
Deciphering Genetic Alterations of Hairy Cell Leukemia and Hairy Cell Leukemia-like Disorders in 98 Patients

 , , , , , , ,
, , , , , , ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Samples
2.2. Next Generation Sequencing (NGS)
2.3. Statistical Analysis and Data Representation
3. Results
3.1. Characteristics of the 98 Patients
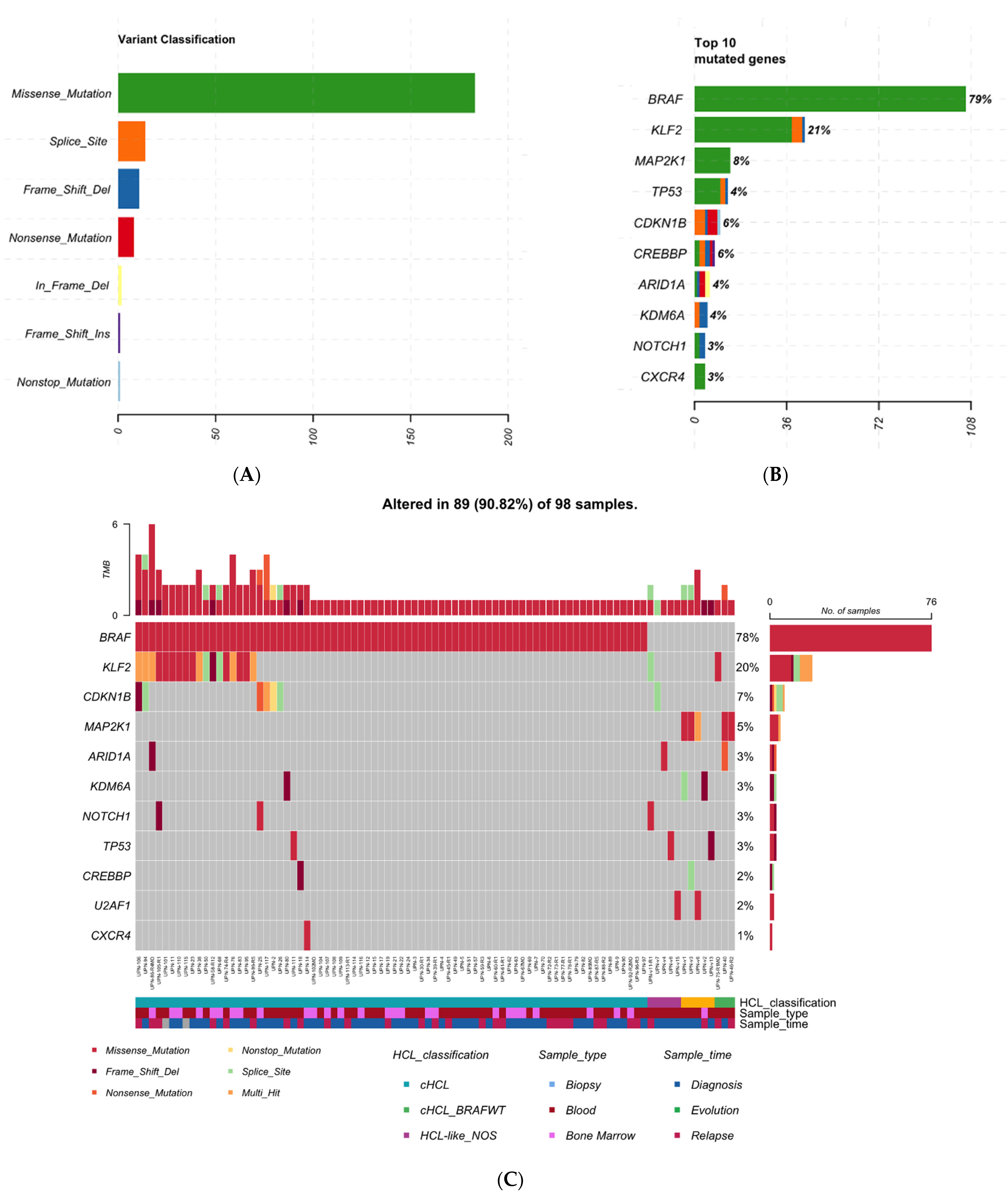
3.2. cHCL Has a Specific Molecular Signature
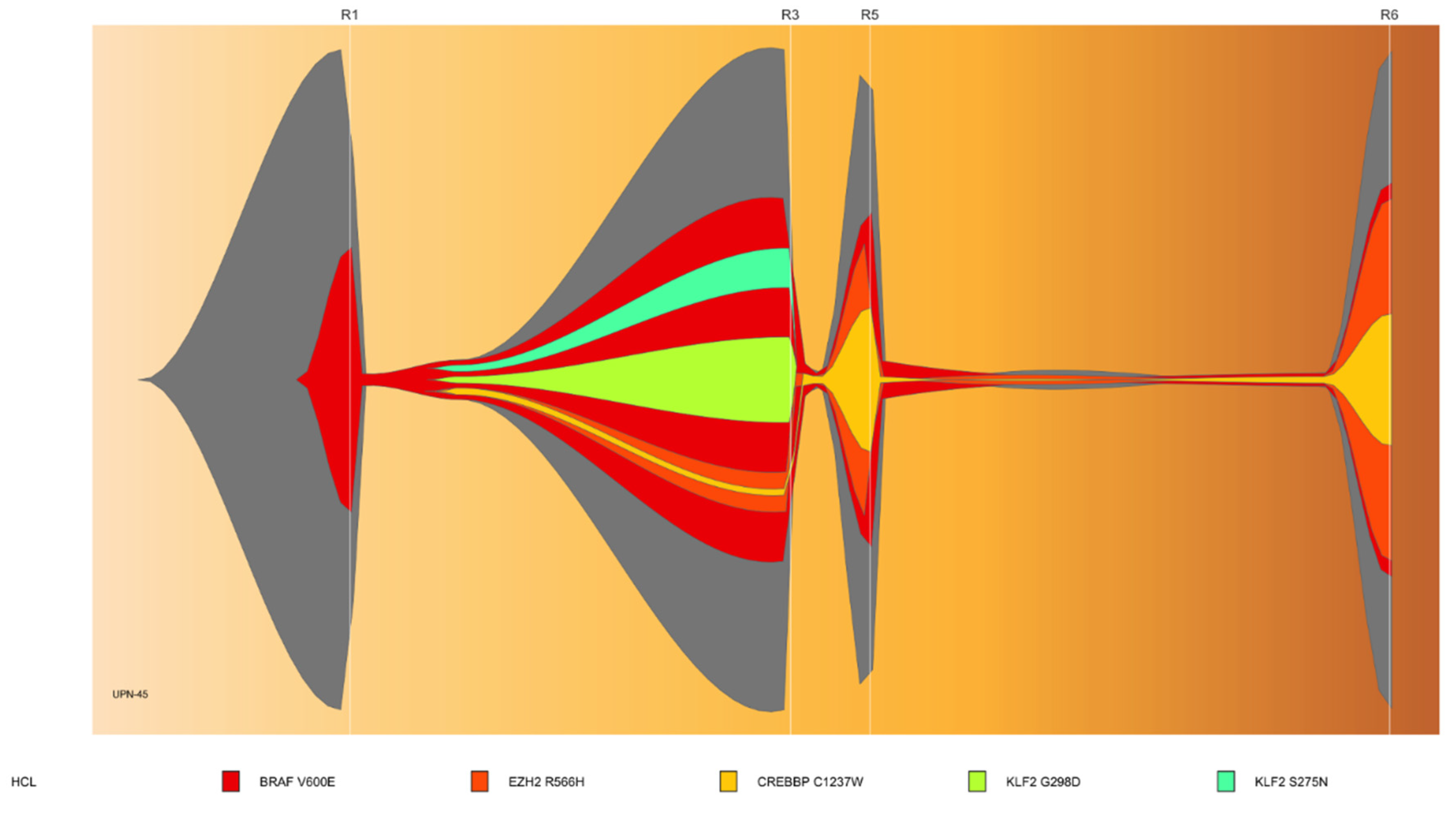
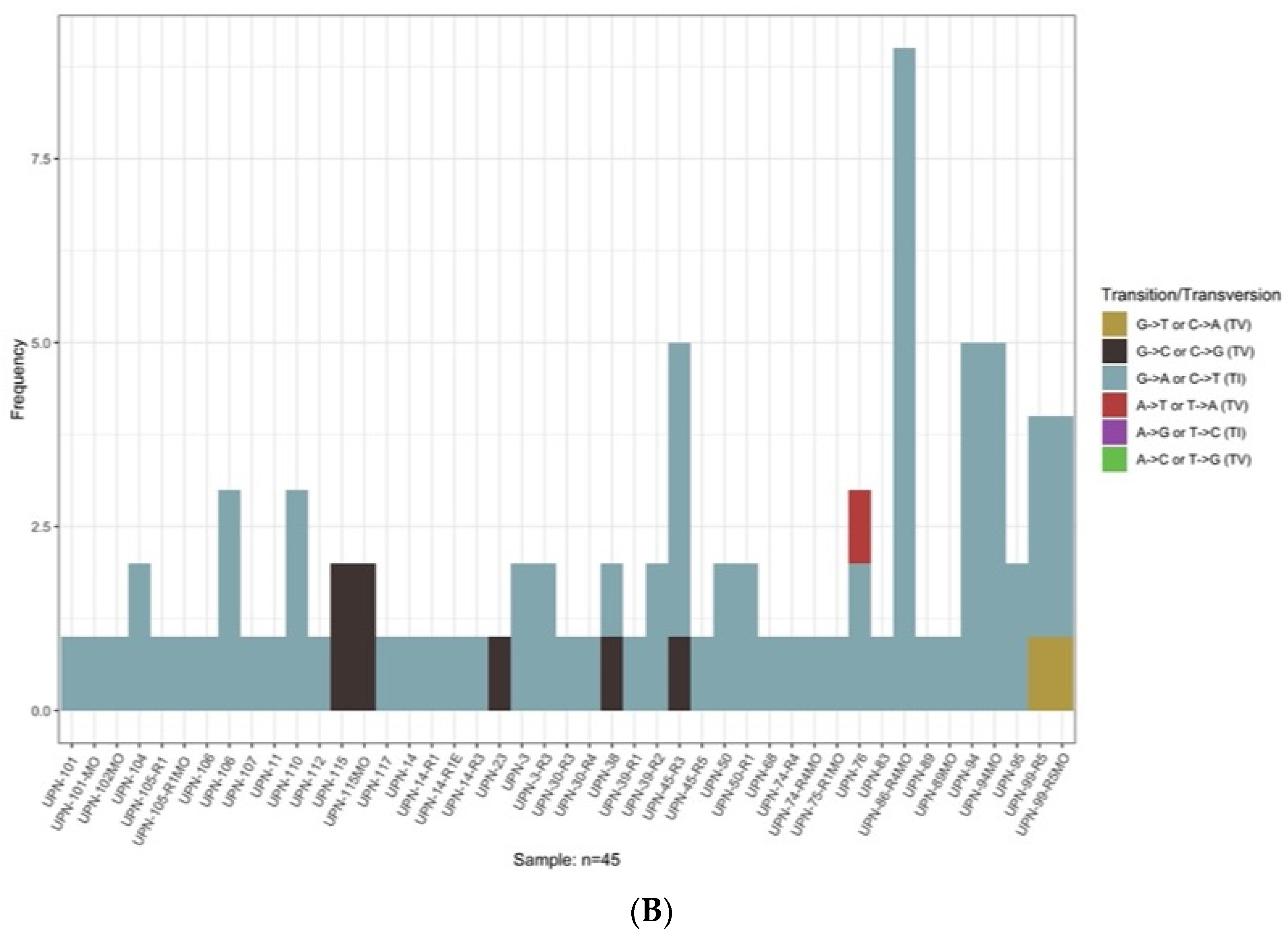
3.3. Sub-Clonal Mutations and Variant Allele Frequency Changes Were Found at Relapse
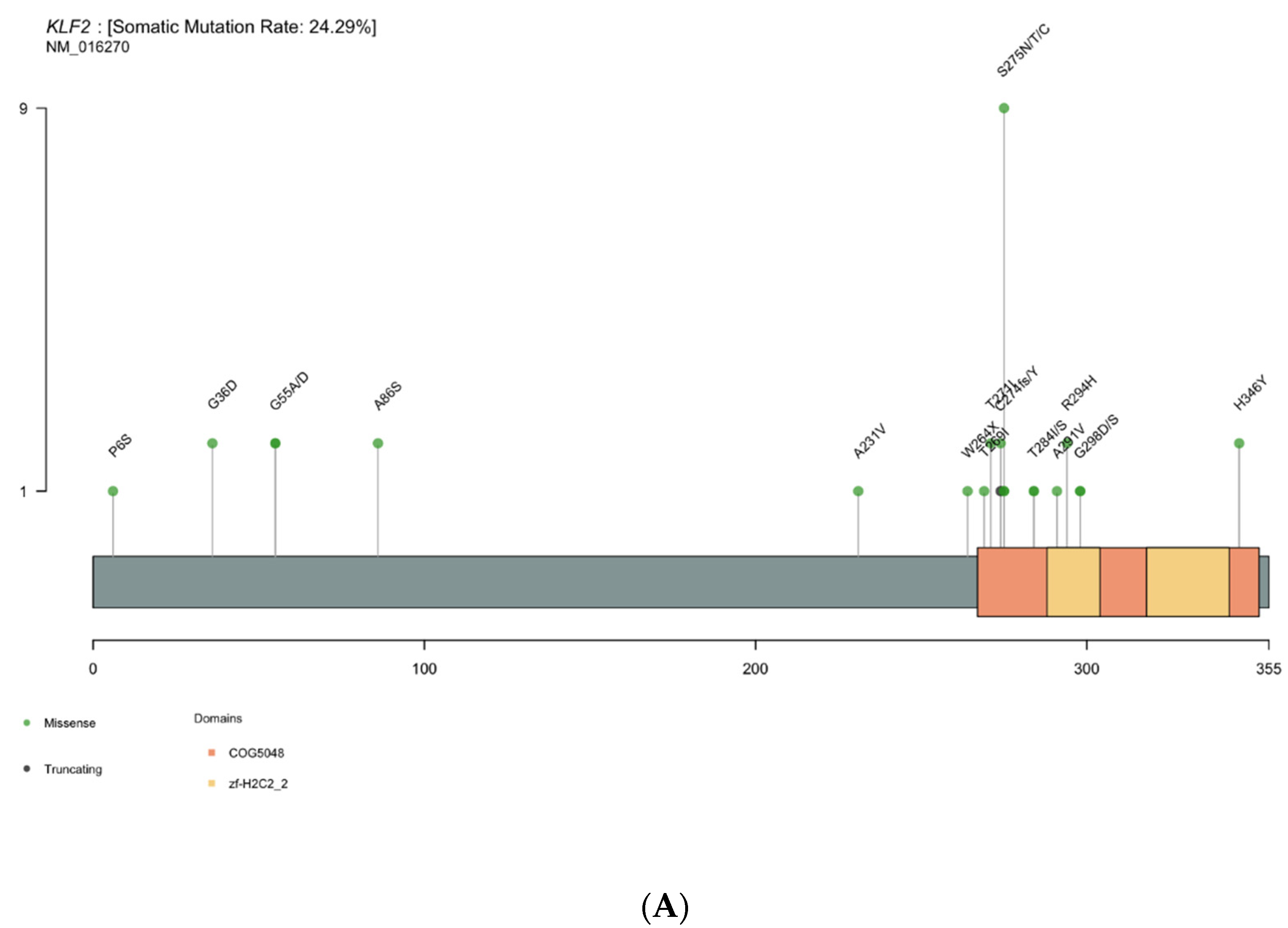
3.4. KLF2 Is a Gene of Interest
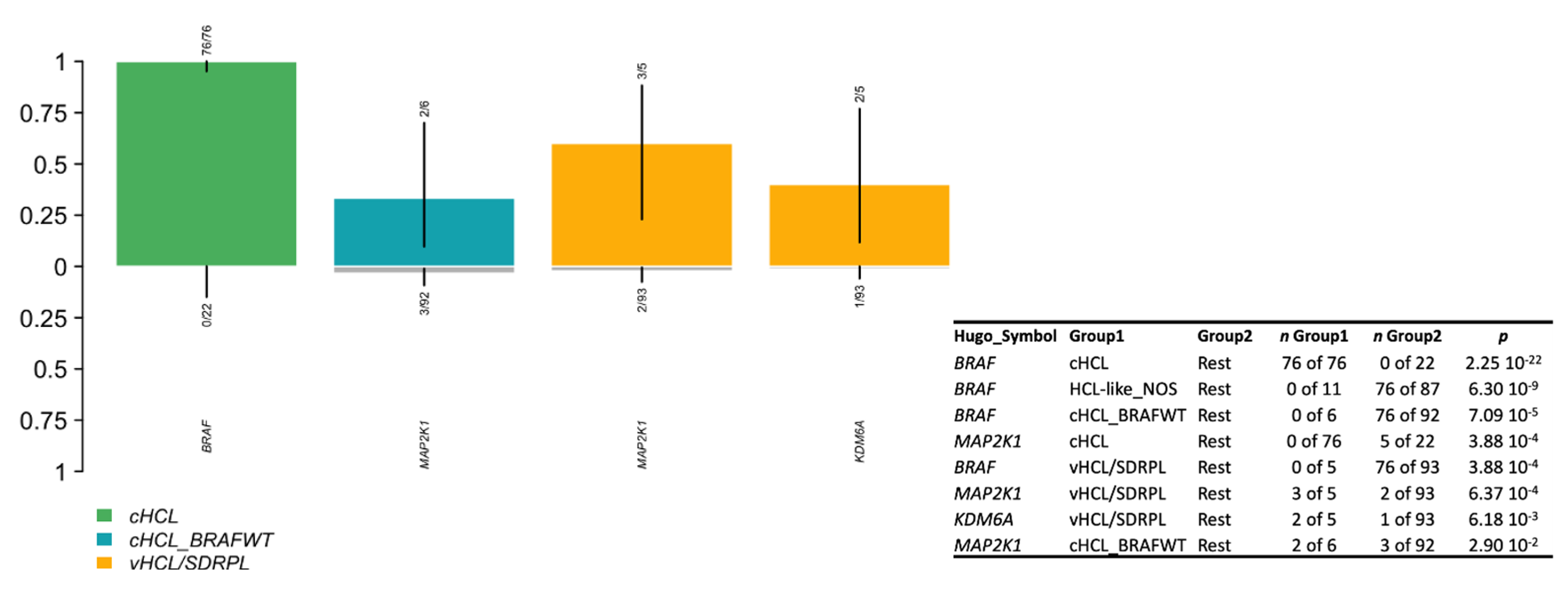
3.5. Genomic Signature in HCL-like Patients
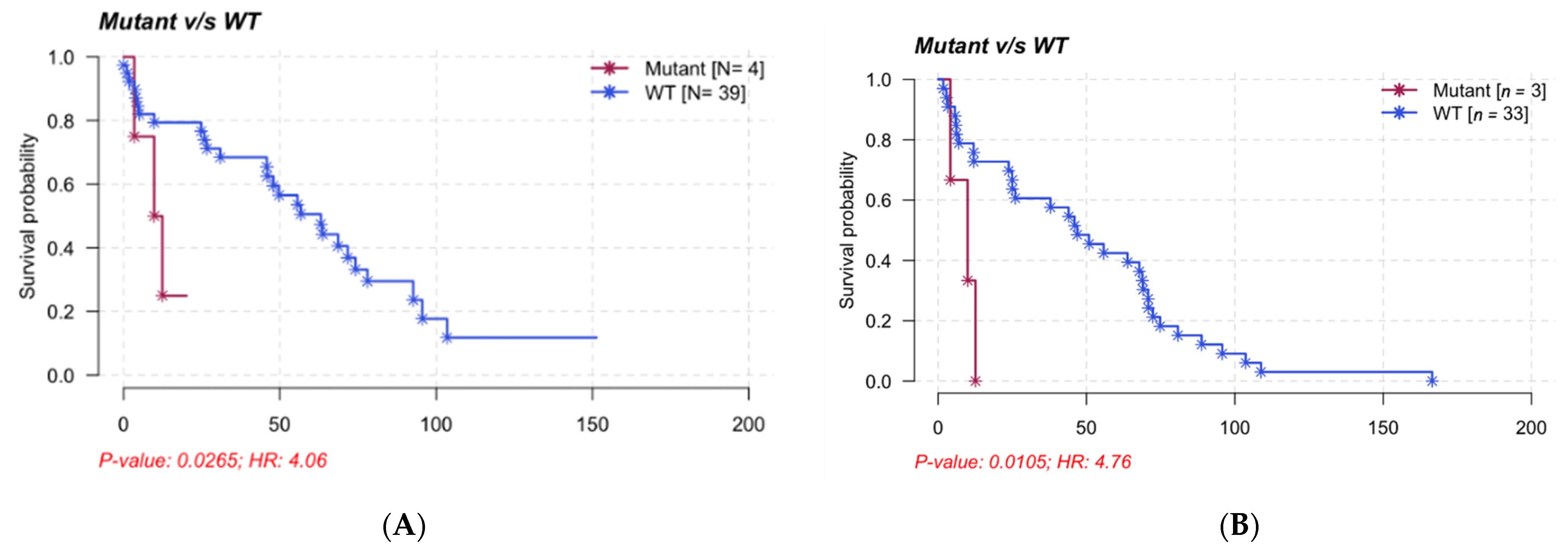
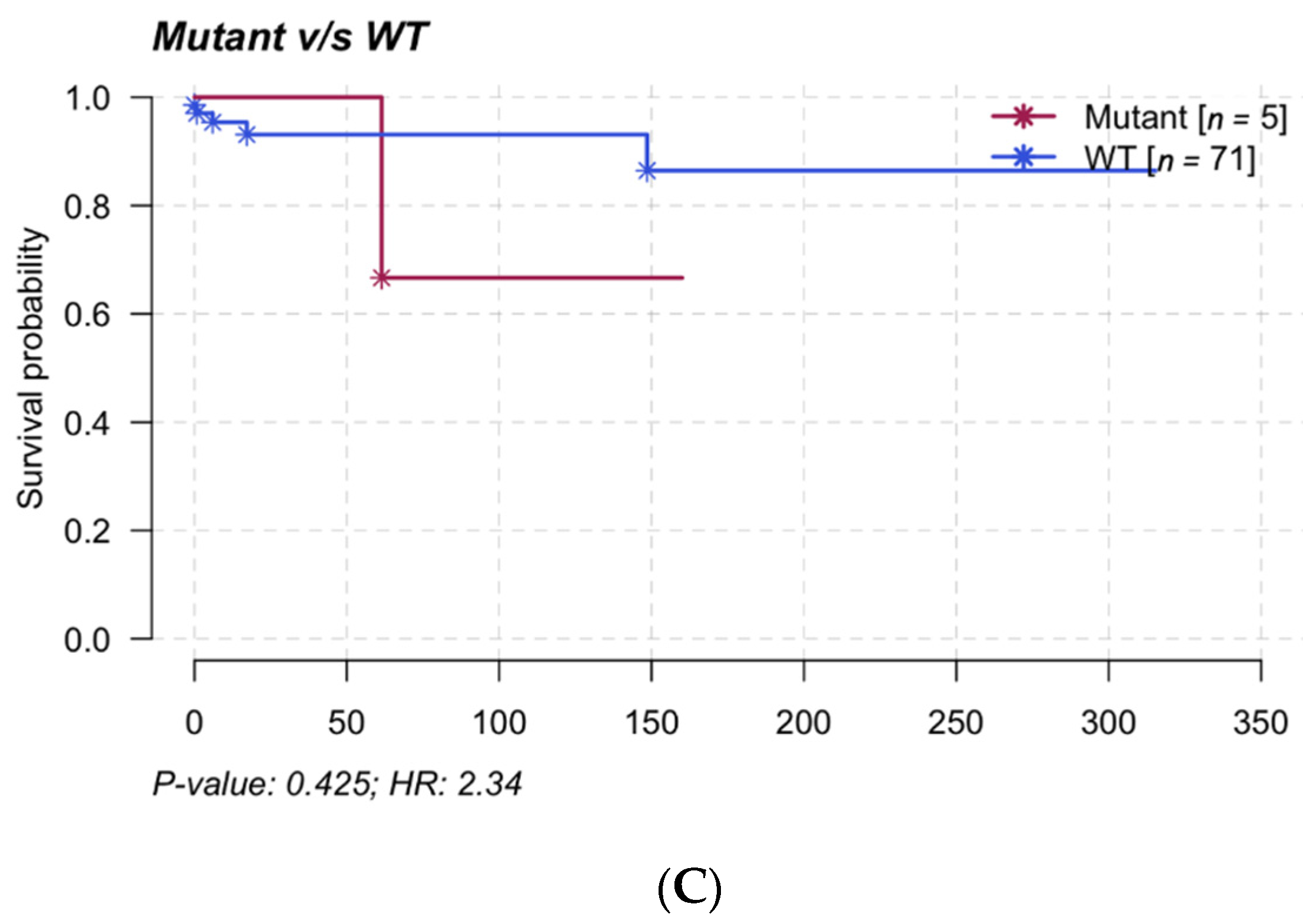
3.6. Clinical Impact and Prognosis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Teras, L.R.; DeSantis, C.E.; Cerhan, J.R.; Morton, L.M.; Jemal, A.; Flowers, C.R. 2016 US lymphoid malignancy statistics by World Health Organization subtypes: 2016 US Lymphoid Malignancy Statistics by World Health Organization Subtypes. CA Cancer J. Clin. 2016, 66, 443–459. [Google Scholar] [CrossRef] [PubMed]
- Troussard, X. Epidemiology of Hairy Cell Leukemia (HCL) and HCL-Like Disorders. Med. Res. Arch. 2020, 8, 1–11. [Google Scholar] [CrossRef]
- Matutes, E.; Morilla, R.; Owusu-Ankomah, K.; Houliham, H.; Meeus, P.; Catovsky, D. The immunophenotype of hairy cell leukemia (HCL). Proposal for a scoring system to distinguish HCL from B-cell disorders with hairy or villous lymphocytes. Leuk. Lymphoma 1994, 14, 57–61. [Google Scholar] [PubMed]
- Xi, L.; Arons, E.; Navarro, W.; Calvo, K.R.; Stetler-Stevenson, M.; Raffeld, M.; Kreitman, R.J. Both variant and IGHV4-34–expressing hairy cell leukemia lack the BRAF V600E mutation. Blood 2012, 119, 3330–3332. [Google Scholar] [CrossRef] [Green Version]
- Forconi, F.; Sozzi, E.; Cencini, E.; Zaja, F.; Intermesoli, T.; Stelitano, C.; Rigacci, L.; Gherlinzoni, F.; Cantaffa, R.; Baraldi, A.; et al. Hairy cell leukemias with unmutated IGHV genes define the minor subset refractory to single-agent cladribine and with more aggressive behavior. Blood 2009, 114, 4696–4702. [Google Scholar] [CrossRef]
- Swerdlow, S.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2017; ISBN 978-92-832-4494-3. [Google Scholar]
- Curiel-Olmo, S.; Mondéjar, R.; Almaraz, C.; Mollejo, M.; Cereceda, L.; Marès, R.; Derdak, S.; Campos-Martín, Y.; Batlle, A.; González de Villambrosía, S.; et al. Splenic diffuse red pulp small B-cell lymphoma displays increased expression of cyclin D3 and recurrent CCND3 mutations. Blood 2017, 129, 1042–1045. [Google Scholar] [CrossRef] [Green Version]
- Jallades, L.; Baseggio, L.; Sujobert, P.; Huet, S.; Chabane, K.; Callet-Bauchu, E.; Verney, A.; Hayette, S.; Desvignes, J.-P.; Salgado, D.; et al. Exome sequencing identifies recurrent BCOR gene alterations and the absence of KLF2, TNFAIP3 and MYD88 mutations in splenic diffuse red pulp small B-cell lymphoma. Haematologica 2017, 102, 1758–1766. [Google Scholar] [CrossRef] [Green Version]
- Mason, E.F.; Brown, R.D.; Szeto, D.P.; Gibson, C.J.; Jia, Y.; Garcia, E.P.; Jacobson, C.A.; Dal Cin, P.; Kuo, F.C.; Pinkus, G.S.; et al. Detection of activating MAP2K1 mutations in atypical hairy cell leukemia and hairy cell leukemia variant. Leuk. Lymphoma 2016, 58, 233–236. [Google Scholar] [CrossRef]
- Waterfall, J.J.; Arons, E.; Walker, R.L.; Pineda, M.; Roth, L.; Killian, J.K.; Abaan, O.D.; Davis, S.R.; Kreitman, R.J.; Meltzer, P.S. High prevalence of MAP2K1 mutations in variant and IGHV4-34–expressing hairy-cell leukemias. Nat. Genet. 2013, 46, 8–10. [Google Scholar] [CrossRef]
- Arons, E.; Kreitman, R.J. Molecular variant of hairy cell leukemia with poor prognosis. Leuk. Lymphoma 2011, 52, 99–102. [Google Scholar] [CrossRef]
- Hockley, S.L.; Giannouli, S.; Morilla, A.; Wotherspoon, A.; Morgan, G.J.; Matutes, E.; Gonzalez, D. Insight into the molecular pathogenesis of hairy cell leukaemia, hairy cell leukaemia variant and splenic marginal zone lymphoma, provided by the analysis of their IGH rearrangements and somatic hypermutation patterns. Br. J. Haematol. 2010, 148, 665–666. [Google Scholar] [CrossRef] [PubMed]
- Hockley, S.L.; Morgan, G.J.; Leone, P.E.; Walker, B.A.; Morilla, A.; Else, M.; Wotherspoon, A.; Dearden, C.; Catovsky, D.; Gonzalez, D.; et al. High-resolution genomic profiling in hairy cell leukemia-variant compared with typical hairy cell leukemia. Leukemia 2011, 25, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Matutes, E.; Martínez-Trillos, A.; Campo, E. Hairy cell leukaemia-variant: Disease features and treatment. Best Pract. Res. Clin. Haematol. 2015, 28, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Traverse-Glehen, A.; Baseggio, L.; Callet-Bauchu, E.; Morel, D.; Gazzo, S.; Ffrench, M.; Verney, A.; Rolland, D.; Thieblemont, C.; Magaud, J.-P.; et al. Splenic red pulp lymphoma with numerous basophilic villous lymphocytes: A distinct clinicopathologic and molecular entity? Blood 2008, 111, 2253–2260. [Google Scholar] [CrossRef] [Green Version]
- Baseggio, L.; Traverse-Glehen, A.; Callet-Bauchu, E.; Morel, D.; Magaud, J.P.; Berger, F.; Salles, G.; Felman, P. Relevance of a scoring system including CD11c expression in the identification of splenic diffuse red pulp small B-cell lymphoma (SRPL). Hematol. Oncol. 2011, 29, 47–51. [Google Scholar] [CrossRef]
- Favre, R.; Manzoni, D.; Traverse-Glehen, A.; Verney, A.; Jallades, L.; Callet-Bauchu, E.; Sujobert, P.; Salles, G.; Baseggio, L. Usefulness of CD200 in the differential diagnosis of SDRPL, SMZL, and HCL. Int. J. Lab. Hematol. 2018, 40, e59. [Google Scholar] [CrossRef]
- Kasar, S.; Brown, J.R. Mutational landscape and underlying mutational processes in chronic lymphocytic leukemia. Mol. Cell. Oncol. 2016, 3, e1157667. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Maitre, E.; Bertrand, P.; Maingonnat, C.; Viailly, P.-J.; Wiber, M.; Naguib, D.; Salaün, V.; Cornet, E.; Damaj, G.; Sola, B.; et al. New generation sequencing of targeted genes in the classical and the variant form of hairy cell leukemia highlights mutations in epigenetic regulation genes. Oncotarget 2018, 9, 11. [Google Scholar] [CrossRef] [Green Version]
- Tiacci, E.; Trifonov, V.; Schiavoni, G.; Holmes, A.; Kern, W.; Martelli, M.P.; Pucciarini, A.; Bigerna, B.; Pacini, R.; Wells, V.A.; et al. BRAF mutations in hairy-cell leukemia. N. Engl. J. Med. 2011, 364, 2305–2315. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, S.; Hüllein, J.; Lee, S.C.-W.; Hutter, B.; Gonzalez, D.; Jayne, S.; Dyer, M.J.; Oleś, M.; Else, M.; Liu, X.; et al. Recurrent CDKN1B (p27) mutations in hairy cell leukemia. Blood 2015, 126, 1005–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weston-Bell, N.J.; Tapper, W.; Gibson, J.; Bryant, D.; Moreno, Y.; John, M.; Ennis, S.; Kluin-Nelemans, H.C.; Collins, A.R.; Sahota, S.S. Exome Sequencing in Classic Hairy Cell Leukaemia Reveals Widespread Variation in Acquired Somatic Mutations between Individual Tumours Apart from the Signature BRAF V(600)E Lesion. PLoS ONE 2016, 11, e0149162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.M.; Datto, M.; Duncavage, E.J.; Kulkarni, S.; Lindeman, N.I.; Roy, S.; Tsimberidou, A.M.; Vnencak-Jones, C.L.; Wolff, D.J.; Younes, A.; et al. Standards and Guidelines for the Interpretation and Reporting of Sequence Variants in Cancer. J. Mol. Diagn. 2017, 19, 4–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [Green Version]
- Skidmore, Z.L.; Wagner, A.H.; Lesurf, R.; Campbell, K.M.; Kunisaki, J.; Griffith, O.L.; Griffith, M. GenVisR: Genomic Visualizations in R. Bioinformatics 2016, 32, 3012–3014. [Google Scholar] [CrossRef] [Green Version]
- Tschernitz, S.; Flossbach, L.; Bonengel, M.; Roth, S.; Rosenwald, A.; Geissinger, E. Alternative BRAF mutations in BRAF V600E-negative hairy cell leukaemias. Br. J. Haematol. 2014, 165, 529–533. [Google Scholar] [CrossRef]
- Thompson, E.R.; Lim, K.J.C.; Kuzich, J.A.; McBean, M.; Westerman, D.; Tam, C.S.; Blombery, P. Detection of an IGH- BRAF fusion in a patient with BRAF Val600Glu negative hairy cell leukemia. Leuk. Lymphoma 2020, 61, 2024–2026. [Google Scholar] [CrossRef]
- Gozzetti, A.; Sammartano, V.; Bacchiarri, F.; Raspadori, D.; Bocchia, M. A BRAF-Negative Classic Hairy Cell Leukemia Patient with LongLasting Complete Remission after Rituximab and Pentostatin. TJH 2020, 37, 286–287. [Google Scholar] [CrossRef]
- Chung, S.S.; Kim, E.; Park, J.H.; Chung, Y.R.; Lito, P.; Teruya-Feldstein, J.; Hu, W.; Beguelin, W.; Monette, S.; Duy, C.; et al. Hematopoietic stem cell origin of BRAFV600E mutations in hairy cell leukemia. Sci. Transl. Med. 2014, 6, ra71–ra238. [Google Scholar] [CrossRef] [Green Version]
- Schuh, A.; Becq, J.; Humphray, S.; Alexa, A.; Burns, A.; Clifford, R.; Feller, S.M.; Grocock, R.; Henderson, S.; Khrebtukova, I.; et al. Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood 2012, 120, 4191–4196. [Google Scholar] [CrossRef] [Green Version]
- Clipson, A.; Wang, M.; de Leval, L.; Ashton-Key, M.; Wotherspoon, A.; Vassiliou, G.; Bolli, N.; Grove, C.; Moody, S.; Escudero-Ibarz, L.; et al. KLF2 mutation is the most frequent somatic change in splenic marginal zone lymphoma and identifies a subset with distinct genotype. Leukemia 2015, 29, 1177–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piva, R.; Deaglio, S.; Famà, R.; Buonincontri, R.; Scarfò, I.; Bruscaggin, A.; Mereu, E.; Serra, S.; Spina, V.; Brusa, D.; et al. The Krüppel-like factor 2 transcription factor gene is recurrently mutated in splenic marginal zone lymphoma. Leukemia 2015, 29, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Tiacci, E.; Pettirossi, V.; Schiavoni, G.; Falini, B. Genomics of Hairy Cell Leukemia. J. Clin. Oncol. 2017, 35, 1002–1010. [Google Scholar] [CrossRef]
- Hart, G.T.; Wang, X.; Hogquist, K.A.; Jameson, S.C. Kruppel-like factor 2 (KLF2) regulates B-cell reactivity, subset differentiation, and trafficking molecule expression. Proc. Natl. Acad. Sci. USA 2011, 108, 716–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkelmann, R.; Sandrock, L.; Porstner, M.; Roth, E.; Mathews, M.; Hobeika, E.; Reth, M.; Kahn, M.L.; Schuh, W.; Jack, H.-M. B cell homeostasis and plasma cell homing controlled by Kruppel-like factor 2. Proc. Natl. Acad. Sci. USA 2011, 108, 710–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, S.; Ehrentraut, S.; Meyer, C.; Kaufmann, M.; Drexler, H.G.; MacLeod, R.A.F. NFkB is activated by multiple mechanisms in hairy cell leukemia: NFkB IS Activated by Multiple Mechanisms. Genes Chromosomes Cancer 2015, 54, 418–432. [Google Scholar] [CrossRef] [PubMed]
- Durandy, A. Mini-review Activation-induced cytidine deaminase: A dual role in class-switch recombination and somatic hypermutation. Eur. J. Immunol. 2003, 33, 2069–2073. [Google Scholar] [CrossRef]
- Saribasak, H.; Saribasak, N.N.; Ipek, F.M.; Ellwart, J.W.; Arakawa, H.; Buerstedde, J.-M. Uracil DNA Glycosylase Disruption Blocks Ig Gene Conversion and Induces Transition Mutations. J. Immunol. 2006, 176, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Hockley, S.L.; Morilla, A.; Else, M.; Dearden, C.; Catovsky, D.; Morgan, G.J.; Matutes, E.; Gonzalez, D. Higher expression levels of activation-induced cytidine deaminase distinguish hairy cell leukemia from hairy cell leukemia-variant and splenic marginal zone lymphoma. Leukemia 2010, 24, 1084–1086. [Google Scholar] [CrossRef] [Green Version]
- Durham, B.H.; Getta, B.; Dietrich, S.; Taylor, J.; Won, H.; Bogenberger, J.M.; Scott, S.; Kim, E.; Chung, Y.R.; Chung, S.S.; et al. Genomic analysis of hairy cell leukemia identifies novel recurrent genetic alterations. Blood 2017, 130, 1644–1648. [Google Scholar] [CrossRef] [Green Version]
- König, E.A.; Kusser, W.C.; Day, C.; Porzsolt, F.; Glickman, B.W.; Messer, G.; Schmid, M.; De Chatel, R.; Marcsek, Z.L.; Demeter, J. p53 mutations in hairy cell leukemia. Leukemia 2000, 14, 706–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hockley, S.L.; Else, M.; Morilla, A.; Wotherspoon, A.; Dearden, C.; Catovsky, D.; Gonzalez, D.; Matutes, E. The prognostic impact of clinical and molecular features in hairy cell leukaemia variant and splenic marginal zone lymphoma. Br. J. Haematol. 2012, 158, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Bromberg-White, J.L.; Andersen, N.J.; Duesbery, N.S. MEK genomics in development and disease. Brief. Funct. Genom. 2012, 11, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Emery, C.M.; Vijayendrana, K.G.; Zipserc, M.C.; Sawyera, A.M.; Niua, L.; Kima, J.J.; Hattona, C.; Choprad, R.; Oberholzera, P.A.; Karpovac, M.B.; et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef] [Green Version]
- Marks, J.L.; Gong, Y.; Chitale, D.; Golas, B.; McLellan, M.D.; Kasai, Y.; Ding, L.; Mardis, E.R.; Wilson, R.K.; Solit, D.; et al. Novel MEK1 Mutation Identified by Mutational Analysis of Epidermal Growth Factor Receptor Signaling Pathway Genes in Lung Adenocarcinoma. Cancer Res. 2008, 68, 5524–5528. [Google Scholar] [CrossRef] [Green Version]
- Wagle, N.; Emery, C.; Berger, M.F.; Davis, M.J.; Sawyer, A.; Pochanard, P.; Kehoe, S.M.; Johannessen, C.M.; MacConaill, L.E.; Hahn, W.C.; et al. Dissecting Therapeutic Resistance to RAF Inhibition in Melanoma by Tumor Genomic Profiling. J. Clin. Oncol. 2011, 29, 3085–3096. [Google Scholar] [CrossRef] [Green Version]
- van Haaften, G.; Dalgliesh, G.L.; Davies, H.; Chen, L.; Bignell, G.; Greenman, C.; Edkins, S.; Hardy, C.; O’Meara, S.; Teague, J.; et al. Somatic mutations of the histone H3K27 demethylase gene UTX in human cancer. Nat. Genet. 2009, 41, 521–523. [Google Scholar] [CrossRef] [Green Version]
- Sengoku, T.; Yokoyama, S. Structural basis for histone H3 Lys 27 demethylation by UTX/KDM6A. Genes Dev. 2011, 25, 2266–2277. [Google Scholar] [CrossRef] [Green Version]
- Dee Ler, L.; Ghosh, S.; Chai, X.; Thike, A.A.; Lee Heng, H.; Yan Siew, E.; Dey, S.; Kai Koh, L.; Quan Lim, J.; Khong Lim, W.; et al. Loss of tumor suppressor KDM6A amplifies PRC2-regulated transcriptional repression in bladder cancer and can be targeted through inhibition of EZH2. Sci. Transl. Med. 2017, 9, eaai8312. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variate | HCL (n = 82) | HCL-like Disorders (n = 16) | |
|---|---|---|---|
| vHCL/SDRPL (n = 5) | HCL-like NOS (n = 11) | ||
| CLINICAL FEATURES | |||
| Sex ratio (M/F) | 2.9 | 5 | 2.7 |
| Median age (years [min-max]) | 55.8 [30–82] | 72 [58–82] | 74 [67–95] |
| Lymphocytosis (>4 G/L) | 12/61 | 3/4 | 5/10 |
| Monocytopenia (<0.2) | 41/56 | 0/4 | 2/10 |
| Diagnosis (n, %), relapse (n, %) sample | 51 (63%), 30 (27%) | 4 (80%), 1 (20%) | 9 (82%), 2 (18%) |
| Median number of treatment lines[min-max] | 2 [1–12] | 1 [0–1] | 0.5 [0–1] |
| BM (n, %), PB (n, %) | 28 (34%), 54 (66%) | 1(20%), 4 (80%) | 1 (9%), 10 (91%) |
| IMMUNOPHENOTYPE | |||
| CD11c | 72/72 | 5/5 | 10/11 |
| CD25 (n) | 72/72 | 1/5 | 2/11 |
| CD103 (n) | 71/72 | 4/5 | 4/11 |
| CD123 (n) | 65/68 | 1/4 | 2/11 |
| GENETIC ALTERATIONS | |||
| IGHV mutated (n, %) | 30/34 (88%) | 2/5 (40%) | 9/10 (90%) |
| IGHV repertory VH4-34 (n) | 3/34 | 3/5 | 1/10 |
| IGHV repertory VH3 (n) | 15/34 | 0/5 | 5/10 |
| IGHV repertory VH1-2 (n) | 2/34 | 0 | 2/10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maitre, E.; Tomowiak, C.; Lebecque, B.; Bijou, F.; Benabed, K.; Naguib, D.; Kerneves, P.; Cornet, E.; Viailly, P.-J.; Arsham, J.; et al. Deciphering Genetic Alterations of Hairy Cell Leukemia and Hairy Cell Leukemia-like Disorders in 98 Patients. Cancers 2022, 14, 1904. https://doi.org/10.3390/cancers14081904
Maitre E, Tomowiak C, Lebecque B, Bijou F, Benabed K, Naguib D, Kerneves P, Cornet E, Viailly P-J, Arsham J, et al. Deciphering Genetic Alterations of Hairy Cell Leukemia and Hairy Cell Leukemia-like Disorders in 98 Patients. Cancers. 2022; 14(8):1904. https://doi.org/10.3390/cancers14081904
Chicago/Turabian StyleMaitre, Elsa, Cécile Tomowiak, Benjamin Lebecque, Fontanet Bijou, Khaled Benabed, Dina Naguib, Pauline Kerneves, Edouard Cornet, Pierre-Julien Viailly, Jeffrey Arsham, and et al. 2022. "Deciphering Genetic Alterations of Hairy Cell Leukemia and Hairy Cell Leukemia-like Disorders in 98 Patients" Cancers 14, no. 8: 1904. https://doi.org/10.3390/cancers14081904