
Long Non-Coding RNA-Based Functional Prediction Reveals Novel Targets in Notch-Upregulated Ovarian Cancer

 , , , ,
, , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Public Databases
2.2. Patients and Specimens
2.3. RNA Isolation and Real-Time PCR
2.4. Cell Culture and Reagents
2.5. siRNA Transfection and Western Blot Analysis
2.6. Total RNA Sequencing
2.7. Statistical Analysis
3. Results
3.1. NOTCH1/3 and Their Target Gene Expression in TCGA HGS-OVCA
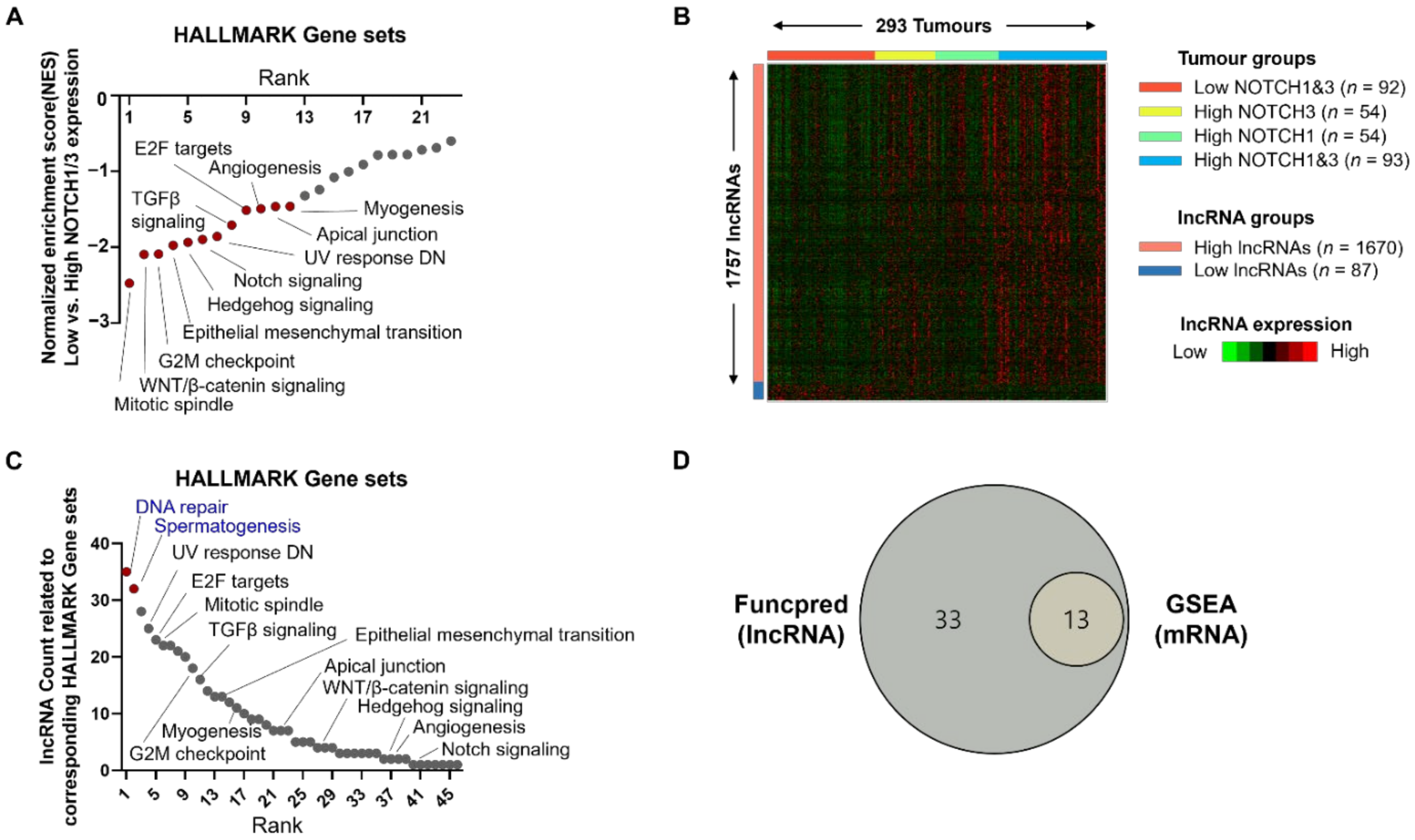
3.2. Discovery of Notch-Related lncRNAs Using TANRIC Database
3.3. Validation of Notch-Related lncRNAs by NOTCH1/3 Silencing and Analysis of Clinical Tissue Samples
3.4. Simultaneous Expression of Common lncRNAs and Genes Related to DNA Repair and Spermatogenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shih, I.M.; Kurman, R.J. Ovarian tumorigenesis: A proposed model based on morphological and molecular genetic analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef]
- Hennessy, B.T.; Coleman, R.L.; Markman, M. Ovarian cancer. Lancet 2009, 374, 1371–1382. [Google Scholar] [CrossRef]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.R.; Kamara, D.; Karlan, B.Y.; Pharoah, P.D.P.; Gayther, S.A. Genetic epidemiology of ovarian cancer and prospects for polygenic risk prediction. Gynecol. Oncol. 2017, 147, 705–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Luo, G.; Li, M.; Guo, P.; Xiao, Y.; Ji, H.; Hao, Y. Global patterns and trends in ovarian cancer incidence: Age, period and birth cohort analysis. BMC Cancer 2019, 19, 984. [Google Scholar] [CrossRef] [PubMed]
- Olivier, R.I.; van Beurden, M.; Lubsen, M.A.; Rookus, M.A.; Mooij, T.M.; van de Vijver, M.J.; van’t Veer, L.J. Clinical outcome of prophylactic oophorectomy in BRCA1/BRCA2 mutation carriers and events during follow-up. Br. J. Cancer 2004, 90, 1492–1497. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.K.; Bundy, B.; Wenzel, L.; Huang, H.Q.; Baergen, R.; Lele, S.; Copeland, L.J.; Walker, J.L.; Burger, R.A.; Gynecologic Oncology, G. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N. Engl. J. Med. 2006, 354, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzanedo, I.; Pereira, F.; Serrano, A.; Perez-Viejo, E.; Martinez-Torres, B.; Carrion, L.; Calzas, J. The use of cisplatin plus doxorubicin or paclitaxel in hyperthermic intraperitoneal chemotherapy (HIPEC) for stage IIIC or IV epithelial ovarian cancer: A comparative study. Clin. Transl. Oncol. 2019, 21, 1357–1363. [Google Scholar] [CrossRef]
- Schmeler, K.M.; Sun, C.C.; Bodurka, D.C.; Deavers, M.T.; Malpica, A.; Coleman, R.L.; Ramirez, P.T.; Gershenson, D.M. Neoadjuvant chemotherapy for low-grade serous carcinoma of the ovary or peritoneum. Gynecol. Oncol. 2008, 108, 510–514. [Google Scholar] [CrossRef]
- Kurman, R.J.; Shih, I.M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer--shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef] [Green Version]
- Cobb, L.P.; Sun, C.C.; Iyer, R.; Nick, A.M.; Fleming, N.D.; Westin, S.N.; Sood, A.K.; Wong, K.K.; Silva, E.G.; Gershenson, D.M. The role of neoadjuvant chemotherapy in the management of low-grade serous carcinoma of the ovary and peritoneum: Further evidence of relative chemoresistance. Gynecol. Oncol. 2020, 158, 653–658. [Google Scholar] [CrossRef]
- Giusti, I.; Bianchi, S.; Nottola, S.A.; Macchiarelli, G.; Dolo, V. Clinical electron microscopy in the study of human ovarian tissues. EuroMediterr. Biomed. J. 2019, 14, 145–151. [Google Scholar]
- Pinato, D.J.; Graham, J.; Gabra, H.; Sharma, R. Evolving concepts in the management of drug resistant ovarian cancer: Dose dense chemotherapy and the reversal of clinical platinum resistance. Cancer Treat. Rev. 2013, 39, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.-Z.; Guan, N.-N.; Qu, J.; Huang, Z.-A.; Zhu, Z.-X.; Li, J.-Q. Computational models for lncRNA function prediction and functional similarity calculation. Brief. Funct. Genom. 2019, 18, 58–82. [Google Scholar] [CrossRef]
- Perron, U.; Provero, P.; Molineris, I. In silico prediction of lncRNA function using tissue specific and evolutionary conserved expression. BMC Bioinform. 2017, 18, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akrami, R.; Jacobsen, A.; Hoell, J.; Schultz, N.; Sander, C.; Larsson, E. Comprehensive analysis of long non-coding RNAs in ovarian cancer reveals global patterns and targeted DNA amplification. PLoS ONE 2013, 8, e80306. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.Y.; Jin, S.M.; Luo, H.Q.; Leng, H.L.; Fang, J.D. LncRNA HOTAIR regulates anoikis-resistance capacity and spheroid formation of ovarian cancer cells by recruiting EZH2 and influencing H3K27 methylation. Neoplasma 2021, 68, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef]
- Shawber, C.; Boulter, J.; Lindsell, C.E.; Weinmaster, G. Jagged2: A serrate-like gene expressed during rat embryogenesis. Dev. Biol. 1996, 180, 370–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeuchi, T.; Sisodia, S.S. The Notch ligands, Delta1 and Jagged2, are substrates for presenilin-dependent “gamma-secretase” cleavage. J. Biol. Chem. 2003, 278, 7751–7754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinoza, I.; Miele, L. Notch inhibitors for cancer treatment. Pharmacol. Ther. 2013, 139, 95–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groeneweg, J.W.; Foster, R.; Growdon, W.B.; Verheijen, R.H.; Rueda, B.R. Notch signaling in serous ovarian cancer. J. Ovarian Res. 2014, 7, 95. [Google Scholar] [CrossRef]
- Locatelli, M.; Curigliano, G. Notch inhibitors and their role in the treatment of triple negative breast cancer: Promises and failures. Curr. Opin. Oncol. 2017, 29, 411–427. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, G.; Qiu, H.; Yang, J.; Bu, X.; Zhu, S.; Zheng, J.; Dang, C.; Wang, W.; Chu, D. The Novel Notch-induced Long Noncoding RNA LUNAR1 Determines the Proliferation and Prognosis of Colorectal Cancer. Sci. Rep. 2019, 9, 19915. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Zhang, Q.; Burdette, J.E.; Wang, J.P. Integrative network analysis of TCGA data for ovarian cancer. BMC Syst. Biol. 2014, 8, 1338. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Han, L.; Roebuck, P.; Diao, L.; Liu, L.; Yuan, Y.; Weinstein, J.N.; Liang, H. TANRIC: An Interactive Open Platform to Explore the Function of lncRNAs in Cancer. Cancer Res. 2015, 75, 3728–3737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guigo, R.; Flicek, P.; Abril, J.F.; Reymond, A.; Lagarde, J.; Denoeud, F.; Antonarakis, S.; Ashburner, M.; Bajic, V.B.; Birney, E.; et al. EGASP: The human ENCODE Genome Annotation Assessment Project. Genome Biol. 2006, 7, S2. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Lu, J.; Kulbokas, E.J.; Golub, T.R.; Mootha, V.; Lindblad-Toh, K.; Lander, E.S.; Kellis, M. Systematic discovery of regulatory motifs in human promoters and 3’ UTRs by comparison of several mammals. Nature 2005, 434, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.M.; Jan, Y.N. Roles for proteolysis and trafficking in notch maturation and signal transduction. Cell 1998, 94, 423–426. [Google Scholar] [CrossRef] [Green Version]
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998, 393, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Rand, M.D.; Grimm, L.M.; Artavanis-Tsakonas, S.; Patriub, V.; Blacklow, S.C.; Sklar, J.; Aster, J.C. Calcium depletion dissociates and activates heterodimeric notch receptors. Mol. Cell Biol. 2000, 20, 1825–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.G.; Kwon, Y.D.; Song, J.A.; Back, M.J.; Lee, S.Y.; Lee, C.; Hwang, Y.Y.; An, H.J. Prognostic significance of Notch 3 gene expression in ovarian serous carcinoma. Cancer Sci. 2010, 101, 1977–1983. [Google Scholar] [CrossRef] [PubMed]
- Iso, T.; Kedes, L.; Hamamori, Y. HES and HERP families: Multiple effectors of the Notch signaling pathway. J. Cell Physiol. 2003, 194, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Oswald, F.; Liptay, S.; Adler, G.; Schmid, R.M. NF-kappaB2 is a putative target gene of activated Notch-1 via RBP-Jkappa. Mol. Cell Biol. 1998, 18, 2077–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronchini, C.; Capobianco, A.J. Induction of cyclin D1 transcription and CDK2 activity by Notch(ic): Implication for cell cycle disruption in transformation by Notch(ic). Mol. Cell Biol. 2001, 21, 5925–5934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.W.; Pampeno, C.; Vukmanovic, S.; Meruelo, D. Characterization of the transcriptional expression of Notch-1 signaling pathway members, Deltex and HES-1, in developing mouse thymocytes. Dev. Comp. Immunol. 2002, 26, 575–588. [Google Scholar] [CrossRef]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; Del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurlbut, G.D.; Kankel, M.W.; Lake, R.J.; Artavanis-Tsakonas, S. Crossing paths with Notch in the hyper-network. Curr. Opin. Cell Biol. 2007, 19, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Brechbiel, J.; Miller-Moslin, K.; Adjei, A.A. Crosstalk between hedgehog and other signaling pathways as a basis for combination therapies in cancer. Cancer Treat. Rev. 2014, 40, 750–759. [Google Scholar] [CrossRef]
- Duncan, A.W.; Rattis, F.M.; DiMascio, L.N.; Congdon, K.L.; Pazianos, G.; Zhao, C.; Yoon, K.; Cook, J.M.; Willert, K.; Gaiano, N.; et al. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat. Immunol. 2005, 6, 314–322. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, B.; Miyamoto, A.; Weinmaster, G. The many facets of Notch ligands. Oncogene 2008, 27, 5148–5167. [Google Scholar] [CrossRef] [Green Version]
- Nikpayam, E.; Tasharrofi, B.; Sarrafzadeh, S.; Ghafouri-Fard, S. The Role of Long Non-Coding RNAs in Ovarian Cancer. Iran. Biomed. J. 2017, 21, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.J.; Lin, Y.Y.; Ding, J.X.; Feng, W.W.; Jin, H.Y.; Hua, K.Q. Long non-coding RNA ANRIL predicts poor prognosis and promotes invasion/metastasis in serous ovarian cancer. Int. J. Oncol. 2015, 46, 2497–2505. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Feng, Y.; Zhang, D.; Zhao, S.D.; Hu, Z.; Greshock, J.; Zhang, Y.; Yang, L.; Zhong, X.; Wang, L.P.; et al. A functional genomic approach identifies FAL1 as an oncogenic long noncoding RNA that associates with BMI1 and represses p21 expression in cancer. Cancer Cell 2014, 26, 344–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Z.; Guo, J.; Chen, L.; Luo, N.; Yang, W.; Qu, X. A long noncoding RNA AB073614 promotes tumorigenesis and predicts poor prognosis in ovarian cancer. Oncotarget 2015, 6, 25381–25389. [Google Scholar] [CrossRef] [Green Version]
- Yin, G.; Chen, R.; Alvero, A.B.; Fu, H.H.; Holmberg, J.; Glackin, C.; Rutherford, T.; Mor, G. TWISTing stemness, inflammation and proliferation of epithelial ovarian cancer cells through MIR199A2/214. Oncogene 2010, 29, 3545–3553. [Google Scholar] [CrossRef] [Green Version]
- Tanos, V.; Prus, D.; Ayesh, S.; Weinstein, D.; Tykocinski, M.L.; De-Groot, N.; Hochberg, A.; Ariel, I. Expression of the imprinted H19 oncofetal RNA in epithelial ovarian cancer. Eur. J. Obstet. Gynecol. Reprod. Biol. 1999, 85, 7–11. [Google Scholar] [CrossRef]
- Gao, Y.; Meng, H.; Liu, S.; Hu, J.; Zhang, Y.; Jiao, T.; Liu, Y.; Ou, J.; Wang, D.; Yao, L.; et al. LncRNA-HOST2 regulates cell biological behaviors in epithelial ovarian cancer through a mechanism involving microRNA let-7b. Hum. Mol. Genet. 2015, 24, 841–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.J.; Wang, Y.; Ding, J.X.; Jin, H.Y.; Yang, G.; Hua, K.Q. The long non-coding RNA HOTAIR promotes the proliferation of serous ovarian cancer cells through the regulation of cell cycle arrest and apoptosis. Exp. Cell Res. 2015, 333, 238–248. [Google Scholar] [CrossRef]
- Silva, J.M.; Boczek, N.J.; Berres, M.W.; Ma, X.; Smith, D.I. LSINCT5 is over expressed in breast and ovarian cancer and affects cellular proliferation. RNA Biol. 2011, 8, 496–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinge, C.M. Non-coding RNAs: Long non-coding RNAs and microRNAs in endocrine-related cancers. Endocr. Relat. Cancer 2018, 25, R259–R282. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Li, J.; Yang, L.; Chen, Z.; Zhao, Q.; Tan, L.; Zhou, Y.; Li, J. Promoter hypermethylation influences the suppressive role of maternally expressed 3, a long non-coding RNA, in the development of epithelial ovarian cancer. Oncol. Rep. 2014, 32, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.; Lee, S.G.; Kim, H.; Lee, G.; Park, S.; Kim, I.K.; Lee, J.; Jo, Y.S. Simultaneous Expression of Long Non-Coding RNA FAL1 and Extracellular Matrix Protein 1 Defines Tumour Behaviour in Young Patients with Papillary Thyroid Cancer. Cancers 2021, 13, 3223. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Old, L.J. Cancer/testis (CT) antigens—A new link between gametogenesis and cancer. Cancer Immun. 2001, 1, 1. [Google Scholar] [PubMed]
- Simpson, A.J.; Caballero, O.L.; Jungbluth, A.; Chen, Y.T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, S.; Park, S.; Jo, Y.S.; Choi, M.J.; Lee, G.; Lee, S.G.; Choi, M.C.; Park, H.; Joo, W.D.; Jung, S.G.; et al. Long Non-Coding RNA-Based Functional Prediction Reveals Novel Targets in Notch-Upregulated Ovarian Cancer. Cancers 2022, 14, 1557. https://doi.org/10.3390/cancers14061557
Jeong S, Park S, Jo YS, Choi MJ, Lee G, Lee SG, Choi MC, Park H, Joo WD, Jung SG, et al. Long Non-Coding RNA-Based Functional Prediction Reveals Novel Targets in Notch-Upregulated Ovarian Cancer. Cancers. 2022; 14(6):1557. https://doi.org/10.3390/cancers14061557
Chicago/Turabian StyleJeong, Seonhyang, Sunmi Park, Young Suk Jo, Moon Jung Choi, Gibbeum Lee, Seul Gi Lee, Min Chul Choi, Hyun Park, Won Duk Joo, Sang Geun Jung, and et al. 2022. "Long Non-Coding RNA-Based Functional Prediction Reveals Novel Targets in Notch-Upregulated Ovarian Cancer" Cancers 14, no. 6: 1557. https://doi.org/10.3390/cancers14061557