
Harnessing the Heterogeneity of Prostate Cancer for Target Discovery Using Patient-Derived Explants

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Consent and Tissue Collection
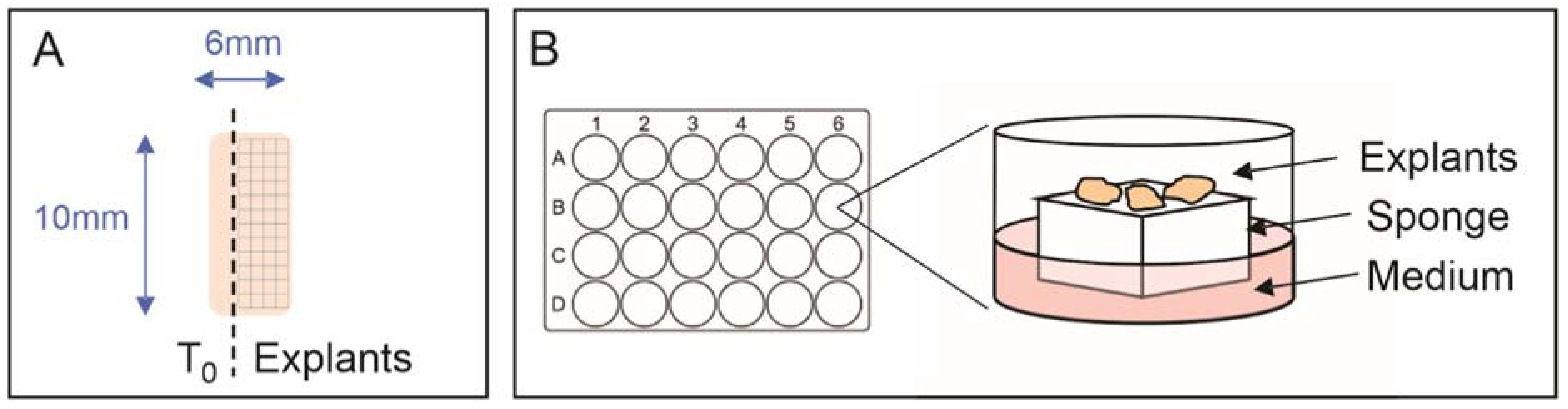
2.2. Explant Tissue Culture
2.3. Histology
2.4. Immunohistochemistry
2.5. Statistical Modeling of Ki67 Immunostaining Cell Positivity
2.6. RNA Sequencing Analysis
3. Results
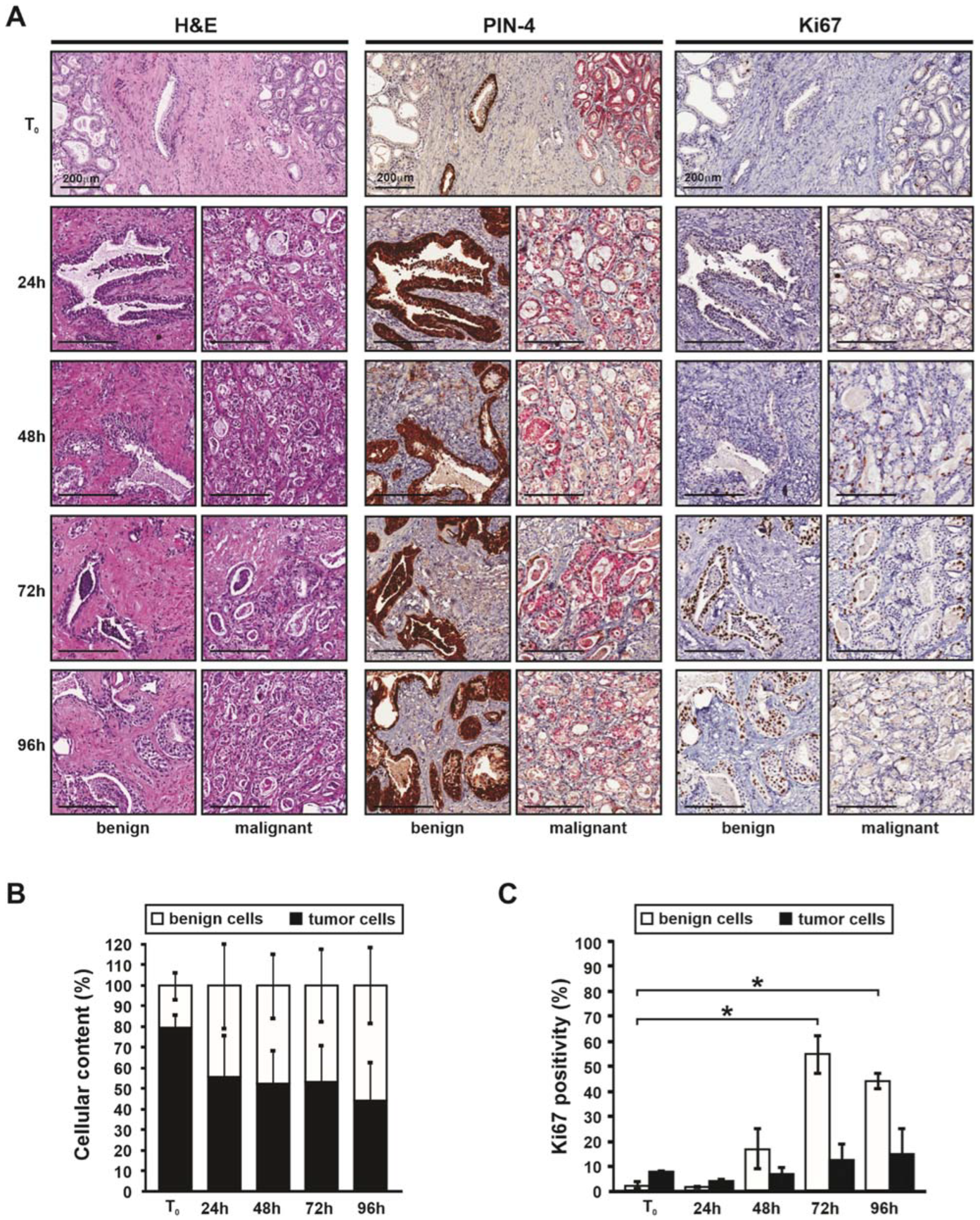
3.1. Histology of Patient-Derived Explants Enables Visualization of the Tumor Microenvironment
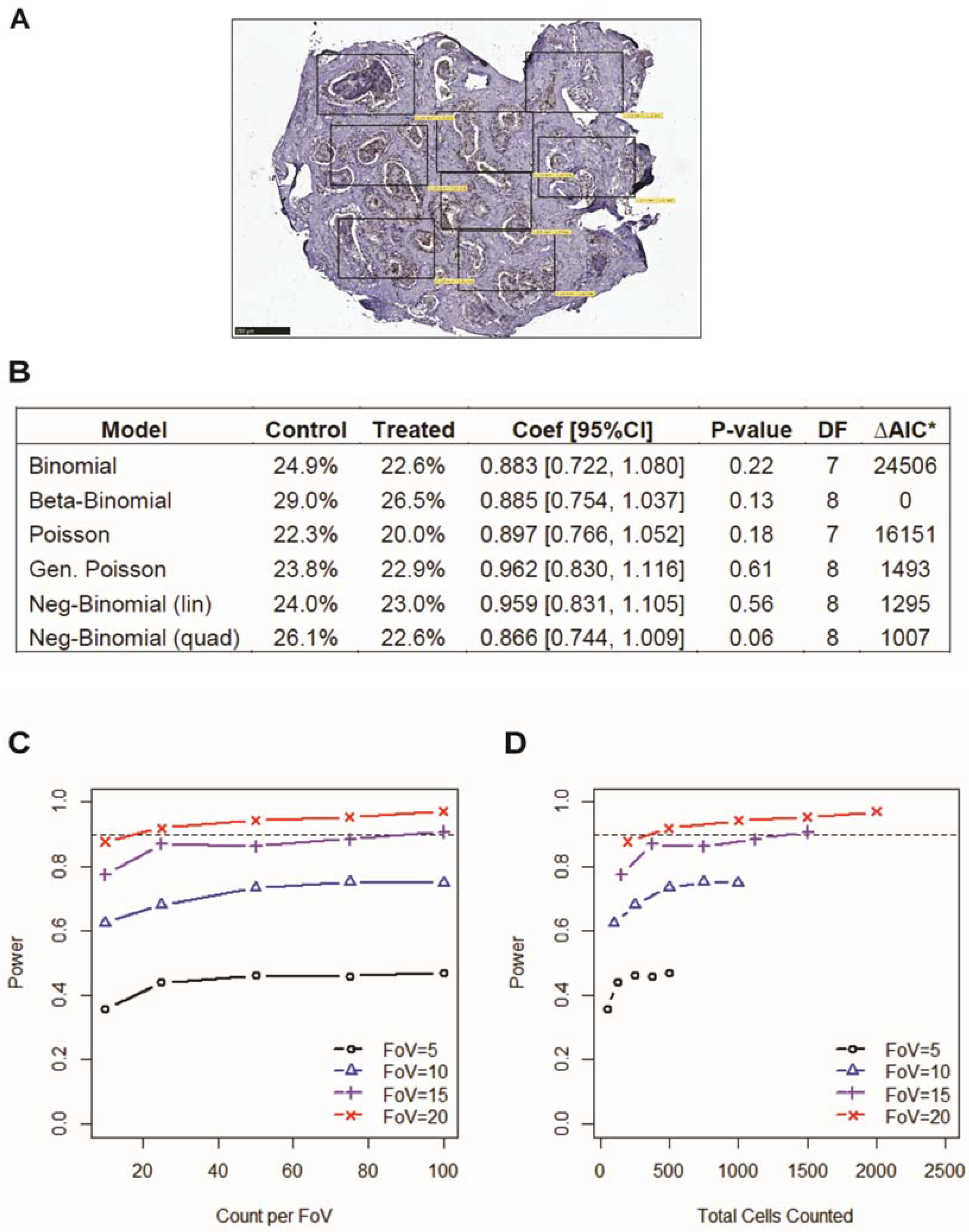
3.2. Incorporating Field-of-View Cell Counts Provides Accuracy in Immunostaining Quantification
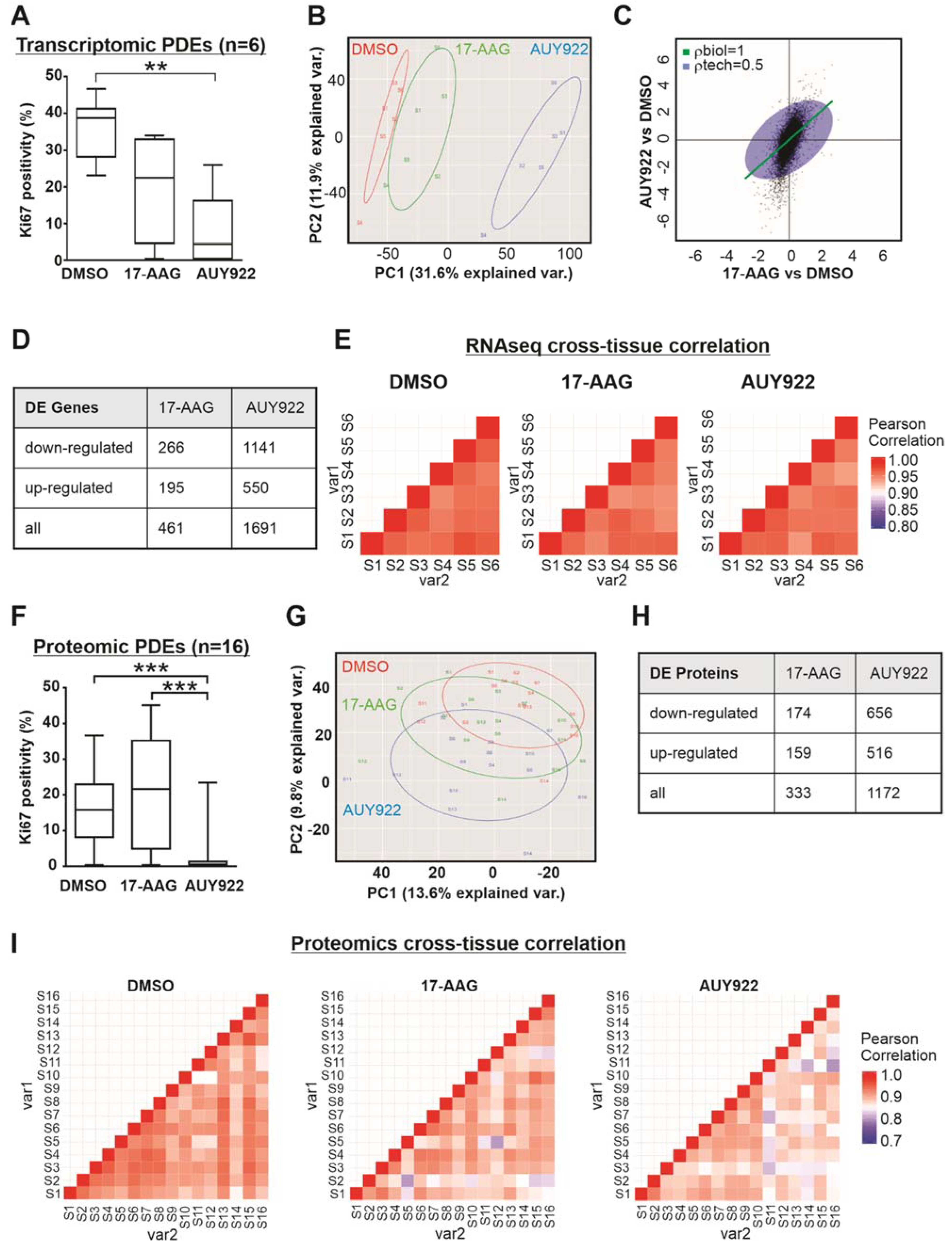
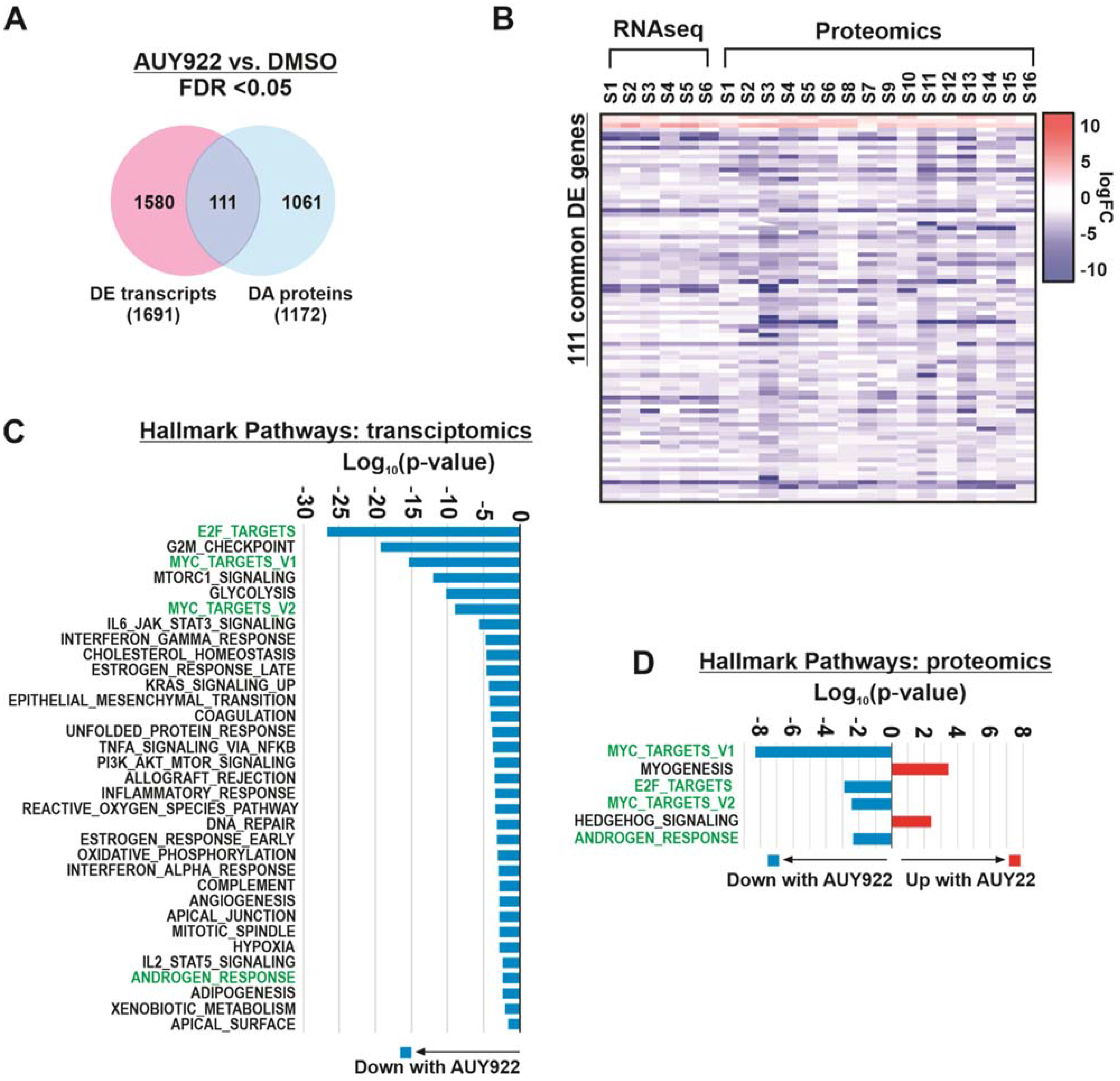
3.3. Transcriptomic and Proteomic Analysis of Patient-Derived Explants Identifies Consistent Expression Changes Irrespective of Heterogeneity
3.4. Integration of Transcript and Protein Expression Profiles Identifies Key Pathways of Interest
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moreno, L.; Pearson, A.D. How can attrition rates be reduced in cancer drug discovery? Expert Opin. Drug Discov. 2013, 8, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Peehl, D.M. Primary cell cultures as models of prostate cancer development. Endocr. Relat. Cancer 2005, 12, 19–47. [Google Scholar] [CrossRef] [PubMed]
- Namekawa, T.; Ikeda, K.; Horie-Inoue, K.; Inoue, S. Application of prostate cancer models for preclinical study: Advantages and limitations of cell lines, patient-derived xenografts, and three-dimensional culture of patient-derived cells. Cells 2019, 8, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiao, S.L.; Chu, G.C.; Chung, L.W. Regulation of prostate cancer progression by the tumor microenvironment. Cancer Lett. 2016, 380, 340–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Karthaus, W.R.; Lee, Y.S.; Gao, V.R.; Wu, C.; Russo, J.W.; Liu, M.; Mota, J.M.; Abida, W.; Linton, E.; et al. Tumor microenvironment-derived nrg1 promotes antiandrogen resistance in prostate cancer. Cancer Cell 2020, 38, e279. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent spop, foxa1 and med12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [Green Version]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 162, 454. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [Green Version]
- Ledford, H. Us cancer institute to overhaul tumour cell lines. Nature 2016, 530, 391. [Google Scholar] [CrossRef] [Green Version]
- Risbridger, G.P.; Toivanen, R.; Taylor, R.A. Preclinical models of prostate cancer: Patient-derived xenografts, organoids, and other explant models. Cold Spring Harb. Perspect. Med. 2018, 8, a030536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef]
- Lawrence, M.G.; Taylor, R.A.; Toivanen, R.; Pedersen, J.; Norden, S.; Pook, D.W.; Frydenberg, M.; Papargiris, M.M.; Niranjan, B.; Richards, M.G.; et al. A preclinical xenograft model of prostate cancer using human tumors. Nat. Protoc. 2013, 8, 836–848. [Google Scholar] [CrossRef]
- Izumchenko, E.; Paz, K.; Ciznadija, D.; Sloma, I.; Katz, A.; Vasquez-Dunddel, D.; Ben-Zvi, I.; Stebbing, J.; McGuire, W.; Harris, W.; et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. 2017, 28, 2595–2605. [Google Scholar] [CrossRef]
- Porter, L.H.; Bakshi, A.; Pook, D.; Clark, A.; Clouston, D.; Kourambas, J.; Goode, D.L.; Risbridger, G.P.; Taylor, R.A.; Lawrence, M.G.; et al. Androgen receptor enhancer amplification in matched patient-derived xenografts of primary and castrate-resistant prostate cancer. J. Pathol. 2021, 254, 121–134. [Google Scholar] [CrossRef]
- Topp, M.D.; Hartley, L.; Cook, M.; Heong, V.; Boehm, E.; McShane, L.; Pyman, J.; McNally, O.; Ananda, S.; Harrell, M.; et al. Molecular correlates of platinum response in human high-grade serous ovarian cancer patient-derived xenografts. Mol. Oncol. 2014, 8, 656–668. [Google Scholar] [CrossRef]
- Zhang, X.; Claerhout, S.; Prat, A.; Dobrolecki, L.E.; Petrovic, I.; Lai, Q.; Landis, M.D.; Wiechmann, L.; Schiff, R.; Giuliano, M.; et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013, 73, 4885–4897. [Google Scholar] [CrossRef] [Green Version]
- Powley, I.R.; Patel, M.; Miles, G.; Pringle, H.; Howells, L.; Thomas, A.; Kettleborough, C.; Bryans, J.; Hammonds, T.; MacFarlane, M.; et al. Patient-derived explants (pdes) as a powerful preclinical platform for anti-cancer drug and biomarker discovery. Br. J. Cancer 2020, 122, 735–744. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, H.K.; Koay, Y.C.; Irani, S.; Das, R.; Nassar, Z.D.; Australian Prostate Cancer Bioresource; Selth, L.A.; Centenera, M.M.; McAlpine, S.R.; Butler, L.M. A novel class of hsp90 c-terminal modulators have pre-clinical efficacy in prostate tumor cells without induction of a heat shock response. Prostate 2016, 76, 1546–1559. [Google Scholar] [CrossRef] [PubMed]
- Centenera, M.M.; Gillis, J.L.; Hanson, A.R.; Jindal, S.; Taylor, R.A.; Risbridger, G.P.; Sutherland, P.D.; Scher, H.I.; Raj, G.V.; Knudsen, K.E.; et al. Evidence for efficacy of new hsp90 inhibitors revealed by ex vivo culture of human prostate tumors. Clin. Cancer Res. 2012, 18, 3562–3570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comstock, C.E.; Augello, M.A.; Goodwin, J.F.; de Leeuw, R.; Schiewer, M.J.; Ostrander, W.F., Jr.; Burkhart, R.A.; McClendon, A.K.; McCue, P.A.; Trabulsi, E.J.; et al. Targeting cell cycle and hormone receptor pathways in cancer. Oncogene 2013, 32, 5481–5491. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.M.; Lee, B.Y.; Castillo, L.; Spielman, C.; Grogan, J.; Yeung, N.K.; Kench, J.G.; Stricker, P.D.; Haynes, A.M.; Centenera, M.M.; et al. Effect of fak inhibitor vs-6063 (defactinib) on docetaxel efficacy in prostate cancer. Prostate 2018, 78, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Ravindranathan, P.; Lee, T.K.; Yang, L.; Centenera, M.M.; Butler, L.; Tilley, W.D.; Hsieh, J.T.; Ahn, J.M.; Raj, G.V. Peptidomimetic targeting of critical androgen receptor-coregulator interactions in prostate cancer. Nat. Commun. 2013, 4, 1923. [Google Scholar] [CrossRef]
- Schiewer, M.J.; Goodwin, J.F.; Han, S.; Brenner, J.C.; Augello, M.A.; Dean, J.L.; Liu, F.; Planck, J.L.; Ravindranathan, P.; Chinnaiyan, A.M.; et al. Dual roles of parp-1 promote cancer growth and progression. Cancer Discov. 2012, 2, 1134–1149. [Google Scholar] [CrossRef] [Green Version]
- Tran, L.N.K.; Kichenadasse, G.; Butler, L.M.; Centenera, M.M.; Morel, K.L.; Ormsby, R.J.; Michael, M.Z.; Lower, K.M.; Sykes, P.J. The combination of metformin and valproic acid induces synergistic apoptosis in the presence of p53 and androgen signaling in prostate cancer. Mol. Cancer Ther. 2017, 16, 2689–2700. [Google Scholar] [CrossRef] [Green Version]
- Centenera, M.M.; Scott, J.S.; Machiels, J.; Nassar, Z.D.; Miller, D.C.; Zinonos, I.; Dehairs, J.; Burvenich, I.J.G.; Zadra, G.; Chetta, P.M.; et al. Elovl5 is a critical and targetable fatty acid elongase in prostate cancer. Cancer Res. 2021, 81, 1704–1718. [Google Scholar] [CrossRef]
- Gillis, J.L.; Hinneh, J.A.; Ryan, N.K.; Irani, S.; Moldovan, M.; Quek, L.E.; Shrestha, R.K.; Hanson, A.R.; Xie, J.; Hoy, A.J.; et al. A feedback loop between the androgen receptor and 6-phosphogluoconate dehydrogenase (6pgd) drives prostate cancer growth. Elife 2021, 10, e62592. [Google Scholar] [CrossRef]
- Scheinberg, T.; Kench, J.; Stockler, M.; Mahon, K.L.; Sebastian, L.; Stricker, P.; Joshua, A.M.; Woo, H.; Thanigasalam, R.; Ahmadi, N.; et al. Pharmacodynamics effects of cdk4/6 inhibitor lee011 (ribociclib) in high-risk, localised prostate cancer: A study protocol for a randomised controlled phase ii trial (leep study: Lee011 in high-risk, localised prostate cancer). BMJ Open 2020, 10, e033667. [Google Scholar] [CrossRef] [Green Version]
- Epstein, J.I.; Egevad, L.; Humphrey, P.A.; Montironi, R. Best practices recommendations in the application of immunohistochemistry in the prostate: Report from the international society of urologic pathology consensus conference. Am. J. Surg. Pathol. 2014, 38, e6–e19. [Google Scholar] [CrossRef] [PubMed]
- Rathod, S.G.; Jaiswal, D.G.; Bindu, R.S. Diagnostic utility of triple antibody (amacr, hmwck and p63) stain in prostate neoplasm. J. Fam. Med. Prim. Care 2019, 8, 2651–2655. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, H.K.; Gillis, J.L.; Johnson, I.R.D.; Nassar, Z.D.; Moldovan, M.; Levrier, C.; Sadowski, M.C.; Chin, M.Y.; Tomlinson Guns, E.S.; Tarulli, G.; et al. Dysregulated fibronectin trafficking by hsp90 inhibition restricts prostate cancer cell invasion. Sci. Rep. 2018, 8, 2090. [Google Scholar] [CrossRef]
- Nguyen, E.V.; Centenera, M.M.; Moldovan, M.; Das, R.; Irani, S.; Vincent, A.D.; Chan, H.; Horvath, L.G.; Lynn, D.J.; Daly, R.J.; et al. Identification of novel response and predictive biomarkers to hsp90 inhibitors through proteomic profiling of patient-derived prostate tumor explants. Mol. Cell. Proteom. 2018, 17, 1470–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (msigdb) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef]
- Butler, L.M.; Mah, C.Y.; Machiels, J.; Vincent, A.D.; Irani, S.; Mutuku, S.M.; Spotbeen, X.; Bagadi, M.; Waltregny, D.; Moldovan, M.; et al. Lipidomic profiling of clinical prostate cancer reveals targetable alterations in membrane lipid composition. Cancer Res. 2021, 81, 4981–4993. [Google Scholar] [CrossRef]
- Busby, M.A.; Stewart, C.; Miller, C.A.; Grzeda, K.R.; Marth, G.T. Scotty: A web tool for designing rna-seq experiments to measure differential gene expression. Bioinformatics 2013, 29, 656–657. [Google Scholar] [CrossRef] [Green Version]
- Yamaki, H.; Nakajima, M.; Shimotohno, K.W.; Tanaka, N. Molecular basis for the actions of hsp90 inhibitors and cancer therapy. J. Antibiot. 2011, 64, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Hoter, A.; Rizk, S.; Naim, H.Y. The multiple roles and therapeutic potential of molecular chaperones in prostate cancer. Cancers 2019, 11, 1194. [Google Scholar] [CrossRef] [Green Version]
- Butler, L.M.; Ferraldeschi, R.; Armstrong, H.K.; Centenera, M.M.; Workman, P. Maximizing the therapeutic potential of hsp90 inhibitors. Mol. Cancer Res. MCR 2015, 13, 1445–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slovin, S.; Hussain, S.; Saad, F.; Garcia, J.; Picus, J.; Ferraldeschi, R.; Crespo, M.; Flohr, P.; Riisnaes, R.; Lin, C.; et al. Pharmacodynamic and clinical results from a phase i/ii study of the hsp90 inhibitor onalespib in combination with abiraterone acetate in prostate cancer. Clin. Cancer Res. 2019, 25, 4624–4633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraldeschi, R.; Welti, J.; Luo, J.; Attard, G.; de Bono, J.S. Targeting the androgen receptor pathway in castration-resistant prostate cancer: Progresses and prospects. Oncogene 2015, 34, 1745–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebello, R.J.; Pearson, R.B.; Hannan, R.D.; Furic, L. Therapeutic approaches targeting myc-driven prostate cancer. Genes 2017, 8, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, J.N.; Cho, M.; Park, S.; So, I.; Jeon, J.-H. The conflicting role of e2f1 in prostate cancer: A matter of cell context or interpretational flexibility? Biochim. Biophys. Acta BBA Rev. Cancer 2020, 1873, 188336. [Google Scholar] [CrossRef]
- Liang, Y.X.; Lu, J.M.; Mo, R.J.; He, H.C.; Xie, J.; Jiang, F.N.; Lin, Z.Y.; Chen, Y.R.; Wu, Y.D.; Luo, H.W.; et al. E2f1 promotes tumor cell invasion and migration through regulating cd147 in prostate cancer. Int. J. Oncol. 2016, 48, 1650–1658. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Tang, W.; Zhang, P.; Liu, Z.; Lyu, F.; Xiao, Y.; Ni, D.; Zhang, P. Comprehensive analysis of the functional and prognostic value of e2f transcription factors in human prostate cancer through data mining and experimental validation. Transl. Cancer Res. 2021, 10, 5095–5109. [Google Scholar] [CrossRef]
- Guan, L.; Zou, Q.; Liu, Q.; Lin, Y.; Chen, S. Hsp90 inhibitor ganetespib (sta-9090) inhibits tumor growth in c-myc-dependent esophageal squamous cell carcinoma. OncoTargets Ther. 2020, 13, 2997–3011. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Zhang, L.L.; Wu, W.; Guo, H.; Li, Y.; Sukhanova, M.; Venkataraman, G.; Huang, S.; Zhang, H.; Alikhan, M.; et al. Activation of myc, a bona fide client of hsp90, contributes to intrinsic ibrutinib resistance in mantle cell lymphoma. Blood Adv. 2018, 2, 2039–2051. [Google Scholar] [CrossRef]
- Poole, C.J.; Zheng, W.; Lee, H.; Young, D.; Lodh, A.; Chadli, A.; van Riggelen, J. Targeting the myc oncogene in burkitt lymphoma through hsp90 inhibition. Cancers 2018, 10, 448. [Google Scholar] [CrossRef] [Green Version]
- Regan, P.L.; Jacobs, J.; Wang, G.; Torres, J.; Edo, R.; Friedmann, J.; Tang, X.X. Hsp90 inhibition increases p53 expression and destabilizes mycn and myc in neuroblastoma. Int. J. Oncol. 2011, 38, 105–112. [Google Scholar] [PubMed]
- Kotwal, A.; Suran, S.; Amere Subbarao, S. Hsp90 chaperone facilitates e2f1/2-dependent gene transcription in human breast cancer cells. Eur. J. Cell. Biol. 2021, 100, 151148. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lu, Z.; Shi, X.; Liu, L.; Zhang, P.; Golemis, E.A.; Tu, Z. Hsp90 inhibition downregulates DNA replication and repair genes via e2f1 repression. J. Biol. Chem. 2021, 297, 100996. [Google Scholar] [CrossRef] [PubMed]
- Jung, P.S.; Kim, D.Y.; Kim, M.B.; Lee, S.W.; Kim, J.H.; Kim, Y.M.; Kim, Y.T.; Hoffman, R.M.; Nam, J.H. Progression-free survival is accurately predicted in patients treated with chemotherapy for epithelial ovarian cancer by the histoculture drug response assay in a prospective correlative clinical trial at a single institution. Anticancer Res. 2013, 33, 1029–1034. [Google Scholar] [PubMed]
- Karekla, E.; Liao, W.J.; Sharp, B.; Pugh, J.; Reid, H.; Le Quesne, J.; Moore, D.; Pritchard, C.; MacFarlane, M.; Pringle, J.H. Ex vivo explant cultures of non-small cell lung carcinoma enable evaluation of primary tumor responses to anticancer therapy. Cancer Res. 2017, 77, 2029–2039. [Google Scholar] [CrossRef] [Green Version]
- Templeton, A.R.; Jeffery, P.L.; Thomas, P.B.; Perera, M.P.J.; Ng, G.; Calabrese, A.R.; Nicholls, C.; Mackenzie, N.J.; Wood, J.; Bray, L.J.; et al. Patient-derived explants as a precision medicine patient-proximal testing platform informing cancer management. Front. Oncol. 2021, 11, 767697. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Centenera, M.M.; Vincent, A.D.; Moldovan, M.; Lin, H.-M.; Lynn, D.J.; Horvath, L.G.; Butler, L.M. Harnessing the Heterogeneity of Prostate Cancer for Target Discovery Using Patient-Derived Explants. Cancers 2022, 14, 1708. https://doi.org/10.3390/cancers14071708
Centenera MM, Vincent AD, Moldovan M, Lin H-M, Lynn DJ, Horvath LG, Butler LM. Harnessing the Heterogeneity of Prostate Cancer for Target Discovery Using Patient-Derived Explants. Cancers. 2022; 14(7):1708. https://doi.org/10.3390/cancers14071708
Chicago/Turabian StyleCentenera, Margaret M., Andrew D. Vincent, Max Moldovan, Hui-Ming Lin, David J. Lynn, Lisa G. Horvath, and Lisa M. Butler. 2022. "Harnessing the Heterogeneity of Prostate Cancer for Target Discovery Using Patient-Derived Explants" Cancers 14, no. 7: 1708. https://doi.org/10.3390/cancers14071708
APA StyleCentenera, M. M., Vincent, A. D., Moldovan, M., Lin, H.-M., Lynn, D. J., Horvath, L. G., & Butler, L. M. (2022). Harnessing the Heterogeneity of Prostate Cancer for Target Discovery Using Patient-Derived Explants. Cancers, 14(7), 1708. https://doi.org/10.3390/cancers14071708