
Platelet-Derived PDGFB Promotes Recruitment of Cancer-Associated Fibroblasts, Deposition of Extracellular Matrix and Tgfβ Signaling in the Tumor Microenvironment

 ,
,  , ,
, ,  , and
, and 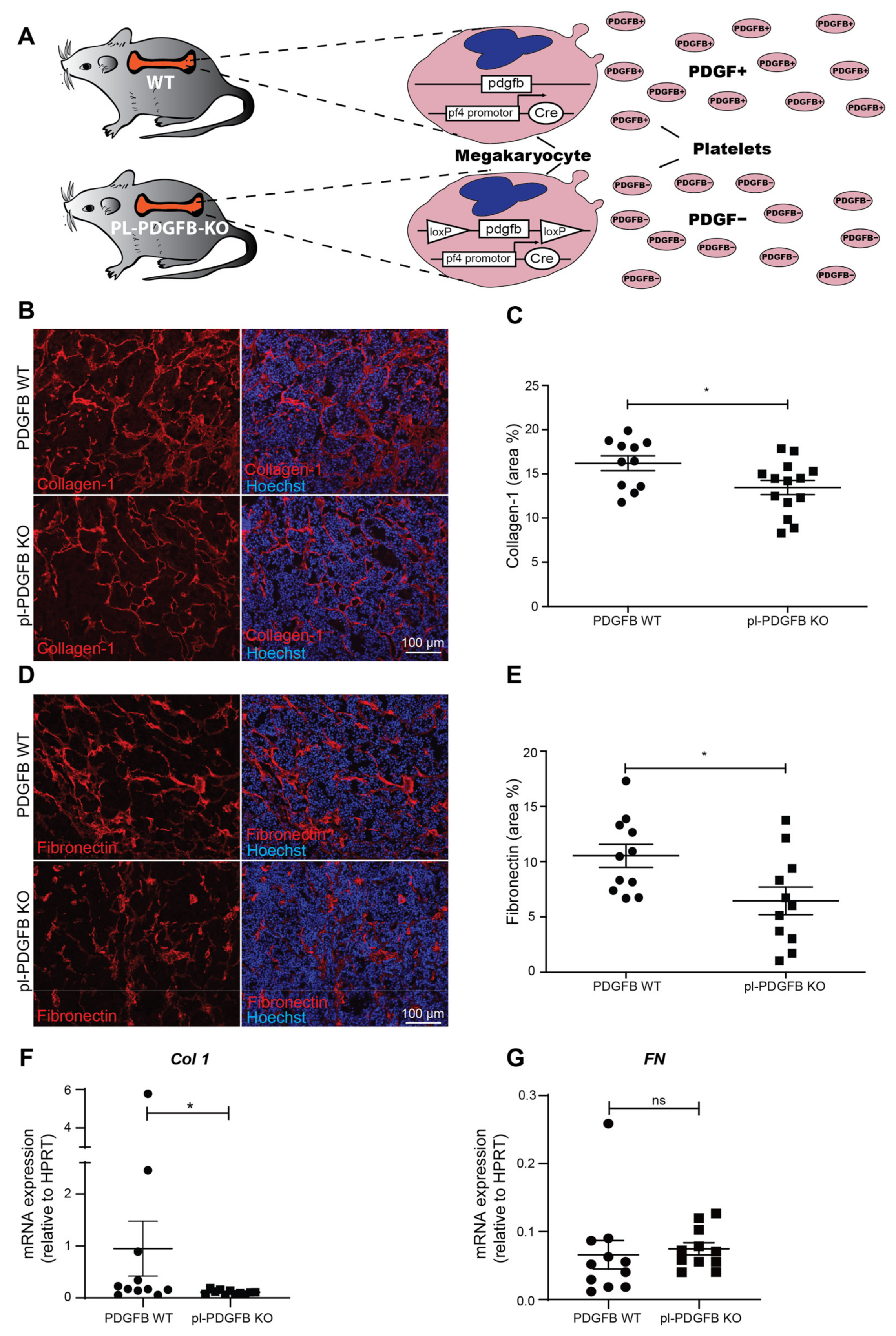{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Immunostainings
2.3. Antibodies
2.4. Masson Trichrome Staining
2.5. Tumor RNA Extraction and qPCR
2.6. Isolation and RNA Extraction of Endothelial Cells from RT2 Tumors
2.7. Proximity Extension Assay
2.8. Image Analysis
2.9. Statistics
3. Results
3.1. Platelet-Specific Ablation of PDGFB Reduced Extracellular Matrix Formation in the Tumor Microenvironment
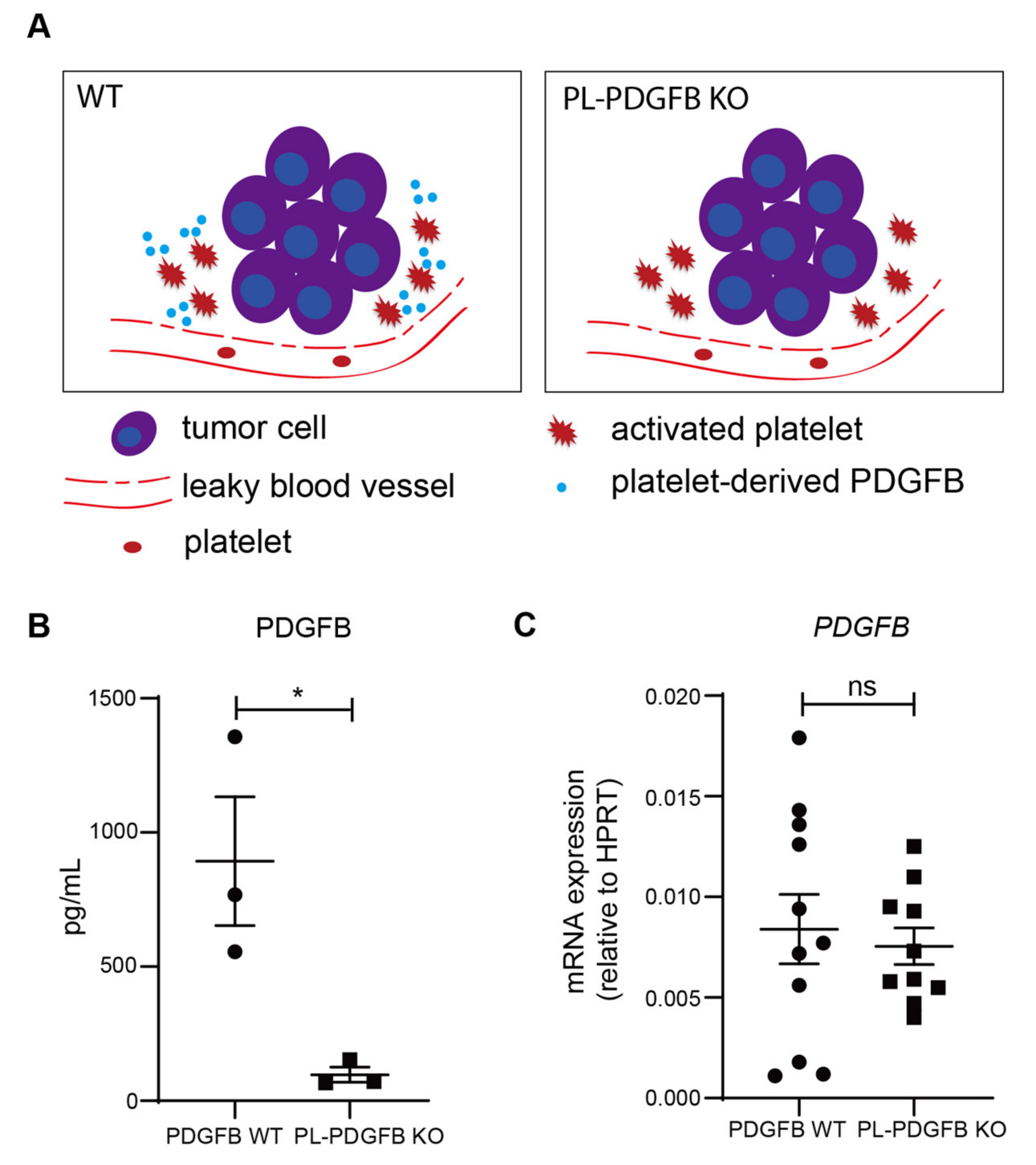
3.2. The Amount of PDGFB Is Reduced in Tumors from pl-PDGFB KO Mice
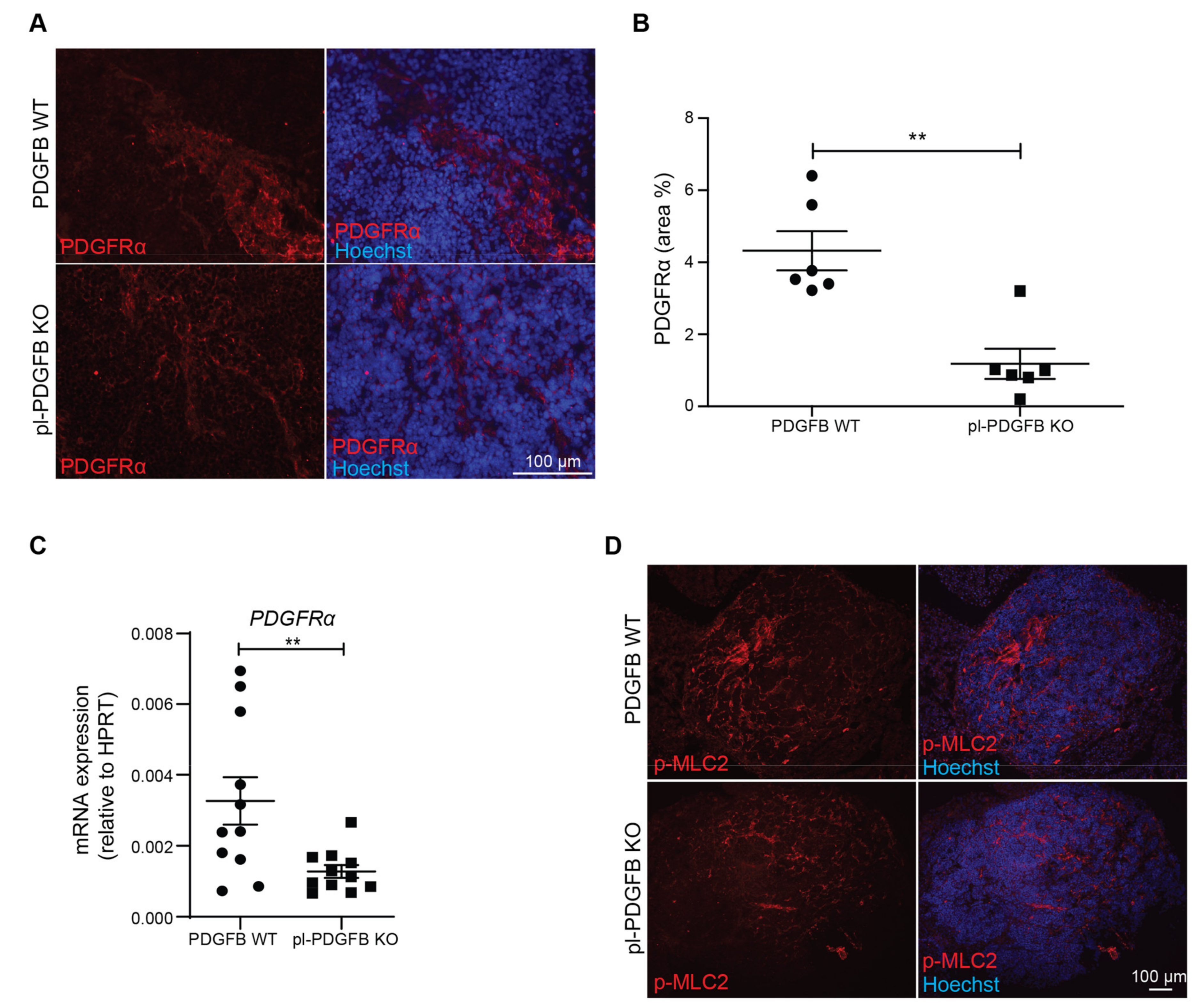
3.3. Tumors in pl-PDGFB KO Mice Have Fewer Cancer-Associated Fibroblasts
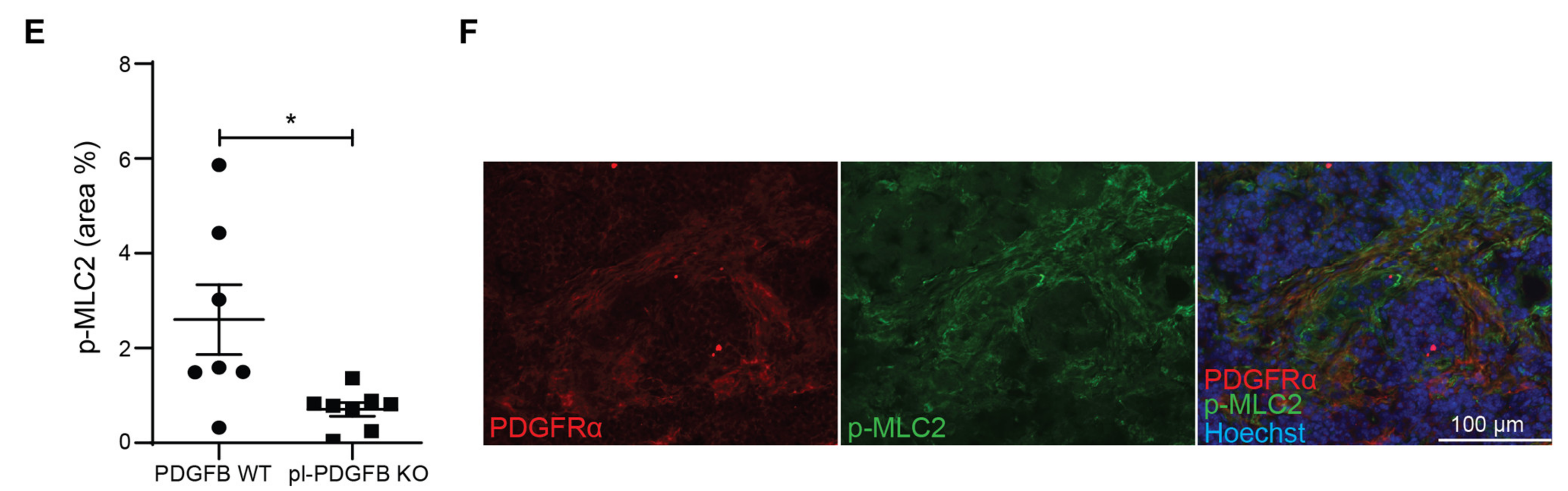
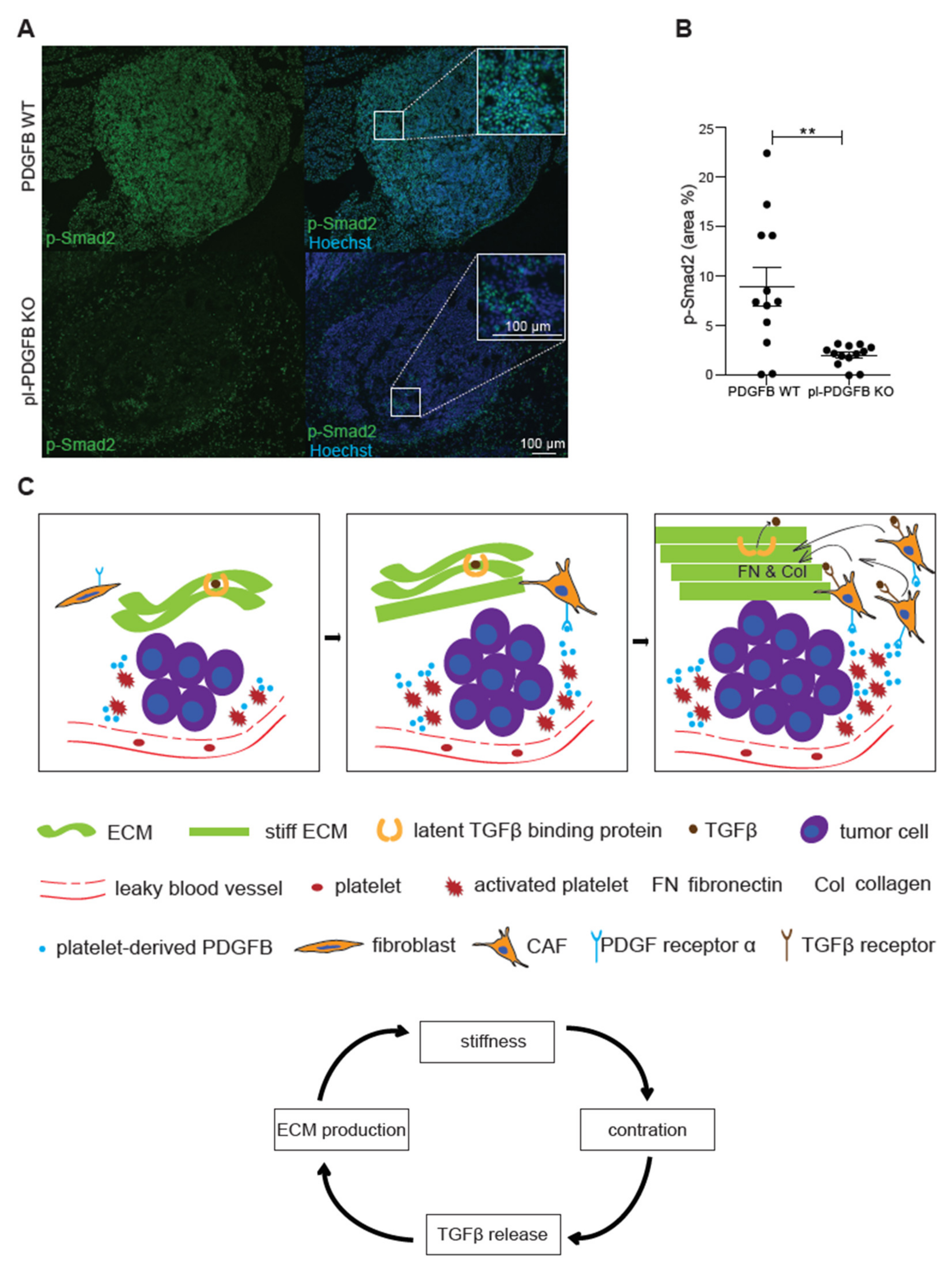
3.4. TGFβ-Dependent Signaling Is Reduced in Tumors from pl-PDGFB KO Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jurk, K.; Kehrel, B.E. Platelets: Physiology and Biochemistry. Semin. Thromb. Hemost. 2005, 31, 381–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, S.P. The growing complexity of platelet aggregation. Blood 2007, 109, 5087–5095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timp, J.F.; Braekkan, S.K.; Versteeg, H.; Cannegieter, S.C. Epidemiology of cancer-associated venous thrombosis. Blood 2013, 122, 1712–1723. [Google Scholar] [CrossRef] [Green Version]
- Falanga, A.; Le Gal, G.; Carrier, M.; Abdel-Razeq, H.; Ay, C.; Martin, A.J.M.; Rocha, A.T.C.; Agnelli, G.; Elalamy, I.; Brenner, B. Management of Cancer-Associated Thrombosis: Unmet Needs and Future Perspectives. TH Open 2021, 05, e376–e386. [Google Scholar] [CrossRef] [PubMed]
- Lip, G.Y.; Chin, B.S.; Blann, A.D. Cancer and the prothrombotic state. Lancet Oncol. 2002, 3, 27–34. [Google Scholar] [CrossRef]
- Versteeg, H.H. Tissue Factor: Old and New Links with Cancer Biology. Semin. Thromb. Hemost. 2015, 41, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Hisada, Y.; Mackman, N. Tissue Factor and Cancer: Regulation, Tumor Growth, and Metastasis. Semin. Thromb. Hemost. 2019, 45, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [Green Version]
- Braun, L.J.; Stegmeyer, R.I.; Schäfer, K.; Volkery, S.; Currie, S.M.; Kempe, B.; Nottebaum, A.F.; Vestweber, D. Platelets docking to VWF prevent leaks during leukocyte extravasation by stimulating Tie-2. Blood 2020, 136, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cedervall, J.; Hamidi, A.; Herre, M.; Viitaniemi, K.; D’Amico, G.; Miao, Z.; Unnithan, R.V.M.; Vaccaro, A.; van Hooren, L.; et al. Platelet-Specific PDGFB Ablation Impairs Tumor Vessel Integrity and Promotes Metastasis. Cancer Res. 2020, 80, 3345–3358. [Google Scholar] [CrossRef]
- Workman, P.; Balmain, A.; Hickman, J.A.; McNally, N.J.; Rohas, A.M.; Mitchison, N.A.; Pierrepoint, C.G.; Raymond, R.; Rowlatt, C.; Stephens, T.C. UKCCCR guidelines for the welfare of animals in experimental neoplasia. Lab. Anim. 1988, 22, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Tiedt, R.; Schomber, T.; Hao-Shen, H.; Skoda, R.C. Pf4-Cre transgenic mice allow the generation of lineage-restricted gene knockouts for studying megakaryocyte and platelet function in vivo. Blood 2007, 109, 1503–1506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enge, M.; Bjarnegård, M.; Gerhardt, H.; Gustafsson, E.; Kalén, M.; Asker, N.; Hammes, H.; Shani, M.; Fässler, R.; Betsholtz, C. Endothelium-specific platelet-derived growth factor-B ablation mimics diabetic retinopathy. EMBO J. 2002, 21, 4307–4316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Björkesten, J.; Thorsen, S.B.; Ekman, D.; Eriksson, A.; Dickens, E.R.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-Plex PEA Immunoassay Exhibiting High Sensitivity, Specificity, and Excellent Scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Q.; Björkesten, J.; Galli, J.; Ekman, D.; Broberg, J.; Nordberg, N.; Tillander, A.; Kamali-Moghaddam, M.; Tybring, G.; Landegren, U. Strong impact on plasma protein profiles by precentrifugation delay but not by repeated freeze-thaw cycles, as analyzed using multiplex proximity extension assays. Clin. Chem. Lab. Med. (CCLM) 2017, 56, 582–594. [Google Scholar] [CrossRef]
- Bergers, G.; Javaherian, K.; Lo, K.-M.; Folkman, J.; Hanahan, D. Effects of Angiogenesis Inhibitors on Multistage Carcinogenesis in Mice. Science 1999, 284, 808–812. [Google Scholar] [CrossRef] [Green Version]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123 Pt 24, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Ren, X.; Li, L.; Wu, J.; Lin, K.; He, Y.; Bian, L. PDGF-BB regulates the transformation of fibroblasts into cancer-associated fibroblasts via the lncRNA LU-RAP1L-AS1/LURAP1L/IKK/IkappaB/NF-kappaB signaling pathway. Oncol. Lett. 2021, 22, 537. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N. Transforming growth factor-beta in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef] [PubMed]
- Martino, F.; Perestrelo, A.R.; Vinarsky, V.; Pagliari, S.; Forte, G. Cellular Mechanotransduction: From Tension to Function. Front. Physiol. 2018, 9, 824. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell. Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Moustakas, A.; Heldin, C.H. The regulation of TGFbeta signal transduction. Development 2009, 136, 3699–3714. [Google Scholar] [CrossRef] [Green Version]
- Robertson, I.B.; Rifkin, D.B. Regulation of the Bioavailability of TGF-beta and TGF-beta-Related Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a021907. [Google Scholar] [CrossRef]
- Sheppard, D. Integrin-mediated activation of latent transforming growth factor beta. Cancer Metastasis. Rev. 2005, 24, 395–402. [Google Scholar] [CrossRef]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Grauel, A.L.; Nguyen, B.; Ruddy, D.; Laszewski, T.; Schwartz, S.; Chang, J.; Chen, J.; Piquet, M.; Pelletier, M.; Yan, Z.; et al. TGFbeta-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat. Commun. 2020, 11, 6315. [Google Scholar] [CrossRef]
- Seppä, H.; Grotendorst, G.; Seppä, S.; Schiffmann, E.; Martin, G.R. JBC 1981. Platelet-derived growth factor in chemotactic for fibroblasts. J. Cell. Biol. 1982, 92, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Monypenny, J.; Zicha, D.; Higashida, C.; Oceguera-Yanez, F.; Narumiya, S.; Watanabe, N. Cdc42 and Rac Family GTPases Regulate Mode and Speed but Not Direction of Primary Fibroblast Migration during Platelet-Derived Growth Factor-Dependent Chemotaxis. Mol. Cell. Biol. 2009, 29, 2730–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegbahn, A.; Johnell, M.; Rorsman, C.; Ezban, M.; Heldin, C.-H.; Ronnstrand, L. Binding of factor VIIa to tissue factor on human fibroblasts leads to activation of phospholipase C and enhanced PDGF-BB-stimulated chemotaxis. Blood 2000, 96, 3452–3458. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H.; Lennartsson, J.; Westermark, B. Involvement of platelet-derived growth factor ligands and receptors in tumorigenesis. J. Intern. Med. 2018, 283, 16–44. [Google Scholar] [CrossRef] [Green Version]
- Hammer, A.M.; Sizemore, G.M.; Shukla, V.C.; Avendano, A.; Sizemore, S.T.; Chang, J.J.; Kladney, R.D.; Cuitino, M.C.; Thies, K.A.; Verfurth, Q.; et al. Stromal PDGFR-α Activation Enhances Matrix Stiffness, Impedes Mammary Ductal Devel-opment, and Accelerates Tumor Growth. Neoplasia 2017, 19, 496–508. [Google Scholar] [CrossRef]
- Pietras, K.; Ostman, A.; Sjoquist, M.; Buchdunger, E.; Reed, R.K.; Heldin, C.-H.; Rubin, K. Inhibition of platelet-derived growth factor receptors reduces interstitial hypertension and in-creases transcapillary transport in tumors. Cancer Res. 2001, 61, 2929–2934. [Google Scholar]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of Fibronectin Extracellular Matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef] [Green Version]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.-H. Targeting the PDGF signaling pathway in tumor treatment. Cell Commun. Signal. 2013, 11, 97. [Google Scholar] [CrossRef] [Green Version]
- Verheul, H.; Hoekman, K.; Bakker, S.L.-D.; A Eekman, C.; Folman, C.C.; Broxterman, H.J.; Pinedo, H.M. Platelet: Transporter of vascular endothelial growth factor. Clin. Cancer Res. 1997, 3, 2187–2190. [Google Scholar]
- Karolczak, K.; Watala, C. Blood Platelets as an Important but Underrated Circulating Source of TGFbeta. Int. J. Mol. Sci. 2021, 22, 4492. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Manouchehri Doulabi, E.; Herre, M.; Cedervall, J.; Qiao, Q.; Miao, Z.; Hamidi, A.; Hellman, L.; Kamali-Moghaddam, M.; Olsson, A.-K. Platelet-Derived PDGFB Promotes Recruitment of Cancer-Associated Fibroblasts, Deposition of Extracellular Matrix and Tgfβ Signaling in the Tumor Microenvironment. Cancers 2022, 14, 1947. https://doi.org/10.3390/cancers14081947
Zhang Y, Manouchehri Doulabi E, Herre M, Cedervall J, Qiao Q, Miao Z, Hamidi A, Hellman L, Kamali-Moghaddam M, Olsson A-K. Platelet-Derived PDGFB Promotes Recruitment of Cancer-Associated Fibroblasts, Deposition of Extracellular Matrix and Tgfβ Signaling in the Tumor Microenvironment. Cancers. 2022; 14(8):1947. https://doi.org/10.3390/cancers14081947
Chicago/Turabian StyleZhang, Yanyu, Ehsan Manouchehri Doulabi, Melanie Herre, Jessica Cedervall, Qi Qiao, Zuoxiu Miao, Anahita Hamidi, Lars Hellman, Masood Kamali-Moghaddam, and Anna-Karin Olsson. 2022. "Platelet-Derived PDGFB Promotes Recruitment of Cancer-Associated Fibroblasts, Deposition of Extracellular Matrix and Tgfβ Signaling in the Tumor Microenvironment" Cancers 14, no. 8: 1947. https://doi.org/10.3390/cancers14081947
APA StyleZhang, Y., Manouchehri Doulabi, E., Herre, M., Cedervall, J., Qiao, Q., Miao, Z., Hamidi, A., Hellman, L., Kamali-Moghaddam, M., & Olsson, A.-K. (2022). Platelet-Derived PDGFB Promotes Recruitment of Cancer-Associated Fibroblasts, Deposition of Extracellular Matrix and Tgfβ Signaling in the Tumor Microenvironment. Cancers, 14(8), 1947. https://doi.org/10.3390/cancers14081947