
ERα36-High Cancer-Associated Fibroblasts as an Unfavorable Factor in Triple-Negative Breast Cancer

 , , , , , , , and
, , , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines, Antibodies, and Reagents
2.2. Cancer-Associated Fibroblast Isolation and Conditioned Media Preparation
2.3. Immunofluorescence Staining
2.4. Western Blotting
2.5. Cancer-Associated Fibroblasts Secretome Analysis
2.6. Cell Migration Assay
2.7. Analysis of Cell Growth in Three-Dimensional Cultures
2.8. Stimulation with CAF-Conditioned Media and HGF, Signaling Analysis, and Inhibitory Effects
2.9. Clinical Data Analysis
2.10. ERα36 Protein Levels in Tumor Stroma
2.11. Hormone Receptors and HER2 Status Analysis in Breast Cancer Samples
2.12. Statistical Analysis
2.13. RNA Extraction
2.14. nCounter Gene Expression Assay
2.15. NanoString Data Processing
3. Results
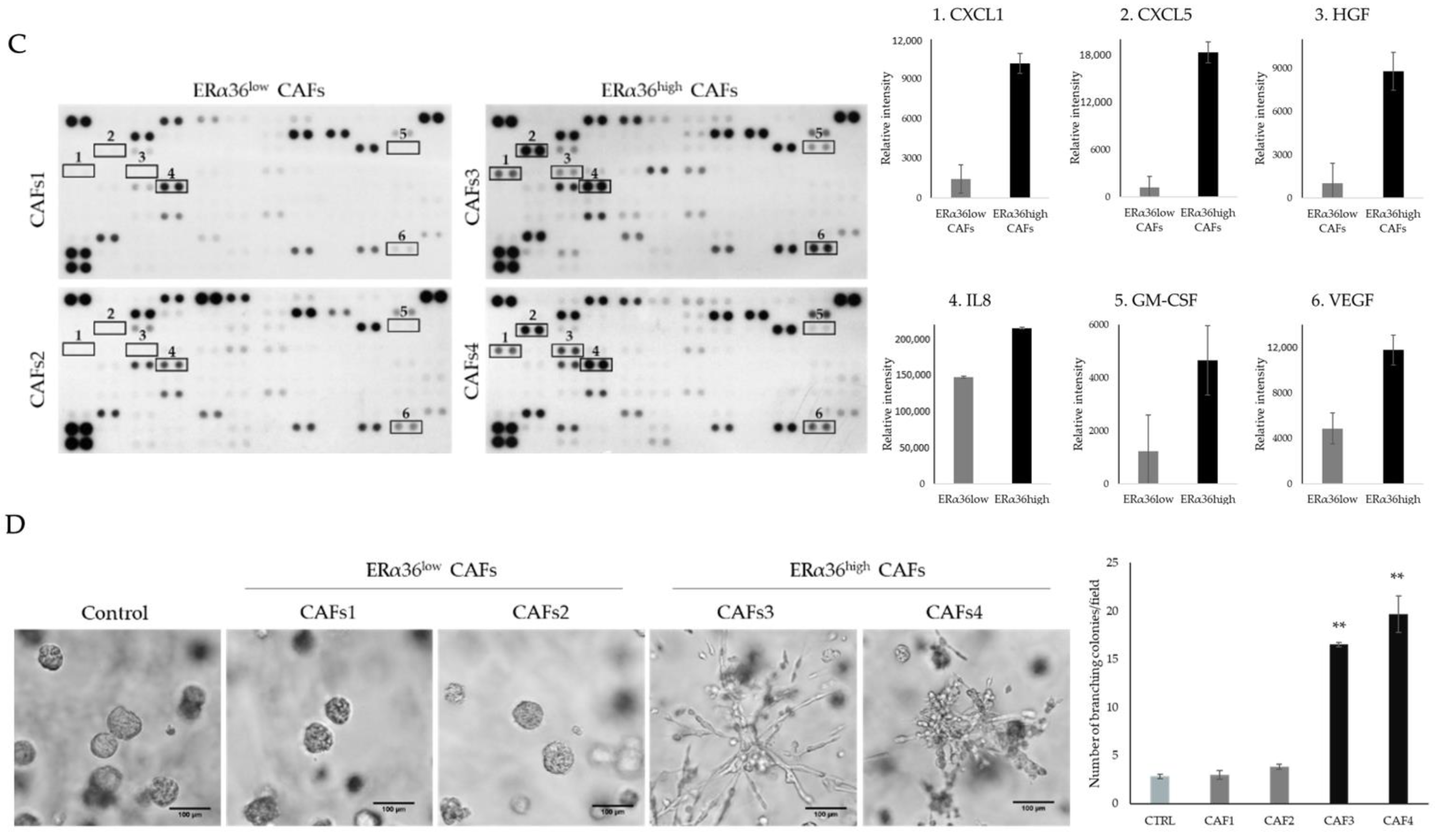
3.1. CAFs Characterized by Different ERα36 Expression Represent Distinct Subpopulations with Diverse Influence on TNBC Cells
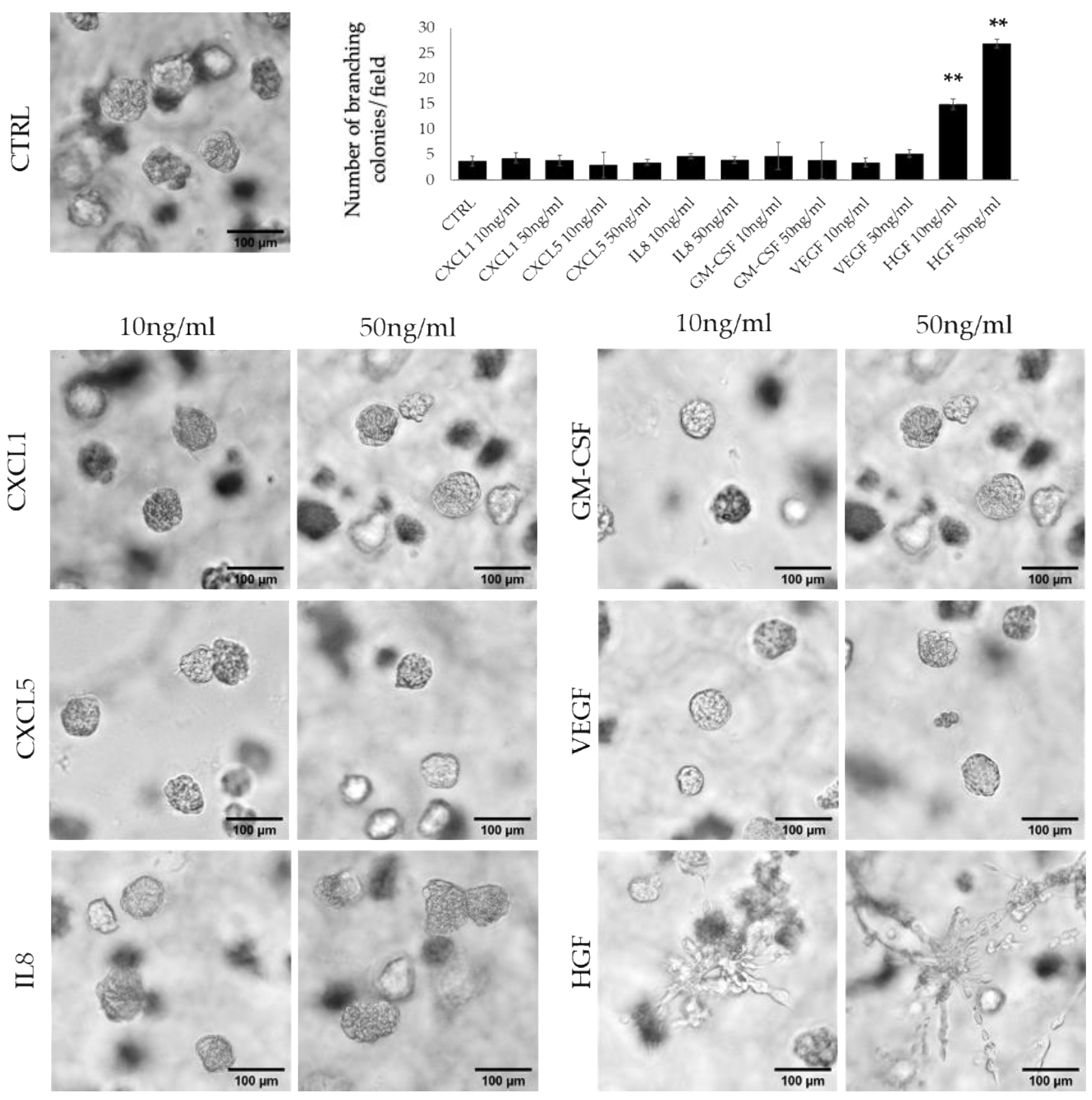
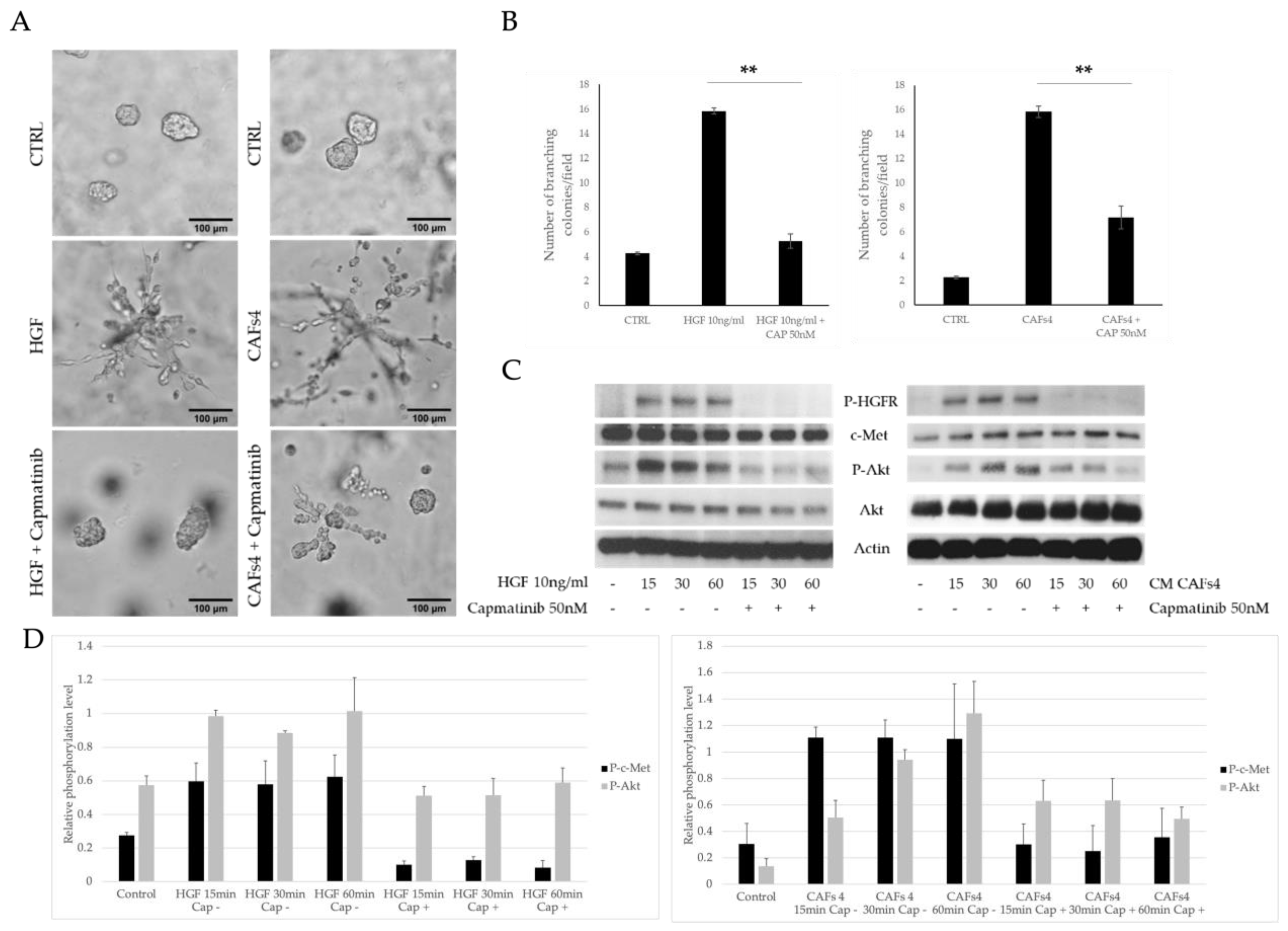
3.2. HFG Secreted by ERα36high CAFs Induces Invasive Phenotype of TNBC Cells
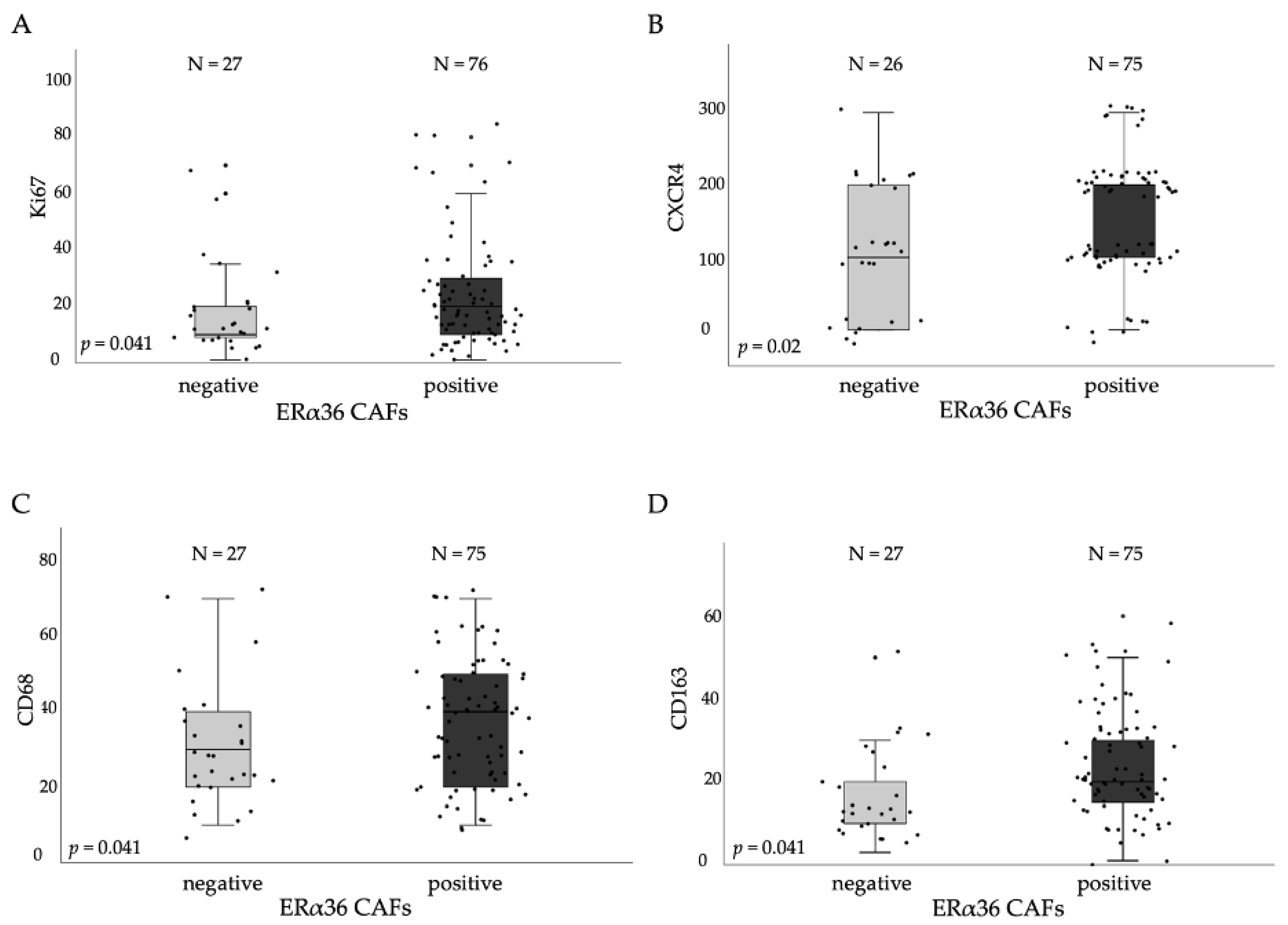
3.3. ERα36high CAFs in Tumor Stroma Correlates with Proliferation and TAMs Markers
3.4. Cytokines Produced by ERα36high CAFs Confer Poor Prognosis of TNBC Patients
3.5. ERα36 Expression Affects Transcription of Matrix Disassembly and Immune Response Genes in BC Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Yang, J.; Peng, L.; Sahin, A.A.; Huo, L.; Ward, K.C.; O’Regan, R.; Torres, M.A.; Meisel, J.L. Triple-negative breast cancer has worse overall survival and cause-specific survival than non-triple-negative breast cancer. Breast Cancer Res. Treat. 2017, 161, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Cleator, S.; Heller, W.; Coombes, R.C. Triple-negative breast cancer: Therapeutic options. Lancet Oncol. 2007, 8, 235–244. [Google Scholar] [CrossRef]
- De Laurentiis, M.; Cianniello, D.; Caputo, R.; Stanzione, B.; Arpino, G.; Cinieri, S.; Lorusso, V.; de Placido, S. Treatment of triple negative breast cancer (TNBC): Current options and future perspectives. Cancer Treat. Rev. 2010, 36 (Suppl. 3), S80–S86. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thurlimann, B.; Senn, H.J. Strategies for subtypes—Dealing with the diversity of breast cancer: Highlights of the St. Gallen International Expert Consensus on the primary therapy of early breast cancer 2011. Ann. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef]
- Micke, P.; Ostman, A. Exploring the tumour environment: Cancer-associated fibroblasts as targets in cancer therapy. Expert Opin. Ther. Targets 2005, 9, 1217–1233. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef] [Green Version]
- Takai, K.; Le, A.; Weaver, V.M.; Werb, Z. Targeting the cancer-associated fibroblasts as a treatment in triple-negative breast cancer. Oncotarget 2016, 7, 82889–82901. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Tu, G.; Liu, Z.; Liu, M. Cancer-associated fibroblasts: A multifaceted driver of breast cancer progression. Cancer Lett. 2015, 361, 155–163. [Google Scholar] [CrossRef]
- Kikuchi, K.; McNamara, K.M.; Miki, Y.; Moon, J.Y.; Choi, M.H.; Omata, F.; Sakurai, M.; Onodera, Y.; Rai, Y.; Ohi, Y.; et al. Effects of cytokines derived from cancer-associated fibroblasts on androgen synthetic enzymes in estrogen receptor-negative breast carcinoma. Breast Cancer Res. Treat. 2017, 166, 709–723. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brechbuhl, H.M.; Finlay-Schultz, J.; Yamamoto, T.M.; Gillen, A.E.; Cittelly, D.M.; Tan, A.C.; Sams, S.B.; Pillai, M.M.; Elias, A.D.; Robinson, W.A.; et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin. Cancer Res. 2017, 23, 1710–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Kruijf, E.M.; van Nes, J.G.; van de Velde, C.J.; Putter, H.; Smit, V.T.; Liefers, G.J.; Kuppen, P.J.; Tollenaar, R.A.; Mesker, W.E. Tumor-stroma ratio in the primary tumor is a prognostic factor in early breast cancer patients, especially in triple-negative carcinoma patients. Breast Cancer Res. Treat. 2011, 125, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Slavin, S.; Yeh, C.R.; Da, J.; Yu, S.; Miyamoto, H.; Messing, E.M.; Guancial, E.; Yeh, S. Estrogen receptor α in cancer-associated fibroblasts suppresses prostate cancer invasion via modulation of thrombospondin 2 and matrix metalloproteinase 3. Carcinogenesis 2014, 35, 1301–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.R.; Slavin, S.; Da, J.; Hsu, I.; Luo, J.; Xiao, G.Q.; Ding, J.; Chou, F.J.; Yeh, S. Estrogen receptor α in cancer associated fibroblasts suppresses prostate cancer invasion via reducing CCL5, IL6 and macrophage infiltration in the tumor microenvironment. Mol. Cancer 2016, 15, 7. [Google Scholar] [CrossRef] [Green Version]
- Da, J.; Lu, M.; Wang, Z. Estrogen receptor alpha (ERα)-associated fibroblasts promote cell growth in prostate cancer. Cell Biochem. Biophys. 2015, 73, 793–798. [Google Scholar] [CrossRef]
- Kumar, M.M.; Davuluri, S.; Poojar, S.; Mukherjee, G.; Bajpai, A.K.; Bafna, U.D.; Devi, U.K.; Kallur, P.P.; Kshitish, A.K.; Jayshree, R.S. Role of estrogen receptor alpha in human cervical cancer-associated fibroblasts: A transcriptomic study. Tumor Biol. 2016, 37, 4409–4420. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Yin, L. Estrogen receptor alpha-36 (ER-α36): A new player in human breast cancer. Mol. Cell. Endocrinol. 2015, 418, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Soltysik, K.; Czekaj, P. ERα36—Another piece of the estrogen puzzle. Eur. J. Cell Biol. 2015, 94, 611–625. [Google Scholar] [CrossRef]
- Chaudhri, R.A.; Schwartz, N.; Elbaradie, K.; Schwartz, Z.; Boyan, B.D. Role of ERα36 in membrane-associated signaling by estrogen. Steroids 2014, 81, 74–80. [Google Scholar] [CrossRef]
- Rao, J.; Jiang, X.; Wang, Y.; Chen, B. Advances in the understanding of the structure and function of ER-α36, a novel variant of human estrogen receptor-alpha. J. Steroid Biochem. Mol. Biol. 2011, 127, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Nagel, A.; Szade, J.; Iliszko, M.; Elzanowska, J.; Welnicka-Jaskiewicz, M.; Skokowski, J.; Stasilojc, G.; Bigda, J.; Sadej, R.; Zaczek, A.; et al. Clinical and biological significance of ESR1 gene alteration and estrogen receptors isoforms expression in breast cancer patients. Int. J. Mol. Sci. 2019, 20, 1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhang, W.; Sun, X.; Lin, Y.; Chen, W. Cancer-associated fibroblasts induce epithelial-mesenchymal transition through secreted cytokines in endometrial cancer cells. Oncol. Lett. 2018, 15, 5694–5702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popeda, M.; Stokowy, T.; Bednarz-Knoll, N.; Jurek, A.; Niemira, M.; Bielska, A.; Kretowski, A.; Kalinowski, L.; Szade, J.; Markiewicz, A.; et al. NF-kappa B signaling-related signatures are connected with the mesenchymal phenotype of circulating tumor cells in non-metastatic breast cancer. Cancers 2019, 11, 1961. [Google Scholar] [CrossRef] [Green Version]
- Liao, D.; Luo, Y.; Markowitz, D.; Xiang, R.; Reisfeld, R.A. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS ONE 2009, 4, e7965. [Google Scholar] [CrossRef] [Green Version]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Finak, G.; Bertos, N.; Pepin, F.; Sadekova, S.; Souleimanova, M.; Zhao, H.; Chen, H.; Omeroglu, G.; Meterissian, S.; Omeroglu, A.; et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat. Med. 2008, 14, 518–527. [Google Scholar] [CrossRef]
- Vastrad, B.; Vastrad, C.; Tengli, A.; Iliger, S. Identification of differentially expressed genes regulated by molecular signature in breast cancer-associated fibroblasts by bioinformatics analysis. Arch. Gynecol. Obstet. 2018, 297, 161–183. [Google Scholar] [CrossRef]
- Dhillon, S. Capmatinib: First approval. Drugs 2020, 80, 1125–1131. [Google Scholar] [CrossRef]
- Deying, W.; Feng, G.; Shumei, L.; Hui, Z.; Ming, L.; Hongqing, W. CAF-derived HGF promotes cell proliferation and drug resistance by up-regulating the c-Met/PI3K/Akt and GRP78 signalling in ovarian cancer cells. Biosci. Rep. 2017, 37, BSR20160470. [Google Scholar] [CrossRef] [Green Version]
- Allinen, M.; Beroukhim, R.; Cai, L.; Brennan, C.; Lahti-Domenici, J.; Huang, H.; Porter, D.; Hu, M.; Chin, L.; Richardson, A.; et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004, 6, 17–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Zou, A.; Lambert, D.; Yeh, H.; Yasukawa, K.; Behbod, F.; Fan, F.; Cheng, N. Elevated CXCL1 expression in breast cancer stroma predicts poor prognosis and is inversely associated with expression of TGF-beta signaling proteins. BMC Cancer 2014, 14, 781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Moreno, R.; Curtis, K.J.; Coughlin, T.R.; Miranda-Vergara, M.C.; Dutta, S.; Natarajan, A.; Facchine, B.A.; Jackson, K.M.; Nystrom, L.; Li, J.; et al. The CXCL5/CXCR2 axis is sufficient to promote breast cancer colonization during bone metastasis. Nat. Commun. 2019, 10, 4404. [Google Scholar] [CrossRef] [Green Version]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef] [Green Version]
- Acharyya, S.; Oskarsson, T.; Vanharanta, S.; Malladi, S.; Kim, J.; Morris, P.G.; Manova-Todorova, K.; Leversha, M.; Hogg, N.; Seshan, V.E.; et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell 2012, 150, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Tawara, K.; Scott, H.; Emathinger, J.; Ide, A.; Fox, R.; Greiner, D.; LaJoie, D.; Hedeen, D.; Nandakumar, M.; Oler, A.J.; et al. Co-expression of VEGF and IL-6 family cytokines is associated with decreased survival in HER2 negative breast cancer patients: Subtype-specific IL-6 family cytokine-mediated VEGF secretion. Transl. Oncol. 2019, 12, 245–255. [Google Scholar] [CrossRef]
- Schirosi, L.; de Summa, S.; Tommasi, S.; Paradiso, A.; Gasparini, G.; Popescu, O.; Simone, G.; Mangia, A. VEGF and TWIST1 in a 16-biomarker immunoprofile useful for prognosis of breast cancer patients. Int. J. Cancer 2017, 141, 1901–1911. [Google Scholar] [CrossRef] [Green Version]
- Casbas-Hernandez, P.; D’Arcy, M.; Roman-Perez, E.; Brauer, H.A.; McNaughton, K.; Miller, S.M.; Chhetri, R.K.; Oldenburg, A.L.; Fleming, J.M.; Amos, K.D.; et al. Role of HGF in epithelial-stromal cell interactions during progression from benign breast disease to ductal carcinoma in situ. Breast Cancer Res. 2013, 15, R82. [Google Scholar] [CrossRef] [Green Version]
- Chin, Y.R.; Toker, A. Function of Akt/PKB signaling to cell motility, invasion and the tumor stroma in cancer. Cell Signal. 2009, 21, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Ridolfi, E.; Matteucci, E.; Maroni, P.; Desiderio, M.A. Inhibitory effect of HGF on invasiveness of aggressive MDA-MB231 breast carcinoma cells, and role of HDACs. Br. J. Cancer 2008, 99, 1623–1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veenstra, C.; Perez-Tenorio, G.; Stelling, A.; Karlsson, E.; Mirwani, S.M.; Nordenskoljd, B.; Fornander, T.; Stal, O. Met and its ligand HGF are associated with clinical outcome in breast cancer. Oncotarget 2016, 7, 37145–37159. [Google Scholar] [CrossRef] [PubMed]
- Ho-Yen, C.M.; Jones, J.L.; Kermorgant, S. The clinical and functional significance of c-Met in breast cancer: A review. Breast Cancer Res. 2015, 17, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Sakakura, K.; Kudo, T.; Toyoda, M.; Kaira, K.; Oyama, T.; Chikamatsu, K. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget 2017, 8, 8633–8647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamiyan, T.; Kuroda, H.; Yamaguchi, R.; Abe, A.; Hayashi, M. CD68- and CD163-positive tumor-associated macrophages in triple negative cancer of the breast. Virchows Arch. 2020, 477, 767–775. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [Green Version]
- Papaccio, F.; della Corte, C.M.; Viscardi, G.; di Liello, R.; Esposito, G.; Sparano, F.; Ciardiello, F.; Morgillo, F. HGF/MET and the immune system: Relevance for cancer immunotherapy. Int. J. Mol. Sci. 2018, 19, 3595. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.K.; Vipparthi, K.; Thatikonda, V.; Arun, I.; Bhattacharjee, S.; Sharan, R.; Arun, P.; Singh, S. A subtype of cancer-associated fibroblasts with lower expression of alpha-smooth muscle actin suppresses stemness through BMP4 in oral carcinoma. Oncogenesis 2018, 7, 78. [Google Scholar] [CrossRef]
- Yamashita, M.; Ogawa, T.; Zhang, X.; Hanamura, N.; Kashikura, Y.; Takamura, M.; Yoneda, M.; Shiraishi, T. Role of stromal myofibroblasts in invasive breast cancer: Stromal expression of alpha-smooth muscle actin correlates with worse clinical outcome. Breast Cancer 2012, 19, 170–176. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | n | Median ERα36 Protein Levels in Tumor Stroma Fibroblasts (25–75th Percentile) | p |
|---|---|---|---|
| T stage | p = 0.151 | ||
| T1 | 44 | 22.25 (15.50–31.00 | |
| T2 | 53 | 18.00 (12.25–28.00) | |
| T3 | 3 | 11.25 (9.79–18.13) | |
| T4 | 2 | 7.50 (7.00–8.00) | |
| N stage | p = 0.425 | ||
| N0 | 43 | 20.00 (13.33–30.00) | |
| N1 | 53 | 20.00 (10.63–26.50) | |
| Grading | p = 0.356 | ||
| 1 | 14 | 13.33 (8.375–32.80) | |
| 2 | 54 | 20.00 (12.50–28.00) | |
| 3 | 40 | 21.00 (12.50–29.50) | |
| Histological subtype | p = 0.960 | ||
| Ductal | 90 | 20.00 (11.81–28.00) | |
| Other | 13 | 17.50 (12.29–33.50) | |
| Molecular type | p = 0.429 | ||
| Luminal A | 30 | 17.75 (9.69–24.50) | |
| Luminal B HER2− | 25 | 17.50 (11.13–28.00) | |
| Luminal B HER2+ | 22 | 20.00 (11.81–24.25) | |
| Non luminal HER2+ | 5 | 22.50 (18.00–45.00) | |
| Triple-negative | 14 | 23.00 (17.00–40.00) | |
| ER status | p = 0.311 | ||
| 0 | 23 | 22.00 (18.00–40.00) | |
| 1 | 73 | 19.00 (11.25–26.50) | |
| PR status | p = 0.118 | ||
| 0 | 26 | 22.50 (19.50–40.00) | |
| 1 | 70 | 17.50 (11.19–28.00) | |
| HER2 status | p = 0.563 | ||
| 0 | 69 | 20.00 (11.13–28.00) | |
| 1 | 27 | 20.00 (14.00–25.00) |
| Cytokine | DFS | OS | ||||||
|---|---|---|---|---|---|---|---|---|
| HR (Hazard Ratio) | 95% Lower Cl | 95% Upper Cl | Log-Rank p-Value | HR (Hazard Ratio) | 95% Lower Cl | 95% Upper Cl | Log-Rank p-Value | |
| CXCL1 | 1.496 | 0.155 | 14.44 | 0.7 | 0.8233 | 0.2375 | 2.855 | 0.8 |
| CXCL5 | 3.98 × 10−9 | 0 | Inf | 0.3 | 1.321 | 0.4677 | 3.731 | 0.6 |
| IL-8 | 4.43 | 0.6219 | 31.56 | 0.1 | 3.157 | 1.226 | 8.133 | 0.03 |
| GM-CSF | 3.83 × 10−9 | 0 | Inf | 0.3 | 0.4564 | 0.1046 | 1.992 | 0.3 |
| VEGF | 1.292 | 0.1339 | 12.46 | 0.8 | 1.121 | 0.3651 | 3.441 | 0.8 |
| HGF | 7.159 | 0.9431 | 54.34 | 0.03 | 2.697 | 1.025 | 7.094 | 0.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagel, A.; Popeda, M.; Muchlinska, A.; Sadej, R.; Szade, J.; Zielinski, J.; Skokowski, J.; Niemira, M.; Kretowski, A.; Markiewicz, A.; et al. ERα36-High Cancer-Associated Fibroblasts as an Unfavorable Factor in Triple-Negative Breast Cancer. Cancers 2022, 14, 2005. https://doi.org/10.3390/cancers14082005
Nagel A, Popeda M, Muchlinska A, Sadej R, Szade J, Zielinski J, Skokowski J, Niemira M, Kretowski A, Markiewicz A, et al. ERα36-High Cancer-Associated Fibroblasts as an Unfavorable Factor in Triple-Negative Breast Cancer. Cancers. 2022; 14(8):2005. https://doi.org/10.3390/cancers14082005
Chicago/Turabian StyleNagel, Anna, Marta Popeda, Anna Muchlinska, Rafal Sadej, Jolanta Szade, Jacek Zielinski, Jaroslaw Skokowski, Magdalena Niemira, Adam Kretowski, Aleksandra Markiewicz, and et al. 2022. "ERα36-High Cancer-Associated Fibroblasts as an Unfavorable Factor in Triple-Negative Breast Cancer" Cancers 14, no. 8: 2005. https://doi.org/10.3390/cancers14082005