
ATRX-Deficient High-Grade Glioma Cells Exhibit Increased Sensitivity to RTK and PDGFR Inhibitors

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Cell Culture
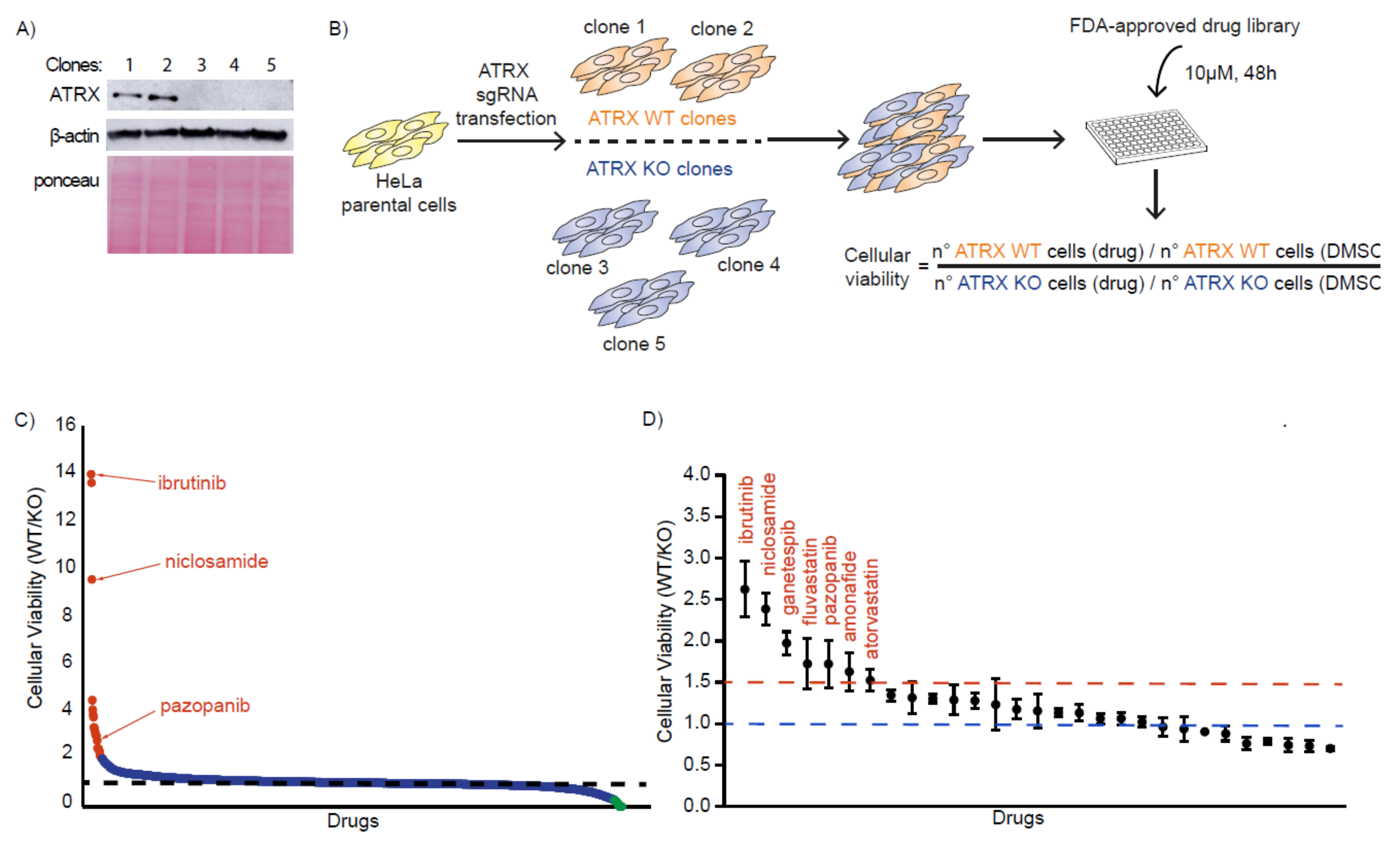
2.2. Generation of ATRX KO Cells
2.3. Western Blot
2.4. High-Throughput Microscopy
2.5. Analysis of the Screen Data
2.6. Immunofluorescence
2.7. Lentivirus Synthesis
2.8. Cell Infection
2.9. Drug Inhibitors
2.10. Cell Viability Analysis
2.11. Dose–Response Curves
2.12. Colony Formation Assay
2.13. Statistical Analysis
3. Results
3.1. Identification of FDA-Approved Compounds That Selectively Kill ATRX-Deficient Cells
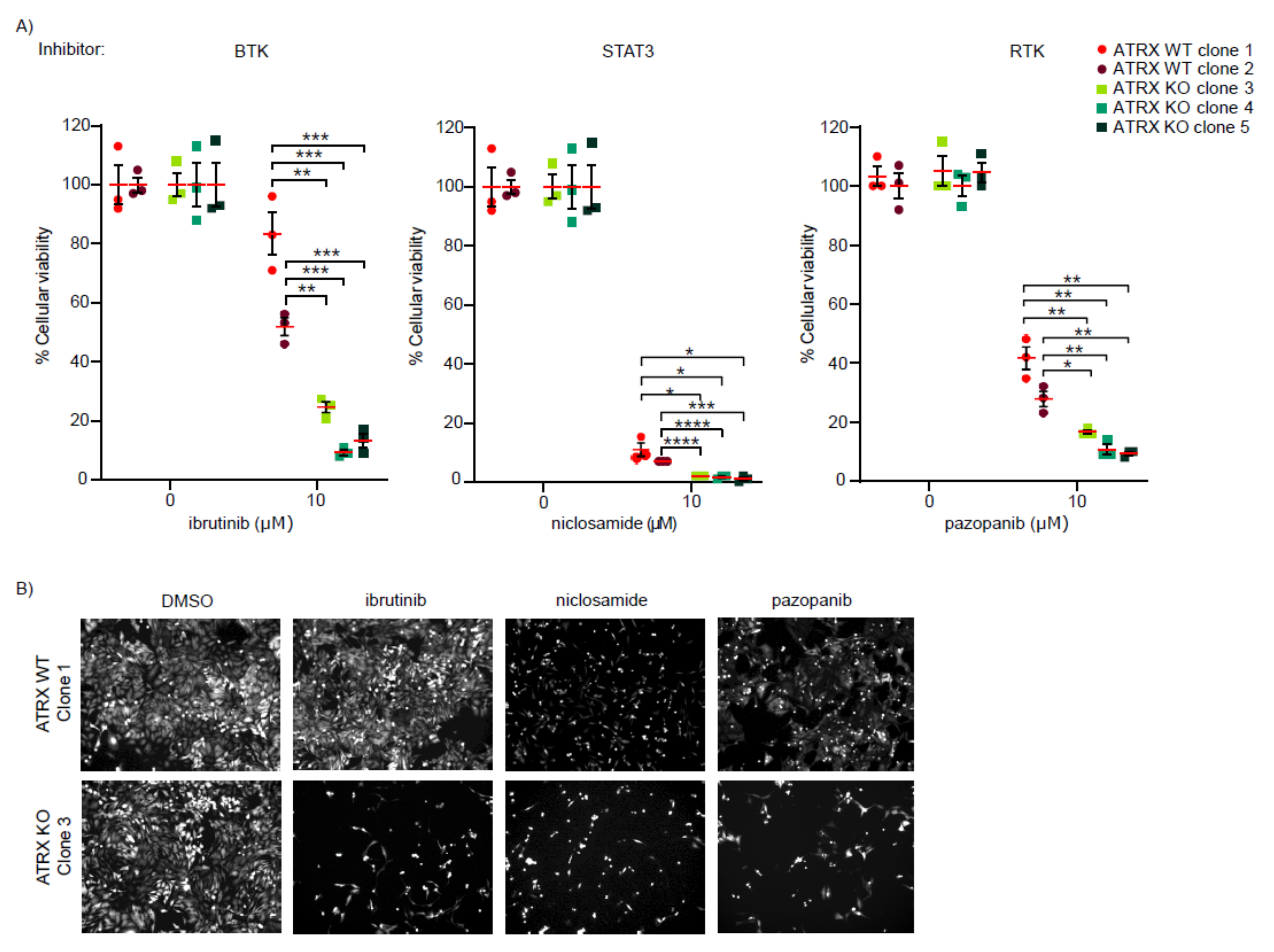
3.2. ATRX-Deficient Cells Show Increased Sensitivity to BTK, STAT3, and RTK Inhibitors
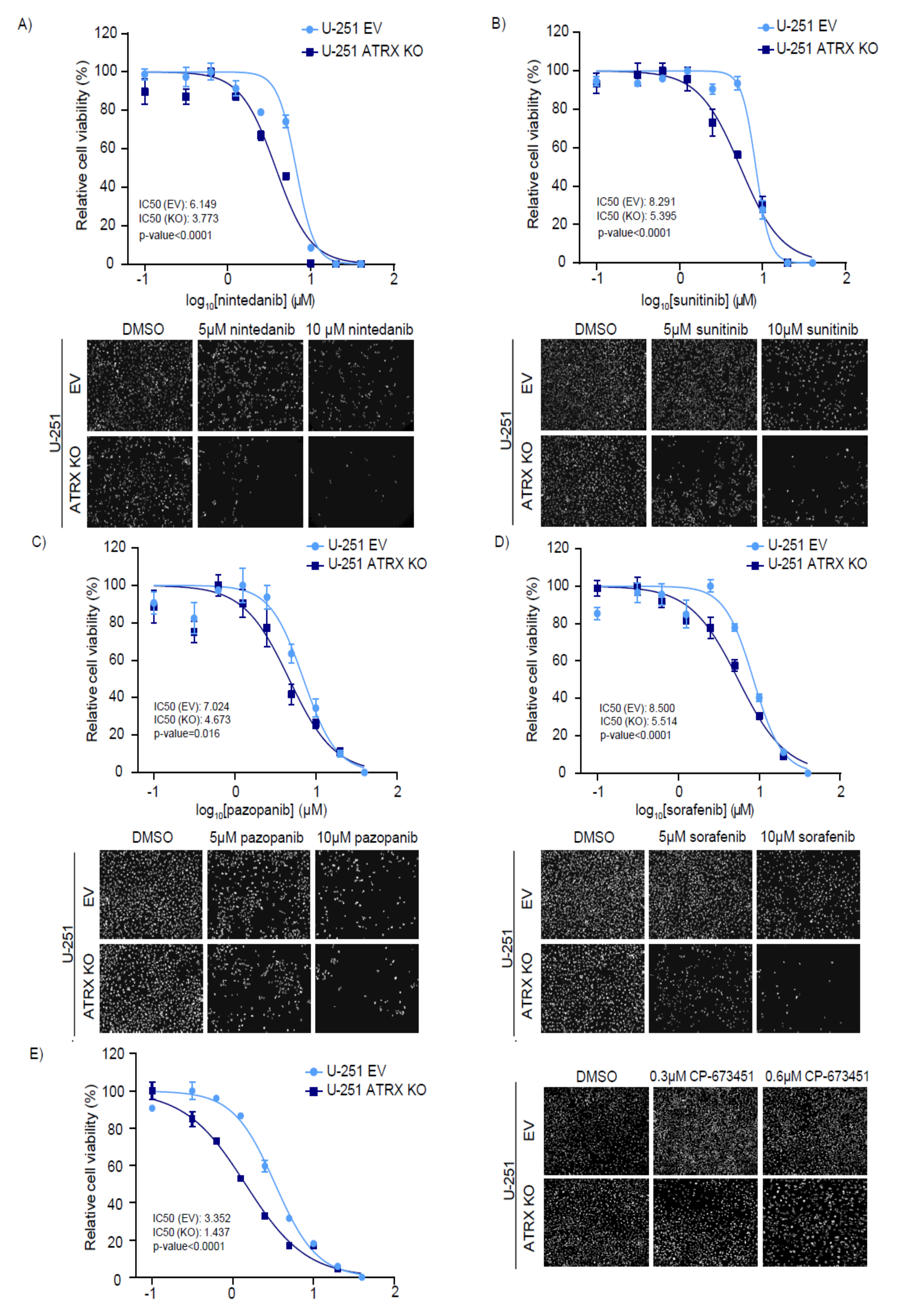
3.3. ATRX KO High-Grade Glioma Cells Are Sensitive to RTK and PDGFR Inhibitors
3.4. ATRX KO High-Grade Glioma Cells Are Sensitive to Combinatorial Treatments of TMZ and PDGFRi
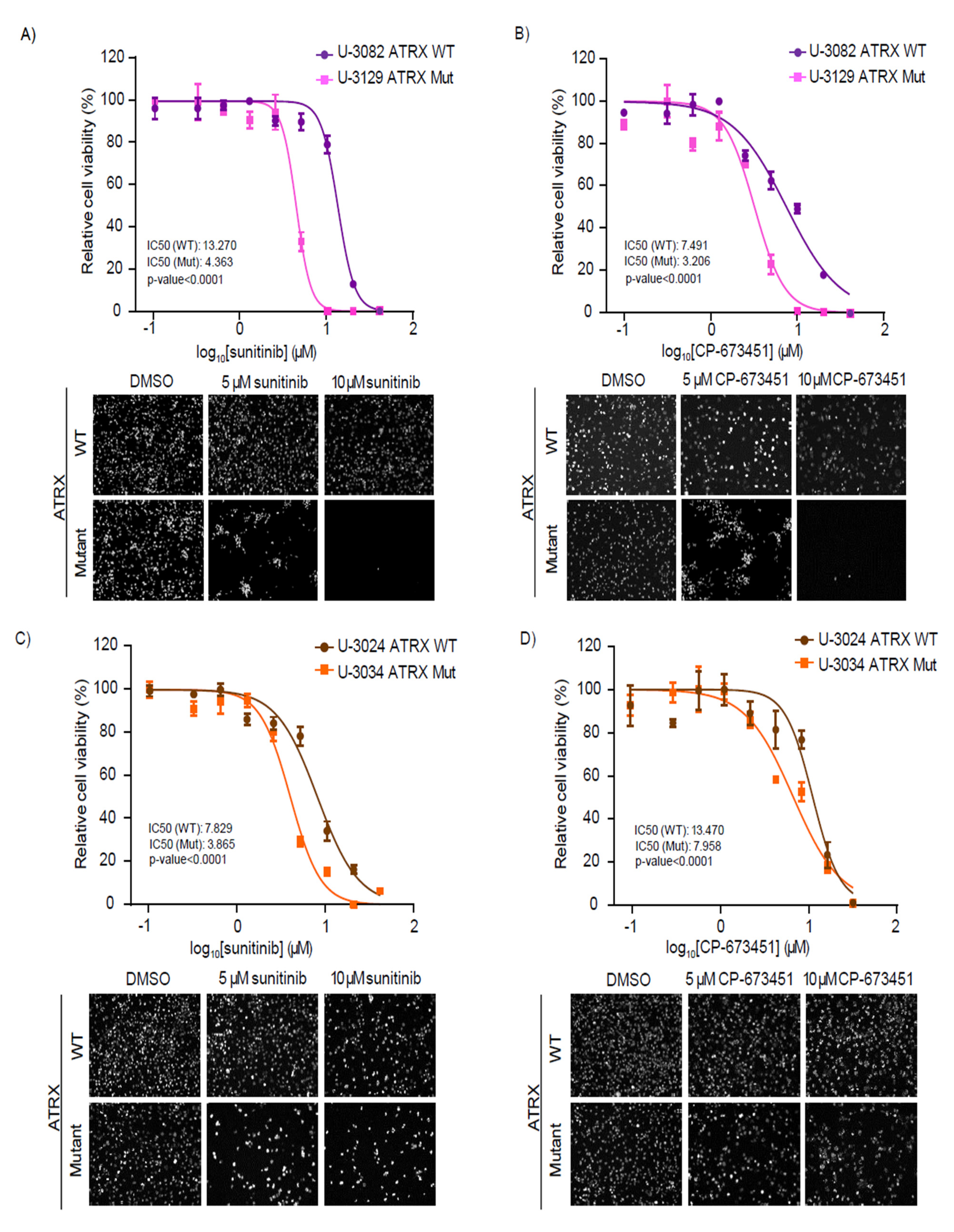
3.5. Patient-Derived GBM Cells with ATRX Mutations Are Sensitive to RTK and PDGFR Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
| ALT | Alternative Lengthening of Telomeres |
| ATRX | alpha thalassemia mental retardation X-linked |
| BTK | Bruton’s tyrosine kinase |
| BTKi | inhibitors targeting BTK |
| CFS | common fragile sites |
| DAXX | death domain-associated protein 6 |
| DMSO | dimethyl sulfoxide |
| DSB | double-strand break |
| EdU | 5-Ethynyl-2′-deoxyuridine |
| EV | empty vector |
| FDA | food and drug administration |
| GBM | glioblastoma multiforme |
| GFP | green fluorescent protein |
| gRNA | guide RNA |
| H3.3 | histone H3.3 |
| HGCC | human glioblastoma cell culture |
| IDH1 | isocitrate dehydrogenase 1 |
| KO | knockout |
| PDGFR | platelet-derived growth factor receptor |
| PDGFRi | inhibitors targeting PDGFR |
| RTK | receptor tyrosine kinase |
| RTKi | inhibitors targeting RTK |
| shRNA | short hairpin RNA |
| STAT3 | signal transducer and activator of transcription 3 |
| STAT3i | inhibitors targeting STAT3 |
| TMZ | temozolomide |
| TP53 | tumor protein 53 |
| WT | wild-type |
| γH2AX | histone H2AX phosphorylation |
References
- Gibbons, R.; Picketts, D.; Villard, L.; Higgs, D. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell 1995, 80, 837–845. [Google Scholar] [CrossRef] [Green Version]
- Dyer, M.; Qadeer, Z.; Valle-Garcia, D.; Bernstein, E. ATRX and DAXX: Mechanisms and Mutations. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.; Banaszynski, L.; Noh, K.; Lewis, P.; Elsaesser, S.; Stadler, S.; Dewell, S.; Law, M.; Guo, X.; Li, X.; et al. Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 2010, 140, 678–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, P.W.; Elsaesser, S.J.; Noh, K.-M.; Stadler, S.C.; Allis, C.D. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 2010, 107, 14075–14080. [Google Scholar] [CrossRef] [Green Version]
- Juhász, S.; Elbakry, A.; Mathes, A.; Löbrich, M. ATRX Promotes DNA Repair Synthesis and Sister Chromatid Exchange during Homologous Recombination. Mol. Cell 2018, 71, 11.e7–24.e7. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.T.; Voon, H.P.J.; Xella, B.; Scott, C.; Clynes, D.; Babbs, C.; Ayyub, H.; Kerry, J.; Sharpe, J.A.; Sloane-Stanley, J.A.; et al. The chromatin remodelling factor ATRX suppresses R-loops in transcribed telomeric repeats. EMBO Rep. 2017, 18, 914–928. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Wild, A.T.; Wu, W.H.; Shah, R.; Danussi, C.; Riggins, G.J.; Kannan, K.; Sulman, E.P.; Chan, T.A.; et al. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun. 2019, 10, 943. [Google Scholar] [CrossRef] [Green Version]
- Pladevall-Morera, D.; Munk, S.; Ingham, A.; Garribba, L.; Albers, E.; Liu, Y.; Olsen, J.V.; Lopez-Contreras, A.J. Proteomic characterization of chromosomal common fragile site (CFS)-associated proteins uncovers ATRX as a regulator of CFS stability. Nucleic Acids Res. 2019, 47, 8004–8018. [Google Scholar] [CrossRef] [Green Version]
- Amorim, J.P.; Santos, G.; Vinagre, J.; Soares, P. The role of ATRX in the alternative lengthening of telomeres (ALT) phenotype. Genes. 2016, 7, 66. [Google Scholar] [CrossRef]
- Brosnan-Cashman, J.A.; Yuan, M.; Graham, M.K.; Rizzo, A.J.; Myers, K.M.; Davis, C.; Zhang, R.; Esopi, D.M.; Raabe, E.H.; Eberhart, C.G.; et al. ATRX loss induces multiple hallmarks of the alternative lengthening of telomeres (ALT) phenotype in human glioma cell lines in a cell line-specific manner. PLoS ONE 2018, 13, e0204159. [Google Scholar] [CrossRef] [Green Version]
- Kovatcheva, M.; Liao, W.; Klein, M.E.; Robine, N.; Geiger, H.; Crago, A.M.; Dickson, M.A.; Tap, W.D.; Singer, S.; Koff, A. ATRX is a regulator of therapy induced senescence in human cells. Nat. Commun. 2017, 8, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [Green Version]
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science 2011, 333, 425. [Google Scholar] [CrossRef] [Green Version]
- Haase, S.; Garcia-Fabiani, M.B.; Carney, S.; Altshuler, D.; Núñez, F.J.; Méndez, F.M.; Núñez, F.; Lowenstein, P.R.; Castro, M.G. Mutant ATRX: Uncovering a new therapeutic target for glioma. Expert Opin. Ther. Targets 2018, 22, 599–613. [Google Scholar] [CrossRef]
- Abedalthagafi, M.; Phillips, J.; Kim, G.; Mueller, S.; Haas-Kogen, D.; Marshall, R.; Croul, S.; Santi, M.; Cheng, J.; Zhou, S.; et al. The alternative lengthening of telomere phenotype is significantly associated with loss of ATRX expression in high-grade pediatric and adult astrocytomas: A multi-institutional study of 214 astrocytomas. Mod. Pathol. 2013, 26, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Purkait, S.; Miller, C.A.; Kumar, A.; Sharma, V.; Pathak, P.; Jha, P.; Sharma, M.C.; Suri, V.; Suri, A.; Sharma, B.S.; et al. ATRX in Diffuse Gliomas With its Mosaic/Heterogeneous Expression in a Subset. Brain Pathol. 2017, 27, 138–145. [Google Scholar] [CrossRef]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.A.; Denton, E.L.; Arrowsmith, C.H.; Lupien, M.; Schapira, M. A global assessment of cancer genomic alterations in epigenetic mechanisms. Epigenetics Chromatin 2014, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Zeineldin, M.; Federico, S.; Chen, X.; Fan, Y.; Xu, B.; Stewart, E.; Zhou, X.; Jeon, J.; Griffiths, L.; Nguyen, R.; et al. MYCN amplification and ATRX mutations are incompatible in neuroblastoma. Nat. Commun. 2020, 11, 913. [Google Scholar] [CrossRef] [Green Version]
- Schneider, T.; Mawrin, C.; Scherlach, C.; Skalej, M.; Firsching, R. Gliomas in Adults. Dtsch. Arztebl. Int. 2010, 107, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2009, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Zhao, H.; Diplas, B.H.; Liu, S.; Liu, J.; Wang, D.; Lu, Y.; Zhu, Q.; Wu, J.; Wang, W.; et al. Genome-Wide CRISPR-Cas9 Screen Reveals Selective Vulnerability of ATRX-Mutant Cancers to WEE1 Inhibition. Cancer Res. 2020, 80, 510–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Taylor, S.; Mitson, M.; Bachrati, C.Z.; Higgs, D.R.; Gibbons, R.J. ATRX Dysfunction Induces Replication Defects in Primary Mouse Cells. PLoS ONE 2014, 9, e92915. [Google Scholar] [CrossRef] [Green Version]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef]
- Wang, J.; Liu, X.; Hong, Y.; Wang, S.; Chen, P.; Gu, A.; Guo, X.; Zhao, P. Ibrutinib, a Bruton’s tyrosine kinase inhibitor, exhibits antitumoral activity and induces autophagy in glioblastoma. J. Exp. Clin. Cancer Res. 2017, 36, 96. [Google Scholar] [CrossRef] [Green Version]
- Wieland, A.; Trageser, D.; Gogolok, S.; Reinartz, R.; Höfer, H.; Keller, M.; Leinhaas, A.; Schelle, R.; Normann, S.; Klaas, L.; et al. Anticancer Effects of Niclosamide in Human Glioblastoma. Clin. Cancer Res. 2013, 19, 4124–4136. [Google Scholar] [CrossRef] [Green Version]
- Reardon, D.A.; Groves, M.D.; Wen, P.Y.; Nabors, L.; Mikkelsen, T.; Rosenfeld, S.; Raizer, J.; Barriuso, J.; McLendon, R.E.; Suttle, A.B.; et al. A Phase I/II Trial of Pazopanib in Combination with Lapatinib in Adult Patients with Relapsed Malignant Glioma. Clin. Cancer Res. 2013, 19, 900–908. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.; Morales, L.D.; Zhang, Y.; Mito, S.; Tsin, A. Niclosamide induces protein ubiquitination and inhibits multiple pro-survival signaling pathways in the human glioblastoma U-87 MG cell line. PLoS ONE 2017, 12, e0184324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hojjat-Farsangi, M. Small-Molecule Inhibitors of the Receptor Tyrosine Kinases: Promising Tools for Targeted Cancer Therapies. Int. J. Mol. Sci. 2014, 15, 13768–13801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Cai, J.; Gao, W.; Meng, X.; Gao, F.; Wu, P.; Duan, C.; Wang, R.; Dinislam, M.; Lin, L.; et al. Loss of ATRX suppresses ATM dependent DNA damage repair by modulating H3K9me3 to enhance temozolomide sensitivity in glioma. Cancer Lett. 2018, 419, 280–290. [Google Scholar] [CrossRef]
- Xie, Y.; Bergström, T.; Jiang, Y.; Johansson, P.; Marinescu, V.D.; Lindberg, N.; Segerman, A.; Wicher, G.; Niklasson, M.; Baskaran, S.; et al. The Human Glioblastoma Cell Culture Resource: Validated Cell Models Representing All Molecular Subtypes. EBioMedicine 2015, 2, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, B.A.; Heaphy, C.M.; de Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Obashinjo, S.M.; Marie, S.K.N.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, N.; Maier, D.; Merlo, A.; Tada, M.; Sawamura, Y.; Diserens, A.-C.; Meir, E.G. Van Frequent Co-Alterations of TP53, p16/CDKN2A, p14ARF, PTEN Tumor Suppressor Genes in Human Glioma Cell Lines. Brain Pathol. 1999, 9, 469–479. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, P.M.; Jackman, J.; Bae, I.; Myers, T.G.; Fan, S.; Mutoh, M.; Scudiero, D.A.; Monks, A.; Sausville, E.A.; Weinstein, J.N.; et al. Characterization of the p53 Tumor Suppressor Pathway in Cell Lines of the National Cancer Institute Anticancer Drug Screen and Correlations with the Growth-Inhibitory Potency of 123 Anticancer Agents. Cancer Res. 1997, 57, 4285–4300. [Google Scholar]
- Levitzki, A. PDGF receptor kinase inhibitors for the treatment of PDGF driven diseases. Cytokine Growth Factor Rev. 2004, 15, 229–235. [Google Scholar] [CrossRef]
- George, S.L.; Lorenzi, F.; King, D.; Hartlieb, S.; Campbell, J.; Pemberton, H.; Toprak, U.H.; Barker, K.; Tall, J.; da Costa, B.M.; et al. Therapeutic vulnerabilities in the DNA damage response for the treatment of ATRX mutant neuroblastoma. EBioMedicine 2020, 59, 102971. [Google Scholar] [CrossRef]
- Reardon, D.A.; Vredenburgh, J.J.; Desjardins, A.; Peters, K.; Gururangan, S.; Sampson, J.H.; Marcello, J.; Herndon, J.E.; McLendon, R.E.; Janney, D.; et al. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: Results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J. Neurooncol. 2011, 101, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Hainsworth, J.D.; Ervin, T.; Friedman, E.; Priego, V.; Murphy, P.B.; Clark, B.L.; Lamar, R.E. Concurrent radiotherapy and temozolomide followed by temozolomide and sorafenib in the first-line treatment of patients with glioblastoma multiforme. Cancer 2010, 116, 3663–3669. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Krona, C.; Kundu, S.; Doroszko, M.; Baskaran, S.; Schmidt, L.; Vinel, C.; Almstedt, E.; Elgendy, R.; Elfineh, L.; et al. A Patient-Derived Cell Atlas Informs Precision Targeting of Glioblastoma. Cell Rep. 2020, 32, 107897. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-Y.; Gerges, N.; Korshunov, A.; Sabha, N.; Khuong-Quang, D.-A.; Fontebasso, A.M.; Fleming, A.; Hadjadj, D.; Schwartzentruber, J.; Majewski, J.; et al. Frequent ATRX mutations and loss of expression in adult diffuse astrocytic tumors carrying IDH1/IDH2 and TP53 mutations. Acta Neuropathol. 2012, 124, 615–625. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pladevall-Morera, D.; Castejón-Griñán, M.; Aguilera, P.; Gaardahl, K.; Ingham, A.; Brosnan-Cashman, J.A.; Meeker, A.K.; Lopez-Contreras, A.J. ATRX-Deficient High-Grade Glioma Cells Exhibit Increased Sensitivity to RTK and PDGFR Inhibitors. Cancers 2022, 14, 1790. https://doi.org/10.3390/cancers14071790
Pladevall-Morera D, Castejón-Griñán M, Aguilera P, Gaardahl K, Ingham A, Brosnan-Cashman JA, Meeker AK, Lopez-Contreras AJ. ATRX-Deficient High-Grade Glioma Cells Exhibit Increased Sensitivity to RTK and PDGFR Inhibitors. Cancers. 2022; 14(7):1790. https://doi.org/10.3390/cancers14071790
Chicago/Turabian StylePladevall-Morera, David, María Castejón-Griñán, Paula Aguilera, Karina Gaardahl, Andreas Ingham, Jacqueline A. Brosnan-Cashman, Alan K. Meeker, and Andres J. Lopez-Contreras. 2022. "ATRX-Deficient High-Grade Glioma Cells Exhibit Increased Sensitivity to RTK and PDGFR Inhibitors" Cancers 14, no. 7: 1790. https://doi.org/10.3390/cancers14071790
APA StylePladevall-Morera, D., Castejón-Griñán, M., Aguilera, P., Gaardahl, K., Ingham, A., Brosnan-Cashman, J. A., Meeker, A. K., & Lopez-Contreras, A. J. (2022). ATRX-Deficient High-Grade Glioma Cells Exhibit Increased Sensitivity to RTK and PDGFR Inhibitors. Cancers, 14(7), 1790. https://doi.org/10.3390/cancers14071790