
A Multidisciplinary Approach to Complex Dermal Sarcomas Ensures an Optimal Clinical Outcome

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Types of Dermal Sarcomas
2.1. Dermatofibrosarcoma Protuberans
2.2. Atypical Fibroxanthomas
2.3. Dermal Undifferentiated Pleomorphic Sarcoma
2.4. Cutaneous Leiomyosarcoma
2.5. Vascular Sarcomas
3. Treatment Algorithms and Strategies
3.1. Clear Margins Are Gold Standard
3.2. Adjuvant Therapy for Advanced Dermal Sarcomas
3.3. Biological, Targeted Therapy and Immunotherapy
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Trojani, M.; Contesso, G.; Coindre, J.M.; Rouesse, J.; Bui, N.B.; de Mascarel, A.; Goussot, J.F.; David, M.; Bonichon, F.; Lagarde, C. Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int. J. Cancer 1984, 33, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Rouhani, P.; Fletcher, C.D.M.; Devesa, S.S.; Toro, J.R. Cutaneous soft tissue sarcoma incidence patterns in the U.S.: An analysis of 12,114 cases. Cancer 2008, 113, 616–627. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.J.; Oliveira, A.M.; Nascimento, A.G. The Prognostic Significance of Fibrosarcomatous Transformation in Dermatofibrosarcoma Protuberans. Am. J. Surg. Pathol. 2006, 30, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Criscito, M.C.; Martires, K.J.; Stein, J.A. Prognostic Factors, Treatment, and Survival in Dermatofibrosarcoma Protuberans. JAMA Dermatol. 2016, 152, 1365–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osio, A.; Xu, S.; El Bouchtaoui, M.; Leboeuf, C.; Gapihan, G.; Lemaignan, C.; Bousquet, G.; Lebbé, C.; Janin, A.; Battistella, M. EGFR is involved in dermatofibrosarcoma protuberans progression to high grade sarcoma. Oncotarget 2018, 9, 8478–8488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, I.; Kariya, Y.; Okada, E.; Yasuda, M.; Matori, S.; Ishikawa, O.; Uezato, H.; Takahashi, K. A Novel Chromosomal Translocation Associated with COL1A2-PDGFBGene Fusion in Dermatofibrosarcoma Protuberans: PDGF Expression as a New Diagnostic Tool. JAMA Dermatol. 2015, 151, 1330–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, A.; Ahn, C.; Sangüeza, O.P. Dermatofibrosarcoma Protuberans. Dermatol. Clin. 2019, 37, 483–488. [Google Scholar] [CrossRef]
- Liszewski, W.; Blanchette, D.; Cunningham, A.M.; Miller, D.D. Epidemiology of Bednar tumors in the United States. J. Am. Acad. Dermatol. 2016, 75, 1064–1066. [Google Scholar] [CrossRef] [Green Version]
- Parker, T.L.; Zitelli, J.A. Surgical margins for excision of dermatofibrosarcoma protuberans. J. Am. Acad. Dermatol. 1995, 32, 233–236. [Google Scholar] [CrossRef]
- Rutkowski, P.; Van Glabbeke, M.; Rankin, C.J.; Ruka, W.; Rubin, B.P.; Debiec-Rychter, M.; Lazar, A.; Gelderblom, H.; Sciot, R.; Lopez-Terrada, D.; et al. Imatinib Mesylate in Advanced Dermatofibrosarcoma Protuberans: Pooled Analysis of Two Phase II Clinical Trials. J. Clin. Oncol. 2010, 28, 1772–1779. [Google Scholar] [CrossRef]
- Saiag, P.; Grob, J.-J.; Lebbe, C.; Malvehy, J.; del Marmol, V.; Pehamberger, H.; Peris, K.; Stratigos, A.; Middelton, M.; Bastholt, L.; et al. Diagnosis and treatment of dermatofibrosarcoma protuberans. European consensus-based interdisciplinary guideline. Eur. J. Cancer 2015, 51, 2604–2608. [Google Scholar] [CrossRef] [PubMed]
- Ugurel, S.; Kortmann, R.-D.; Mohr, P.; Mentzel, T.; Garbe, C.; Breuninger, H.; Bauer, S.; Grabbe, S. S1 guidelines for dermatofibrosarcoma protuberans (DFSP)—Update 2018. JDDG J. Dtsch. Dermatol. Ges. 2019, 17, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Bigdeli, A.K.; Thomas, B.; Falkner, F.; Radu, C.A.; Gazyakan, E.; Kneser, U. Microsurgical reconstruction of extensive lower extremity defects with the conjoined parascapular and latissimus dorsi free flap. Microsurgery 2020, 40, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, D.; Bishop, P.W. Sarcoma-like Tumor of Head and Neck Skin. Am. J. Surg. Pathol. 2014, 38, 956–965. [Google Scholar] [CrossRef]
- Beer, T.; Drury, P.; Heenan, P.J. Atypical Fibroxanthoma: A Histological and Immunohistochemical Review of 171 Cases. Am. J. Dermatopathol. 2010, 32, 533–540. [Google Scholar] [CrossRef]
- Mihic-Probst, D.; Zhao, J.; Saremaslani, P.; Baer, A.; Oehlschlegel, C.; Paredes, B.; Komminoth, P.; Heitz, P.U. CGH analysis shows genetic similarities and differences in atypical fibroxanthoma and undifferentiated high. grade pleomorphic sarcoma. Anticancer Res. 2004, 24, 19–26. [Google Scholar]
- Sakamoto, A.; Oda, Y.; Itakura, E.; Oshiro, Y.; Tamiya, S.; Honda, Y.; Ishihara, A.; Iwamoto, Y.; Tsuneyoshi, M. H-, K-, and N-ras gene mutation in atypical fibroxanthoma and malignant fibrous histiocytoma. Hum. Pathol. 2001, 32, 1225–1231. [Google Scholar] [CrossRef]
- Hasegawa, T.; Hasegawa, F.; Hirose, T.; Sano, T.; Matsuno, Y. Expression of smooth muscle markers in so called malignant fibrous histiocytomas. J. Clin. Pathol. 2003, 56, 666–671. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.; Goodlad, J.R.; Brenn, T. Pleomorphic Dermal Sarcoma: Adverse histologic features predict aggressive behavior and allow distinction from atypical fibroxanthoma. Am. J. Surg. Pathol. 2012, 36, 1317–1326. [Google Scholar] [CrossRef]
- Helbig, D.; Mauch, C.; Buettner, R.; Quaas, A. Immunohistochemical expression of melanocytic and myofibroblastic markers and their molecular correlation in atypical fibroxanthomas and pleomorphic dermal sarcomas. J. Cutan. Pathol. 2018, 45, 880–885. [Google Scholar] [CrossRef]
- Griewank, K.G.; Schilling, B.; Murali, R.; Bielefeld, N.; Schwamborn, M.; Sucker, A.; Zimmer, L.; Hillen, U.; Schaller, J.; Brenn, T.; et al. TERT promoter mutations are frequent in atypical fibroxanthomas and pleomorphic dermal sarcomas. Mod. Pathol. 2014, 27, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Griewank, K.G.; Wiesner, T.; Murali, R.; Pischler, C.; Müller, H.; Koelsche, C.; Möller, I.; Franklin, C.; Cosgarea, I.; Sucker, A.; et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma harbor frequent NOTCH1/2 and FAT1 mutations and similar DNA copy number alteration profiles. Mod. Pathol. 2018, 31, 418–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helbig, D.; Ihle, M.A.; Pütz, K.; Tantcheva-Poor, I.; Mauch, C.; Büttner, R.; Quaas, A. Oncogene and therapeutic target analyses in atypical fibroxanthomas and pleomorphic dermal sarcomas. Oncotarget 2016, 7, 21763–21774. [Google Scholar] [CrossRef] [PubMed]
- Winchester, D.; Lehman, J.; Tello, T.; Chimato, N.; Hocker, T.; Kim, S.; Chang, J.; Markey, J.; Yom, S.; Ryan, W.; et al. Undifferentiated pleomorphic sarcoma: Factors predictive of adverse outcomes. J. Am. Acad. Dermatol. 2018, 79, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Persa, O.D.; Loquai, C.; Wobser, M.; Baltaci, M.; Dengler, S.; Kreuter, A.; Volz, A.; Laimer, M.; Emberger, M.; Doerler, M.; et al. Extended surgical safety margins and ulceration are associated with an improved prognosis in pleomorphic dermal sarcomas. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1577–1580. [Google Scholar] [CrossRef]
- Fields, J.P.; Helwig, E.B. Leiomyosarcoma of the skin and subcutaneous tissue. Cancer 1981, 47, 156–169. [Google Scholar] [CrossRef]
- Kaddu, S.; Beham, A.; Cerroni, L.; Humer-Fuchs, U.; Salmhofer, W.; Kerl, H.; Soyer, H.P. Cutaneous Leiomyosarcoma. Am. J. Surg. Pathol. 1997, 21, 979–987. [Google Scholar] [CrossRef]
- Massi, D.; Franchi, A.; Alos, L.; Cook, M.; Di Palma, S.; Enguita, A.B.; Ferrara, G.; Kazakov, D.V.; Mentzel, T.; Michal, M.; et al. Primary cutaneous leiomyosarcoma: Clinicopathological analysis of 36 cases. Histopathology 2010, 56, 251–262. [Google Scholar] [CrossRef]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Prim. 2019, 5, 9. [Google Scholar] [CrossRef]
- Gramolelli, S.; Schulz, T.F. The role of Kaposi sarcoma-associated herpesvirus in the pathogenesis of Kaposi sarcoma. J. Pathol. 2014, 235, 368–380. [Google Scholar] [CrossRef]
- Ackerman, A.B. Subtle clues to diagnosis by conventional microscopy. The patch stage of Kaposi’s sarcoma. Am. J. Dermatopathol. 1979, 1, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.M.; Goldblum, J.R.; Hsi, E.D. Immunohistochemical detection of human herpes virus-8 latent nuclear antigen-1 is useful in the diagnosis of Kaposi sarcoma. Mod. Pathol. 2004, 17, 456–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogt, T.; Brockmeyer, N.; Kutzner, H.; Schöfer, H. Brief S1 guidelines—Cutaneous angiosarcoma and Kaposi sarcoma. JDDG J. Dtsch. Dermatol. Ges. 2013, 11, 2–9. [Google Scholar] [CrossRef]
- James, N.; Coker, R.; Tomlinson, D.; Harris, J.; Gompels, M.; Pinching, A.; Stewart, J. Liposomal doxorubicin (Doxil): An effective new treatment for Kaposi’s sarcoma in AIDS. Clin. Oncol. 1994, 6, 294–296. [Google Scholar] [CrossRef]
- Albores-Saavedra, J.; Schwartz, A.M.; Henson, D.E.; Kostun, L.; Hart, A.; Angeles-Albores, D.; Chablé-Montero, F. Cutaneous angiosarcoma. Analysis of 434 cases from the Surveillance, Epidemiology, and End Results Program, 1973–2007. Ann. Diagn. Pathol. 2011, 15, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Yamamoto, H.; Oda, Y.; Tsuneyoshi, M. Detection and characterization of vascular endothelial growth factors and their receptors in a series of angiosarcomas. J. Surg. Oncol. 2008, 97, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.M.; Billings, S.D. Cutaneous Soft Tissue Tumors That Make You Say, “Oh $*&%!.”. Adv. Anat. Pathol. 2012, 19, 320–330. [Google Scholar] [CrossRef]
- Shustef, E.; Kazlouskaya, V.; Prieto, V.G.; Ivan, D.; Aung, P.P. Cutaneous angiosarcoma: A current update. J. Clin. Pathol. 2017, 70, 917–925. [Google Scholar] [CrossRef]
- Requena, C.; Alsina, M.; Morgado-Carrasco, D.; Cruz, J.; Sanmartín, O.; Serra-Guillén, C.; Llombart, B. Kaposi Sarcoma and Cutaneous Angiosarcoma: Guidelines for Diagnosis and Treatment. Actas Dermo-Sifiliográficas 2018, 109, 878–887. [Google Scholar] [CrossRef]
- Ishida, Y.; Otsuka, A.; Kabashima, K. Cutaneous angiosarcoma: Update on biology and latest treatment. Curr. Opin. Oncol. 2018, 30, 107–112. [Google Scholar] [CrossRef]
- Lee, K.C.; Chuang, S.-K.; Philipone, E.M.; Peters, S.M. Characteristics and Prognosis of Primary Head and Neck Angiosarcomas: A Surveillance, Epidemiology, and End Results Program (SEER) Analysis of 1250 Cases. Head Neck Pathol. 2018, 13, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Ruocco, V.; Schwartz, R.A.; Ruocco, E. Lymphedema: An immunologically vulnerable site for development of neoplasms. J. Am. Acad. Dermatol. 2002, 47, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Billings, S.D.; McKenney, J.K.; Folpe, A.L.; Hardacre, M.C.; Weiss, S.W. Cutaneous Angiosarcoma Following Breast-conserving Surgery and Radiation: An Analysis of 27 Cases. Am. J. Surg. Pathol. 2004, 28, 781–788. [Google Scholar] [CrossRef]
- Li, G.Z.; Fairweather, M.; Wang, J.; Orgill, D.P.; Bertagnolli, M.M.; Raut, C.P. Cutaneous Radiation-associated Breast Angiosarcoma: Radicality of Surgery Impacts Survival. Ann. Surg. 2017, 265, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Motaparthi, K.; Lauer, S.R.; Patel, R.M.; Vidal, C.I.; Linos, K. MYC gene amplification by fluorescence in situ hybridization and MYC protein expression by immunohistochemistry in the diagnosis of cutaneous angiosarcoma: Systematic review and appropriate use criteria. J. Cutan. Pathol. 2021, 48, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Stotter, A.; McLean, N.R.; Fallowfield, M.E.; Breach, N.M.; Westbury, G. Reconstruction after excision of soft tissue sarcomas of the limbs and trunk. Br. J. Surg. 2005, 75, 774–778. [Google Scholar] [CrossRef] [PubMed]
- López, J.F.; Hietanen, K.E.; Kaartinen, I.S.; Kääriäinen, M.T.; Pakarinen, T.-K.; Laitinen, M.; Kuokkanen, H. Primary flap reconstruction of tissue defects after sarcoma surgery enables curative treatment with acceptable functional results: A 7-year review. BMC Surg. 2015, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Youssef, A.; Subramanian, V.; Rogers, B.A.; Pollock, R.E.; Robb, G.L.; Chang, D.W. Upper Extremity Reconstruction Following Resection of Soft Tissue Sarcomas: A Functional Outcomes Analysis. Ann. Surg. Oncol. 2004, 11, 921–927. [Google Scholar] [CrossRef]
- Nishinari, K.; Krutman, M.; Junior, S.A.; Pignataro, B.S.; Yazbek, G.; Bomfim, G.A.Z.; Teivelis, M.P.; Wolosker, N. Surgical outcomes of vascular reconstruction in soft tissue sarcomas of the lower extremities. J. Vasc. Surg. 2015, 62, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Lohman, R.F.; Nabawi, A.S.; Reece, G.P.; Pollock, R.E.; Evans, G.R. Soft tissue sarcoma of the upper extremity: A 5-year ex-perience at two institutions emphasizing the role of soft tissue flap reconstruction. Cancer 2002, 94, 2256–2264. [Google Scholar] [CrossRef]
- Davis, L.A.; Dandachli, F.; Turcotte, R.; Steinmetz, O.K. Limb-sparing surgery with vascular reconstruction for malignant lower extremity soft tissue sarcoma. J. Vasc. Surg. 2017, 65, 151–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, C.; Jacobs, I.; Salti, G. Outcomes of surgery for dermatofibrosarcoma protuberans. Eur. J. Surg. Oncol. (EJSO) 2004, 30, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Farma, J.M.; Ammori, J.B.; Zager, J.S.; Marzban, S.S.; Bui, M.M.; Bichakjian, C.K.; Johnson, T.M.; Lowe, L.; Sabel, M.S.; Wong, S.L.; et al. Dermatofibrosarcoma Protuberans: How Wide Should We Resect? Ann. Surg. Oncol. 2010, 17, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Lowe, G.C.; Onajin, O.; Baum, C.L.; Otley, C.C.; Arpey, C.J.; Roenigk, R.K.; Brewer, J.D. A Comparison of Mohs Micrographic Surgery and Wide Local Excision for Treatment of Dermatofibrosarcoma Protuberans With Long-Term Follow-up: The Mayo Clinic Experience. Dermatol. Surg. 2017, 43, 98–106. [Google Scholar] [CrossRef]
- Gloster, H.M., Jr.; Harris, K.R.; Roenigk, R.K. A comparison between Mohs micrographic surgery and wide surgical excision for the treatment of dermatofibrosarcoma protuberans. J. Am. Acad. Dermatol. 1996, 35, 82–87. [Google Scholar]
- Rutgers, E.J.; Kroon, B.B.; Albus-Lutter, C.E.; Gortzak, E. Dermatofibrosarcoma protuberans: Treatment and prognosis. Eur. J. Surg. Oncol. (EJSO) 1992, 18, 1607035. [Google Scholar]
- Woo, K.-J.; Bang, S.I.; Mun, G.-H.; Oh, K.S.; Pyon, J.-K.; Lim, S.Y. Long-term outcomes of surgical treatment for dermatofibrosarcoma protuberans according to width of gross resection margin. J. Plast. Reconstr. Aesthetic Surg. 2016, 69, 395–401. [Google Scholar] [CrossRef]
- Shi, X.; Tan, Q. Clinical and pathological analysis of dermatofibrosarcoma protuberans with long-term follow-up. J. Plast. Reconstr. Aesthetic Surg. 2020, 73, 1143–1150. [Google Scholar] [CrossRef]
- Tardío, J.C.; Pinedo, F.; Aramburu, J.A.; Suárez-Massa, D.; Pampín-Franco, A.; Requena, L.; Santonja, C. Pleomorphic dermal sarcoma: A more aggressive neoplasm than previously estimated. J. Cutan. Pathol. 2016, 43, 101–112. [Google Scholar] [CrossRef]
- Cesinaro, A.M.; Gallo, G.; Tramontozzi, S.; Migaldi, M. Atypical fibroxanthoma and pleomorphic dermal sarcoma: A reappraisal. J. Cutan. Pathol. 2021, 48, 207–210. [Google Scholar] [CrossRef]
- Jibbe, A.; Worley, B.; Miller, C.H.; Alam, M. Surgical excision margins for fibrohistiocytic tumors, including atypical fibroxanthoma and undifferentiated pleomorphic sarcoma: A probability model based on a systematic review. J. Am. Acad. Dermatol. 2021. [Google Scholar] [CrossRef]
- Crimini, E.; Roberto, M.; Degli Effetti, V.; Marchetti, P.; Botticelli, A.; Schipilliti, F.M.; Arrivi, G.; Mazzuca, F. Electrochemotherapy as Promising Treatment Option in Rare Recurrent Cutaneous Neoplasm of the Scalp: Case Report of an Elderly Patient. Case Rep. Oncol. Med. 2019, 2019, 2507642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llombart, B.; Serra-Guillén, C.; Requena, C.; Alsina, M.; Morgado-Carrasco, D.; Machado, I.; Sanmartín, O. Leiomyosarcoma and Pleomorphic Dermal Sarcoma: Guidelines for Diagnosis and Treatment. Actas Dermo-Sifiliográficas 2018, 110, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Winchester, D.S.; Hocker, T.L.; Brewer, J.D.; Baum, C.L.; Hochwalt, P.C.; Arpey, C.J.; Otley, C.C.; Roenigk, R.K. Leiomyosarcoma of the skin: Clinical, histopathologic, and prognostic factors that influence outcomes. J. Am. Acad. Dermatol. 2014, 71, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Sinnamon, A.J.; Neuwirth, M.G.; BA, M.T.M.; Ecker, B.L.; Bartlett, E.K.; Zhang, P.J.; Kelz, R.R.; Fraker, D.L.; Roses, R.E.; Karakousis, G.C. A prognostic model for resectable soft tissue and cutaneous angiosarcoma. J. Surg. Oncol. 2016, 114, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Young, R.J.; Brown, N.; Reed, M.W.; Hughes, D.J.; Woll, P. Angiosarcoma. Lancet Oncol. 2010, 11, 983–991. [Google Scholar] [CrossRef]
- Wiegand, S.; Dietz, A.; Wichmann, G. Malignant Vascular Tumors of the Head and Neck—Which Type of Therapy Works Best? Cancers 2021, 13, 6201. [Google Scholar] [CrossRef] [PubMed]
- Guadagnolo, B.A.; Zagars, G.K.; Araujo, D.M.; Ravi, V.; Dmd, T.D.S.; Sturgis, E.M. Outcomes after definitive treatment for cutaneous angiosarcoma of the face and scalp. Head Neck 2010, 33, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnhardt, M.; Bohm, J.; Behr, B.; Harati, K.; Hirsch, T.; Daigeler, A. Strahlen-induzierte Angiosarkome der Brust [Radiation-induced angiosarcoma of the breast]. Handchir. Mikrochir. Plast. Chir. 2017, 49, 103–110. [Google Scholar] [CrossRef]
- Styring, E.; Klasson, S.; Rydholm, A.; von Steyern, F.V. Radiation-associated angiosarcoma after breast cancer: Improved survival by excision of all irradiated skin and soft tissue of the thoracic wall? A report of six patients. Acta Oncol. 2014, 54, 1078–1080. [Google Scholar] [CrossRef]
- Seinen, J.M.; Styring, E.; Verstappen, V.; von Steyern, F.V.; Rydholm, A.; Suurmeijer, A.; Hoekstra, H.J. Radiation-Associated Angiosarcoma After Breast Cancer: High Recurrence Rate and Poor Survival Despite Surgical Treatment with R0 Resection. Ann. Surg. Oncol. 2012, 19, 2700–2706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Sullivan, B.; Davis, A.M.; Turcotte, R.; Bell, R.; Catton, C.; Chabot, P.; Wunder, J.; Kandel, R.; Goddard, K.; Sadura, A.; et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: A randomised trial. Lancet 2002, 359, 2235–2241. [Google Scholar] [CrossRef]
- Ibanez, M.A.; Rismiller, K.; Knackstedt, T. Prognostic factors, treatment, and survival in cutaneous pleomorphic sarcoma. J. Am. Acad. Dermatol. 2020, 83, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Edmonson, J.H.; Ryan, L.M.; Blum, R.H.; Brooks, J.S.; Shiraki, M.; Frytak, S.; Parkinson, D.R. Randomized comparison of doxorubicin alone versus ifosfamide plus doxorubicin or mitomycin, doxorubicin, and cisplatin against advanced soft tissue sarcomas. J. Clin. Oncol. 1993, 11, 1269–1275. [Google Scholar] [CrossRef] [Green Version]
- Köstler, W.J.; Brodowicz, T.; Attems, Y.; Hejna, M.; Tomek, S.; Amann, G.; Fiebiger, W.C.C.; Wiltschke, C.; Krainer, M.; Zielinski, C.C. Docetaxel as rescue medication in anthracycline- and ifosfamide-resistant locally advanced or metastatic soft tissue sarcoma: Results of a phase II trial. Ann. Oncol. 2001, 12, 1281–1288. [Google Scholar] [CrossRef]
- Ratan, R.; Patel, S.R. Chemotherapy for soft tissue sarcoma. Cancer 2016, 122, 2952–2960. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.A.; Tepper, J.; Glatstein, E.; Costa, J.; Baker, A.; Brennam, M.; Demoss, E.V.; Seipp, C.; Sindelar, W.F.; Sugarbaker, P.; et al. The Treatment of Soft-tissue Sarcomas of the Extremities: Prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann. Surg. 1982, 196, 305–315. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Tepper, J.; Glatstein, E.; Costa, J.; Young, R.; Baker, A.; Brennan, M.F.; Demoss, E.V.; Seipp, C.; Sindelar, W.F.; et al. Prospective randomized evaluation of adjuvant chemotherapy in adults with soft tissue sarcomas of the extremities. Cancer 1983, 52, 424–434. [Google Scholar] [CrossRef]
- DeLaney, T.F.; Spiro, I.J.; Suit, H.D.; Gebhardt, M.C.; Hornicek, F.J.; Mankin, H.J.; Rosenberg, A.L.; Rosenthal, D.I.; Miryousefi, F.; Ancukiewicz, M.; et al. Neoadjuvant chemotherapy and radiotherapy for large extremity soft-tissue sarcomas. Int. J. Radiat. Oncol. 2003, 56, 1117–1127. [Google Scholar] [CrossRef]
- Mullen, J.T.; Kobayashi, W.; Wang, J.J.; Harmon, D.C.; Choy, E.; Hornicek, F.J.; Rosenberg, A.E.; Chen, Y.-L.; Spiro, I.J.; Delaney, T.F. Long-term follow-up of patients treated with neoadjuvant chemotherapy and radiotherapy for large, extremity soft tissue sarcomas. Cancer 2012, 118, 3758–3765. [Google Scholar] [CrossRef]
- Kérob, D.; Porcher, R.; Vérola, O.; Dalle, S.; Maubec, E.; Aubin, F.; D’Incan, M.; Bodokh, I.; Boulinguez, S.; Madelaine-Chambrin, I.; et al. Imatinib Mesylate as a Preoperative Therapy in Dermatofibrosarcoma: Results of a Multicenter Phase II Study on 25 Patients. Clin. Cancer Res. 2010, 16, 3288–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaziz, J.; Battistella, M.; Delyon, J.; Farges, C.; Marco, O.; Pages, C.; Le Maignan, C.; da Meda, L.; Basset-Seguin, N.; Resche-Rigon, M.; et al. Long-Term Outcome of Neoadjuvant Tyrosine Kinase Inhibitors Followed by Complete Surgery in Locally Advanced Dermatofibrosarcoma Protuberans. Cancers 2021, 13, 2224. [Google Scholar] [CrossRef] [PubMed]
- Birdi, H.K.; Jirovec, A.; Cortés-Kaplan, S.; Werier, J.; Nessim, C.; Diallo, J.-S.; Ardolino, M. Immunotherapy for sarcomas: New frontiers and unveiled opportunities. J. Immunother. Cancer 2021, 9, e001580. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501, Erratum in Lancet Oncol. 2017, 18, e711 and Lancet Oncol. 2018, 19, e8. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.-W.; Sun, C.-M.; Calderaro, J.; Jeng, Y.-M.; Hsiao, L.-P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Florou, V.; Rosenberg, A.E.; Wieder, E.; Komanduri, K.V.; Kolonias, D.; Uduman, M.; Castle, J.C.; Buell, J.S.; Trent, J.C.; Wilky, B.A. Angiosarcoma patients treated with immune checkpoint inhibitors: A case series of seven patients from a single institution. J. Immunother. Cancer 2019, 7, 213. [Google Scholar] [CrossRef] [Green Version]
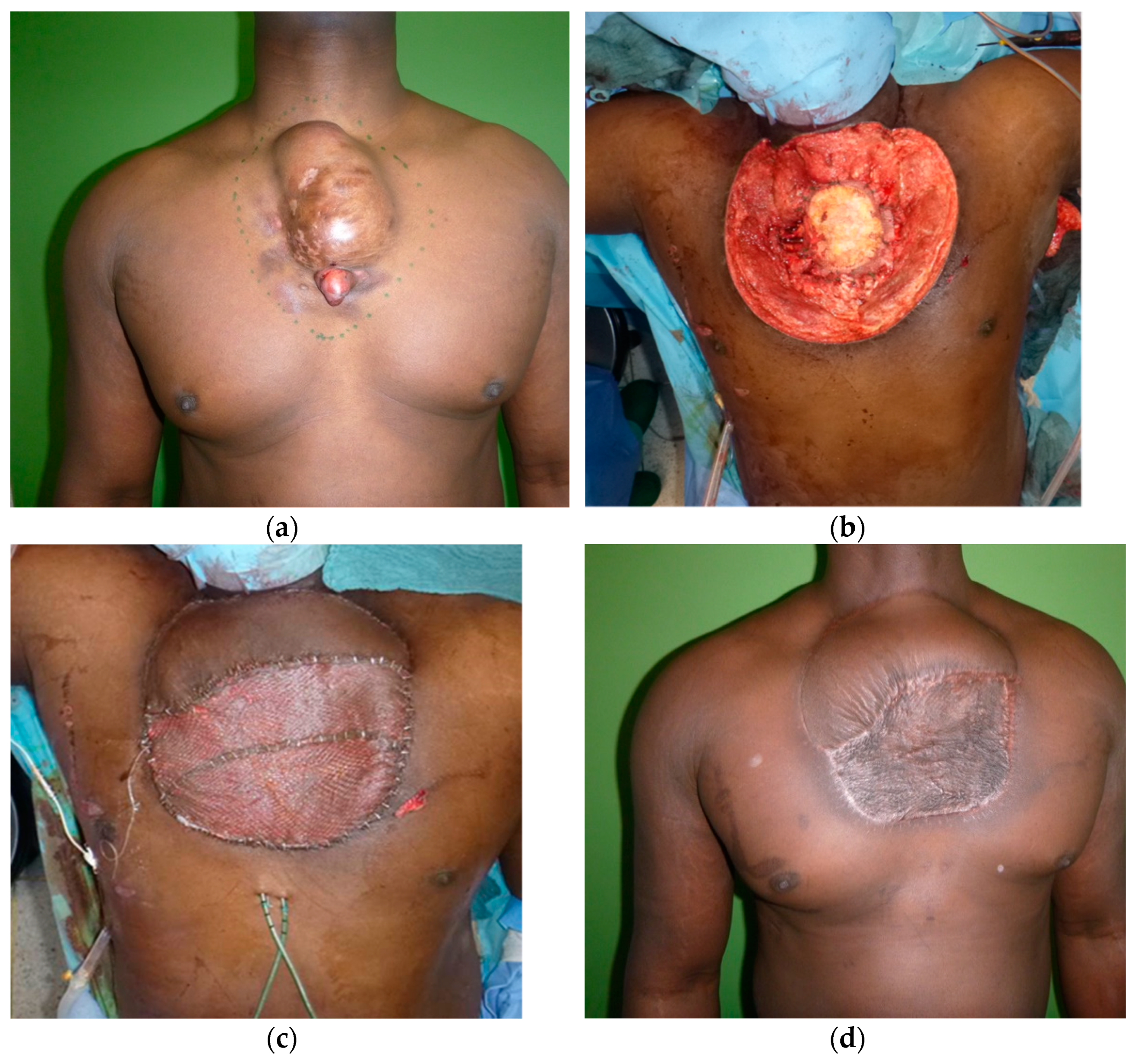



{kind=link}
{kind=link}
{kind=link}
| Type | Clinical Presentation | Pitfalls | Surgery | Adjuvant Therapy | Follow-Up |
|---|---|---|---|---|---|
| Dermatofibrosarcoma Protuberans | Painless pink or violet plaque | Fibrosarcomatous transformation Local destructive growth | Wide local excision: 2–3 cm incl. fascia *** | Imatinib | Every 6 months for 5 years * |
| Atypical Fibroxantoma | Ulcerative, rapidly growing lesion | Previous radiotherapy predisposes | Excision 2 cm | Radiotherapy | Every 6 months for 2 years |
| dermal undifferentiated pleomorphic sarcoma | Rapidly growing | Previous radiotherapy predisposes | Wide local excision: 3 cm incl. fascia *** | Radiotherapy Pembrolizumab | Every 3 months for 2 years + every 6 months for 3 years ** |
| Leiomyosarcoma | Dermal nodule or diffuse growth | Attention to subcutaneous invasion High recurrence rate | Dermal: excision 1–3 cm Subcutaneous: excision 2 cm incl. fascia | Radiotherapy | Every 3 months for 2 years + every 6 months for 3 years |
| Kaposi sarcoma | Violaceous, purple/brown macular plaques/noduli | Can be mistaken for bruising or eczema | None, refer to dermatologic evaluation | Doxorubicin, Paclitaxel HIV-associated: antiretroviral therapy + chemotherapy | Individually planned |
| Primary angiosarcoma | Bruise-like lesions | Aggressive growth | Complete wide resection +/− postoperative radiation | Radiotherapy Doxorubicin | Individually planned, highly specialised |
| Radiation-induced angiosarcoma | Discolouration of the skin, papules | High recurrence rate | Excision of entire radiation field in primary cases | - | Individually planned, highly specialised |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trøstrup, H.; Bigdeli, A.K.; Krogerus, C.; Kneser, U.; Schmidt, G.; Schmidt, V.J. A Multidisciplinary Approach to Complex Dermal Sarcomas Ensures an Optimal Clinical Outcome. Cancers 2022, 14, 1693. https://doi.org/10.3390/cancers14071693
Trøstrup H, Bigdeli AK, Krogerus C, Kneser U, Schmidt G, Schmidt VJ. A Multidisciplinary Approach to Complex Dermal Sarcomas Ensures an Optimal Clinical Outcome. Cancers. 2022; 14(7):1693. https://doi.org/10.3390/cancers14071693
Chicago/Turabian StyleTrøstrup, Hannah, Amir K. Bigdeli, Christina Krogerus, Ulrich Kneser, Grethe Schmidt, and Volker J. Schmidt. 2022. "A Multidisciplinary Approach to Complex Dermal Sarcomas Ensures an Optimal Clinical Outcome" Cancers 14, no. 7: 1693. https://doi.org/10.3390/cancers14071693