Metformin activates AMP kinase through inhibition of AMP deaminase. J. Biol. Chem. 2011, 286, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.W.; Lim, I.K. Sensitization of metformin-cytotoxicity by dichloroacetate via reprogramming glucose metabolism in cancer cells. Cancer Lett. 2014, 346, 300–308. [Google Scholar] [CrossRef]
- Fendt, S.M.; Bell, E.L.; Keibler, M.A.; Davidson, S.M.; Wirth, G.J.; Fiske, B.; Mayers, J.R.; Schwab, M.; Bellinger, G.; Csibi, A.; et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013, 73, 4429–4438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Jump, D.B.; Depner, C.M.; Tripathy, S. Omega-3 fatty acid supplementation and cardiovascular disease. J. Lipid Res. 2012, 53, 2525–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdge, G.C.; Jones, A.E.; Wootton, S.A. Eicosapentaenoic and docosapentaenoic acids are the principal products of alpha-linolenic acid metabolism in young men. Br. J. Nutr. 2002, 88, 355–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dikshit, A.; Hales, K.; Hales, D.B. Whole flaxseed diet alters estrogen metabolism to promote 2-methoxtestradiol-induced apoptosis in hen ovarian cancer. J. Nutr. Biochem. 2017, 42, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Eilati, E.; Bahr, J.M.; Hales, D.B. Long term consumption of flaxseed enriched diet decreased ovarian cancer incidence and prostaglandin E(2)in hens. Gynecol. Oncol. 2013, 130, 620–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.H.; Lii, C.K.; Wang, T.S.; Liu, K.L.; Chen, H.W.; Huang, C.S.; Li, C.C. Docosahexaenoic acid promotes the formation of autophagosomes in MCF-7 breast cancer cells through oxidative stress-induced growth inhibitor 1 mediated activation of AMPK/mTOR pathway. Food Chem. Toxicol. 2021, 154, 112318. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lee, S.D.; Ou, H.C.; Lai, S.C.; Cheng, Y.J. Eicosapentaenoic acid protects against palmitic acid-induced endothelial dysfunction via activation of the AMPK/eNOS pathway. Int. J. Mol. Sci. 2014, 15, 10334–10349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, S.M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Han, L.; Zhang, Y.; Meng, M.; Cheng, D.; Wang, C. Eicosapentaenoic acid induced SKOV-3 cell apoptosis through ERK1/2-mTOR-NF-kappaB pathways. Anticancer Drugs 2016, 27, 635–642. [Google Scholar] [CrossRef]
- Bie, N.; Han, L.; Meng, M.; Zhang, Y.; Guo, M.; Wang, C. Anti-tumor mechanism of eicosapentaenoic acid (EPA) on ovarian tumor model by improving the immunomodulatory activity in F344 rats. J. Funct. Foods 2020, 65, 103739. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, M.F.; Yang, M.L.; Wu, T.Y.; Xu, C.J.; Wang, J.M.; Li, C.J.; Li, X. G Protein-Coupled Receptor 30 Mediates the Anticancer Effects Induced by Eicosapentaenoic Acid in Ovarian Cancer Cells. Cancer Res. Treat. 2020, 52, 815–829. [Google Scholar] [CrossRef] [Green Version]
- Ogo, A.; Miyake, S.; Kubota, H.; Higashida, M.; Matsumoto, H.; Teramoto, F.; Hirai, T. Synergistic Effect of Eicosapentaenoic Acid on Antiproliferative Action of Anticancer Drugs in a Cancer Cell Line Model. Ann. Nutr. Metab. 2017, 71, 247–252. [Google Scholar] [CrossRef]
- Wan, X.H.; Fu, X.; Ababaikeli, G. Docosahexaenoic Acid Induces Growth Suppression on Epithelial Ovarian Cancer Cells More Effectively than Eicosapentaenoic Acid. Nutr. Cancer 2016, 68, 320–327. [Google Scholar] [CrossRef]
- Zajdel, A.; Kalucka, M.; Chodurek, E.; Wilczok, A. DHA but not AA Enhances Cisplatin Cytotoxicity in Ovarian Cancer Cells. Nutr. Cancer 2018, 70, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- West, L.; Yin, Y.; Pierce, S.R.; Fang, Z.; Fan, Y.; Sun, W.; Tucker, K.; Staley, A.; Zhou, C.; Bae-Jump, V. Docosahexaenoic acid (DHA), an omega-3 fatty acid, inhibits tumor growth and metastatic potential of ovarian cancer. Am. J. Cancer Res. 2020, 10, 4450–4463. [Google Scholar] [PubMed]
- Dewey, A.; Baughan, C.; Dean, T.; Higgins, B.; Johnson, I. Eicosapentaenoic acid (EPA, an omega-3 fatty acid from fish oils) for the treatment of cancer cachexia. Cochrane Database Syst. Rev. 2007, 2007, CD004597. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Inhibition of lipolysis and muscle protein degradation by EPA in cancer cachexia. Nutrition 1996, 12, S31–S33. [Google Scholar] [CrossRef]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Smith, T.A.; Phyu, S.M. Metformin Decouples Phospholipid Metabolism in Breast Cancer Cells. PLoS ONE 2016, 11, e0151179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Checkley, L.A.; Rudolph, M.C.; Wellberg, E.A.; Giles, E.D.; Wahdan-Alaswad, R.S.; Houck, J.A.; Edgerton, S.M.; Thor, A.D.; Schedin, P.; Anderson, S.M.; et al. Metformin Accumulation Correlates with Organic Cation Transporter 2 Protein Expression and Predicts Mammary Tumor Regression In Vivo. Cancer Prev. Res. 2017, 10, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T., Jr.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N. Eng. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Lincoff, A.M.; Garcia, M.; Bash, D.; Ballantyne, C.M.; Barter, P.J.; Davidson, M.H.; Kastelein, J.J.P.; Koenig, W.; McGuire, D.K.; et al. Effect of High-Dose Omega-3 Fatty Acids vs Corn Oil on Major Adverse Cardiovascular Events in Patients at High Cardiovascular Risk: The STRENGTH Randomized Clinical Trial. JAMA 2020, 324, 2268–2280. [Google Scholar] [CrossRef] [PubMed]
- Pirillo, A.; Catapano, A.L. Omega-3 for Cardiovascular Diseases: Where Do We Stand After REDUCE-IT and STRENGTH? Circulation 2021, 144, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Preston Mason, R. New Insights into Mechanisms of Action for Omega-3 Fatty Acids in Atherothrombotic Cardiovascular Disease. Curr. Atheroscler. Rep. 2019, 21, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serhan, C.N.; Hong, S.; Gronert, K.; Colgan, S.P.; Devchand, P.R.; Mirick, G.; Moussignac, R.L. Resolvins: A family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002, 196, 1025–1037. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.F.; Pillai, P.S.; Recchiuti, A.; Yang, R.; Serhan, C.N. Pro-resolving actions and stereoselective biosynthesis of 18S E-series resolvins in human leukocytes and murine inflammation. J. Clin. Investig. 2011, 121, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.F.; Tong, W.F.; Ruan, Y.; Sinclair, A.J.; Li, D. Different metabolism of EPA, DPA and DHA in humans: A double-blind cross-over study. Prostaglandins Leukot. Essent. Fatty Acids 2020, 158, 102033. [Google Scholar] [CrossRef] [PubMed]
- Cimen, I.; Astarci, E.; Banerjee, S. 15-lipoxygenase-1 exerts its tumor suppressive role by inhibiting nuclear factor-kappa B via activation of PPAR gamma. J. Cell Biochem. 2011, 112, 2490–2501. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, L.; Chen, J.; Dong, L.; Chen, C.; Wen, Z.; Hu, J.; Fleming, I.; Wang, D.W. Metabolism pathways of arachidonic acids: Mechanisms and potential therapeutic targets. Signal. Transduct. Target. Ther. 2021, 6, 94. [Google Scholar] [CrossRef] [PubMed]
- Ungaro, F.; D’Alessio, S.; Danese, S. The role of pro-resolving lipid mediators in colorectal cancer-associated inflammation: Implications for therapeutic strategies. Cancers 2020, 12, 2060. [Google Scholar] [CrossRef] [PubMed]
- Irun, P.; Lanas, A.; Piazuelo, E. Omega-3 Polyunsaturated Fatty Acids and Their Bioactive Metabolites in Gastrointestinal Malignancies Related to Unresolved Inflammation. A Review. Front. Pharmacol. 2019, 10, 852. [Google Scholar] [CrossRef]
- Sulciner, M.L.; Serhan, C.N.; Gilligan, M.M.; Mudge, D.K.; Chang, J.; Gartung, A.; Lehner, K.A.; Bielenberg, D.R.; Schmidt, B.; Dalli, J.; et al. Resolvins suppress tumor growth and enhance cancer therapy. J. Exp. Med. 2018, 215, 115–140. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Udumula, M.P.; Poisson, L.M.; Dutta, I.; Tiwari, N.; Kim, S.; Chinna-Shankar, J.; Allo, G.; Sakr, S.; Hijaz, M.; Munkarah, A.R.; et al. Divergent Metabolic Effects of Metformin Merge to Enhance Eicosapentaenoic Acid Metabolism and Inhibit Ovarian Cancer In Vivo. Cancers 2022, 14, 1504. https://doi.org/10.3390/cancers14061504
Udumula MP, Poisson LM, Dutta I, Tiwari N, Kim S, Chinna-Shankar J, Allo G, Sakr S, Hijaz M, Munkarah AR, et al. Divergent Metabolic Effects of Metformin Merge to Enhance Eicosapentaenoic Acid Metabolism and Inhibit Ovarian Cancer In Vivo. Cancers. 2022; 14(6):1504. https://doi.org/10.3390/cancers14061504
Chicago/Turabian StyleUdumula, Mary P., Laila M. Poisson, Indrani Dutta, Nivedita Tiwari, Seongho Kim, Jasdeep Chinna-Shankar, Ghassan Allo, Sharif Sakr, Miriana Hijaz, Adnan R. Munkarah, and et al. 2022. "Divergent Metabolic Effects of Metformin Merge to Enhance Eicosapentaenoic Acid Metabolism and Inhibit Ovarian Cancer In Vivo" Cancers 14, no. 6: 1504. https://doi.org/10.3390/cancers14061504
APA StyleUdumula, M. P., Poisson, L. M., Dutta, I., Tiwari, N., Kim, S., Chinna-Shankar, J., Allo, G., Sakr, S., Hijaz, M., Munkarah, A. R., Giri, S., & Rattan, R. (2022). Divergent Metabolic Effects of Metformin Merge to Enhance Eicosapentaenoic Acid Metabolism and Inhibit Ovarian Cancer In Vivo. Cancers, 14(6), 1504. https://doi.org/10.3390/cancers14061504