
Application of an Ultrasensitive NGS-Based Blood Test for the Diagnosis of Early-Stage Lung Cancer: Sensitivity, a Hurdle Still Difficult to Overcome

 , ,
, ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Sample Collection and Processing
2.2.1. AVENIO ctDNA Surveillance Assay
2.2.2. Variant Confirmation
2.3. Statistical Analysis
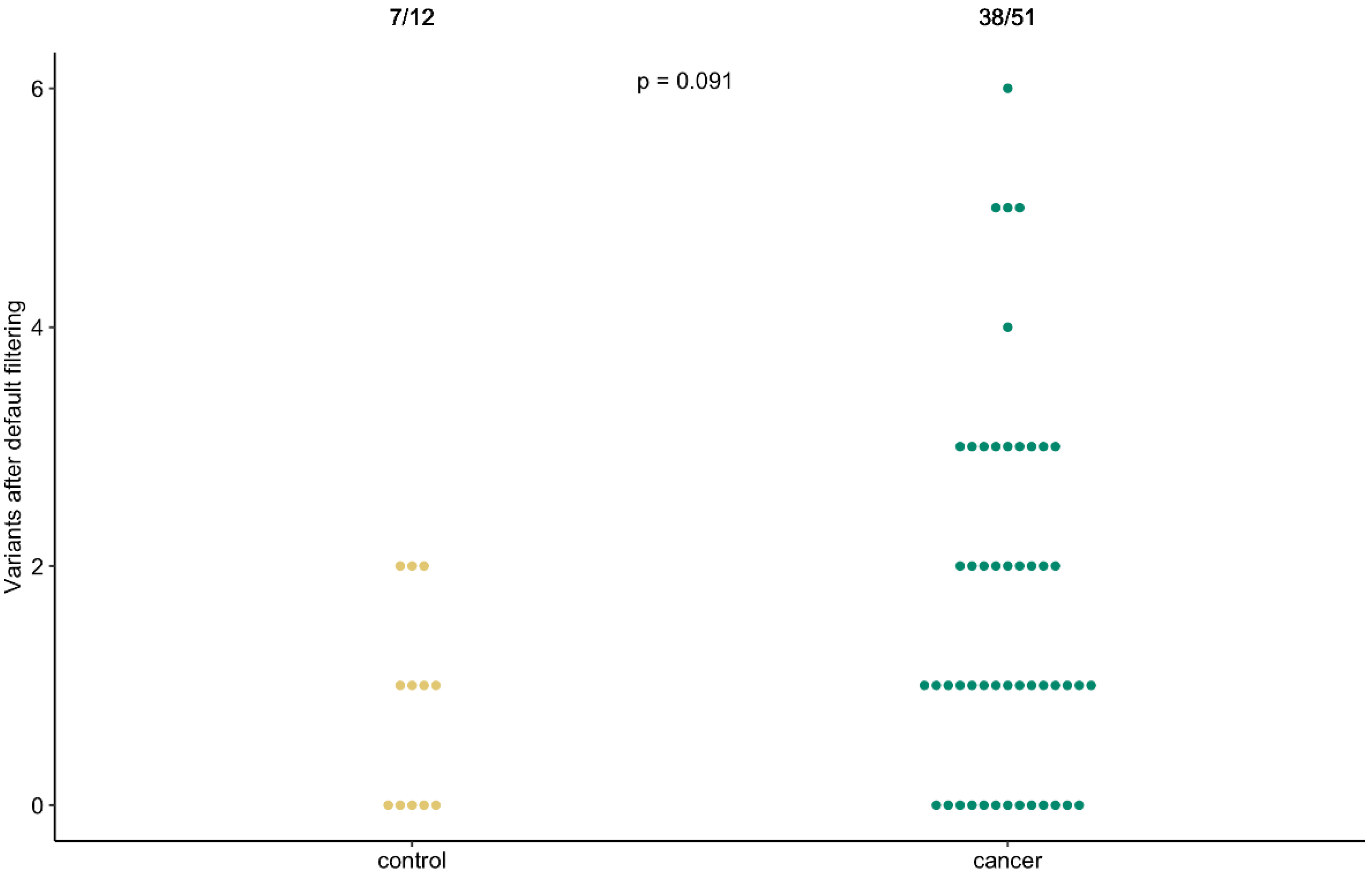
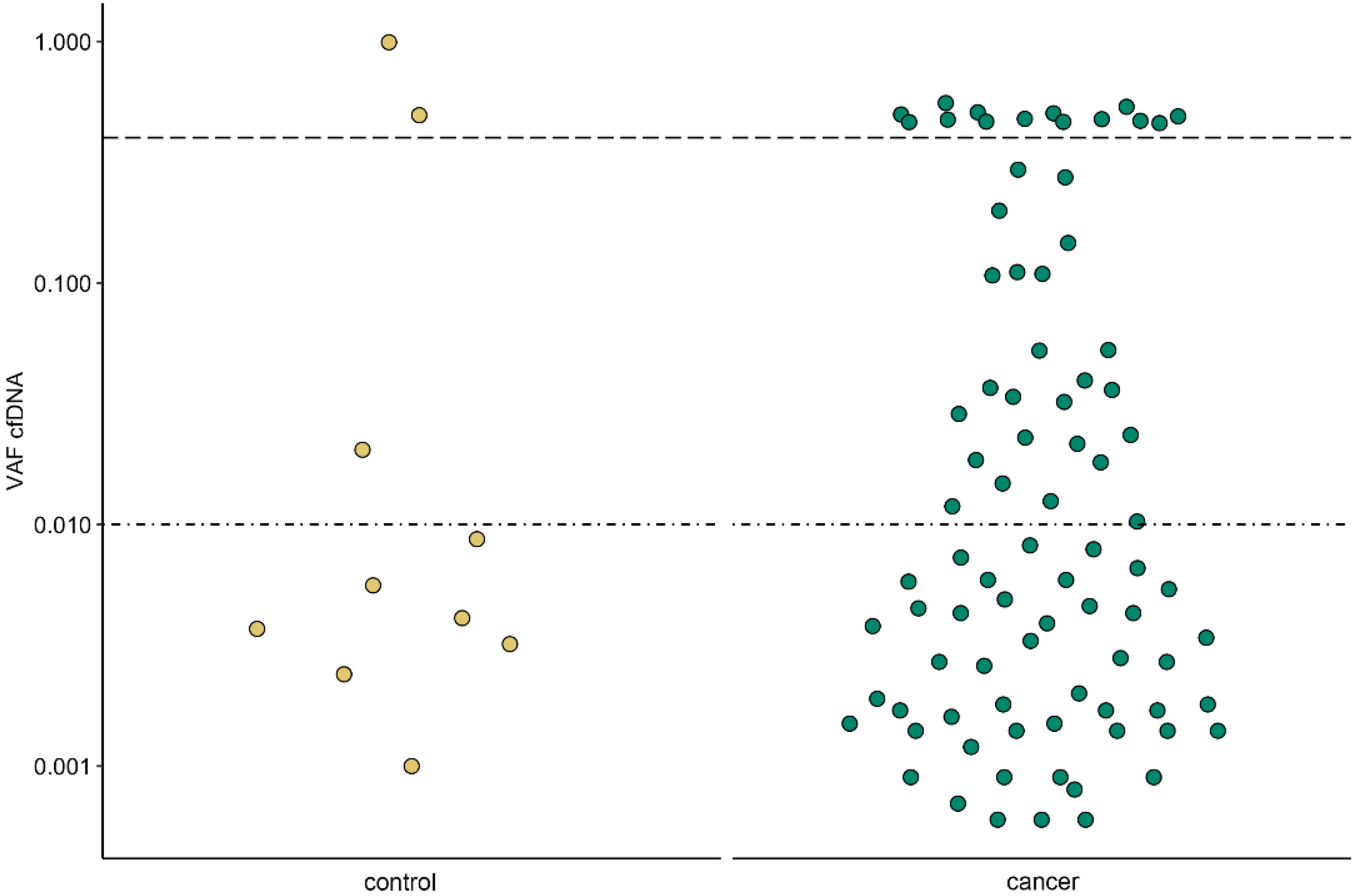
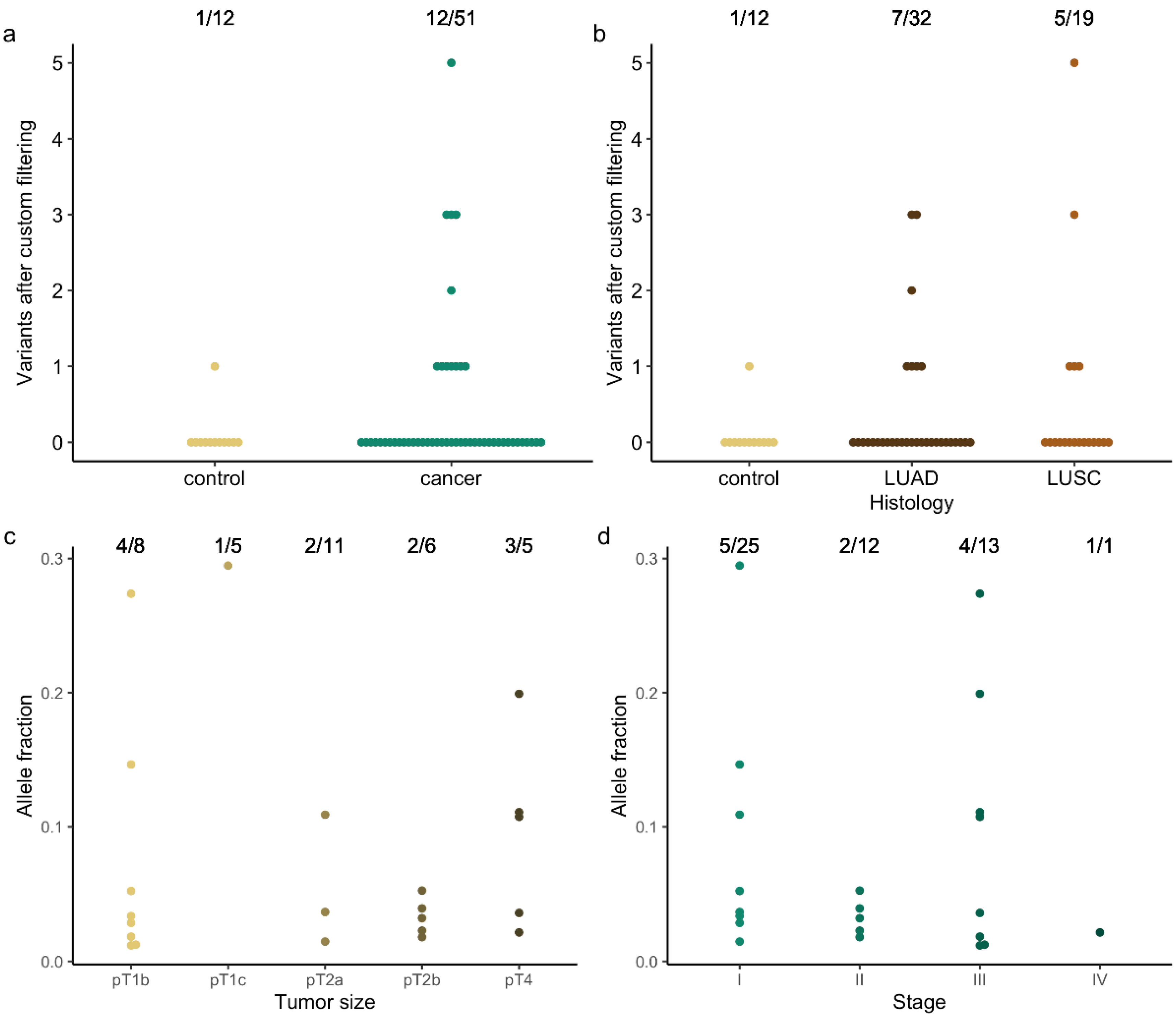
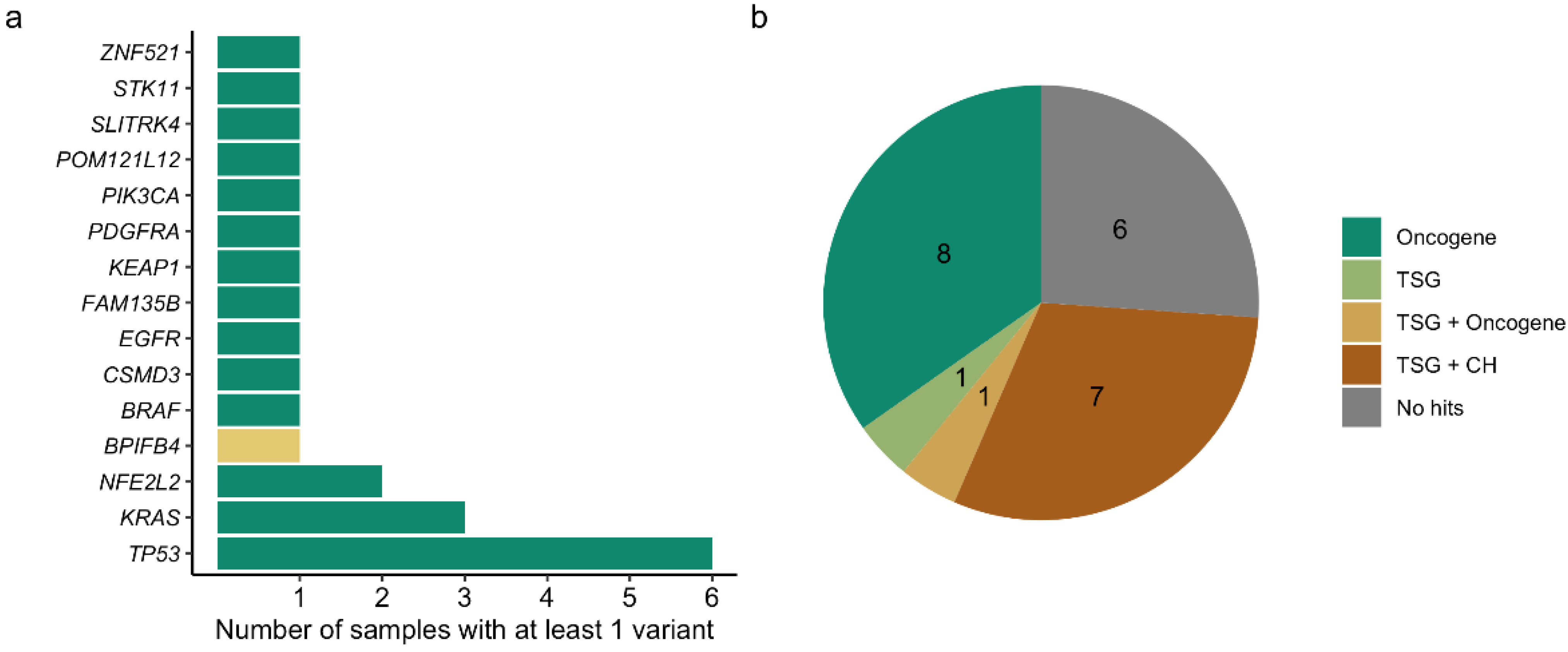
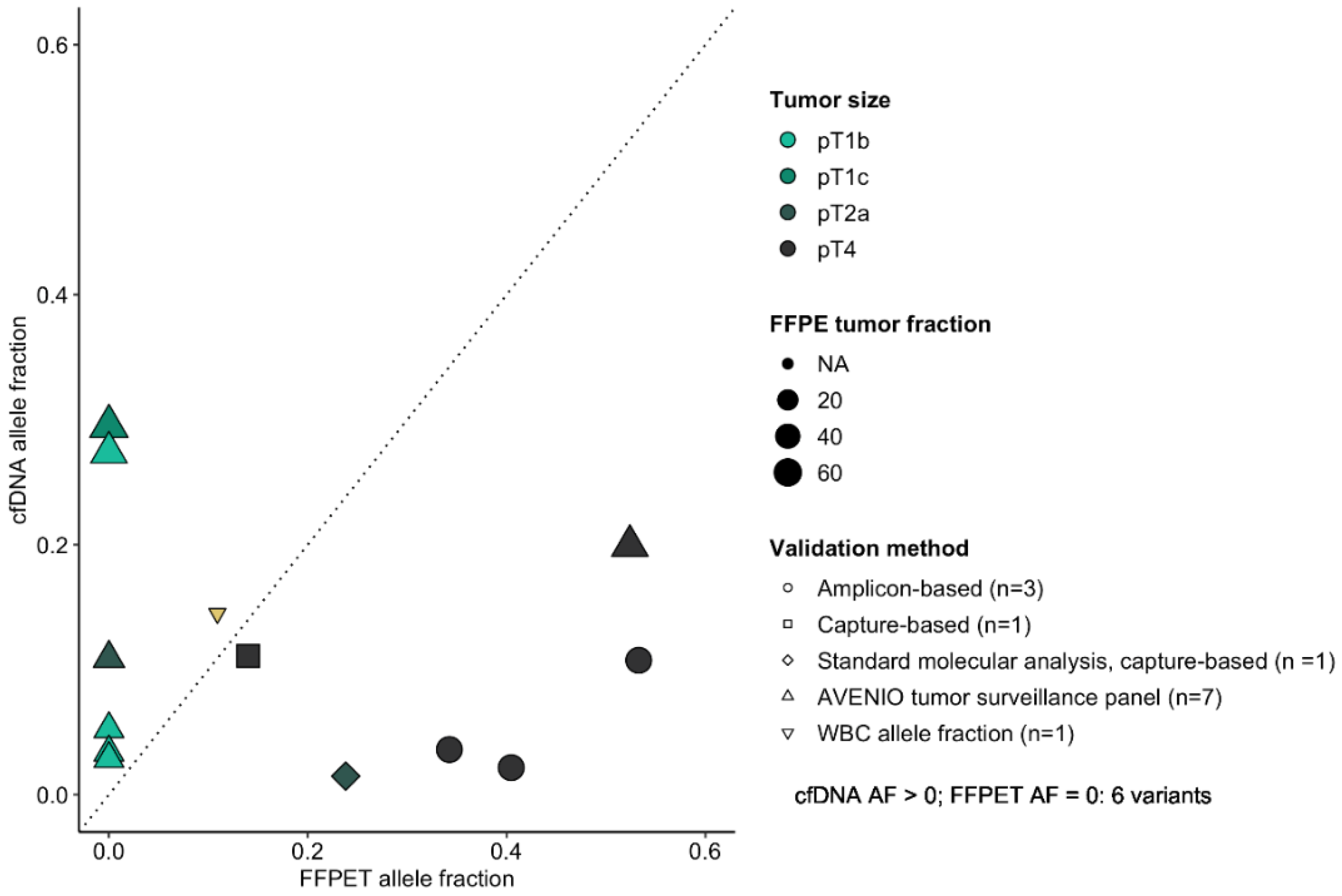
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Zhang, Y.; Shi, L.; Simoff, M.J.; Wagner, O.J.; Lavin, J. Biopsy Frequency and Complications among Lung Cancer Patients in the United States. Lung Cancer Manag. 2020, 9, LMT40. [Google Scholar] [CrossRef] [PubMed]
- Sihoe, A.D.L.; Hiranandani, R.; Wong, H.; Yeung, E.S.L. Operating on a Suspicious Lung Mass without a Preoperative Tissue Diagnosis: Pros and Cons. Eur. J. Cardio-Thorac. Surg. 2013, 44, 231–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaaki, S.; Kidane, B.; Srinathan, S.; Tan, L.; Buduhan, G. Is Tissue Still the Issue? Lobectomy for Suspicious Lung Nodules without Confirmation of Malignancy. J. Surg. Oncol. 2018, 117, 977–984. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Noda, Y.; Shibazaki, T.; Kato, D.; Matsudaira, H.; Hirano, J.; Ohtsuka, T. Definitive Lobectomy without Frozen Section Analysis Is a Treatment Option for Large or Deep Nodules Selected Carefully with Clinical Diagnosis of Malignancy. Thorac. Cancer 2020, 11, 1996–2004. [Google Scholar] [CrossRef] [PubMed]
- Freitas, C.; Sousa, C.; Machado, F.; Serino, M.; Santos, V.; Cruz-Martins, N.; Teixeira, A.; Cunha, A.; Pereira, T.; Oliveira, H.P.; et al. The Role of Liquid Biopsy in Early Diagnosis of Lung Cancer. Front. Oncol. 2021, 11, 634316. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J.; Crinò, L.; Dooms, C.; Douillard, J.Y.; Faivre-Finn, C.; Lim, E.; Rocco, G.; Senan, S.; van Schil, P.; Veronesi, G.; et al. 2nd ESMO Consensus Conference on Lung Cancer: Early-Stage Non-Small-Cell Lung Cancer Consensus on Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2014, 25, 1462–1474. [Google Scholar] [CrossRef]
- Mannelli, C. Tissue vs. Liquid Biopsies for Cancer Detection: Ethical Issues. J. Bioethical Inq. 2019, 16, 551–557. [Google Scholar] [CrossRef]
- Raman, L.; Van Der Linden, M.; Van Der Eecken, K.; Vermaelen, K.; Demedts, I.; Surmont, V.; Himpe, U.; Dedeurwaerdere, F.; Ferdinande, L.; Lievens, Y.; et al. Shallow Whole-Genome Sequencing of Plasma Cell-Free DNA Accurately Differentiates Small from Non-Small Cell Lung Carcinoma. Genome Med. 2020, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Abbosh, C.; Birkbak, N.J.; Swanton, C. Early Stage NSCLC—Challenges to Implementing CtDNA-Based Screening and MRD Detection. Nat. Rev. Clin. Oncol. 2018, 15, 577–586. [Google Scholar] [CrossRef]
- Guo, N.; Lou, F.; Ma, Y.; Li, J.; Yang, B.; Chen, W.; Ye, H.; Zhang, J.B.; Zhao, M.Y.; Wu, W.J.; et al. Circulating Tumor DNA Detection in Lung Cancer Patients before and after Surgery. Sci. Rep. 2016, 6, 33519. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Zhao, H.; Shi, Y.; Yang, F.; Wang, L.T.; Kang, G.; Nie, Y.; Wang, J. Perioperative Dynamic Changes in Circulating Tumor DNA in Patients with Lung Cancer (Dynamic). Clin. Cancer Res. 2019, 25, 7058–7067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An Ultrasensitive Method for Quantitating Circulating Tumor DNA with Broad Patient Coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated Digital Error Suppression for Improved Detection of Circulating Tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Chabon, J.J.; Hamilton, E.G.; Kurtz, D.M.; Esfahani, M.S.; Moding, E.J.; Stehr, H.; Schroers-Martin, J.; Nabet, B.Y.; Chen, B.; Chaudhuri, A.A.; et al. Integrating Genomic Features for Non-Invasive Early Lung Cancer Detection. Nature 2020, 580, 245–251. [Google Scholar] [CrossRef] [PubMed]
- De Leeneer, K.; Hellemans, J.; Steyaert, W.; Lefever, S.; Vereecke, I.; Debals, E.; Crombez, B.; Baetens, M.; Van Heetvelde, M.; Coppieters, F.; et al. Flexible, Scalable, and Efficient Targeted Resequencing on a Benchtop Sequencer for Variant Detection in Clinical Practice. Hum. Mutat. 2015, 36, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Razavi, P.; Li, B.T.; Brown, D.N.; Jung, B.; Hubbell, E.; Shen, R.; Abida, W.; Juluru, K.; De Bruijn, I.; Hou, C.; et al. High-Intensity Sequencing Reveals the Sources of Plasma Circulating Cell-Free DNA Variants. Nat. Med. 2019, 25, 1928–1937. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.e18. [Google Scholar] [CrossRef] [Green Version]
- Brannon, A.R.; Jayakumaran, G.; Diosdado, M.; Patel, J.; Razumova, A.; Hu, Y.; Meng, F.; Haque, M.; Sadowska, J.; Murphy, B.J.; et al. Enhanced Specificity of Clinical High-Sensitivity Tumor Mutation Profiling in Cell-Free DNA via Paired Normal Sequencing Using MSK-ACCESS. Nat. Commun. 2021, 12, 3770. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, J.; Xiao, J.; Wang, L.; Hu, X.; Yu, W.; Song, G.; Lou, J.; Chen, J.F. Heterogeneous Mutation Pattern in Tumor Tissue and Circulating Tumor DNA Warrants Parallel NGS Panel Testing. Mol. Cancer 2018, 17, 4–8. [Google Scholar] [CrossRef]
- Ahrendt, S.A.; Anthony Decker, P.; Alawi, E.A.; Zhu, Y.R.; Sanchez-Cespedes, M.; Yang, S.C.; Haasler, G.B.; Kajdacsy-Balla, A.; Demeure, M.J.; Sidransky, D. Cigarette Smoking Is Strongly Associated with Mutation of the K-Ras Gene in Patients with Primary Adenocarcinoma of the Lung. Cancer 2001, 92, 1525–1530. [Google Scholar] [CrossRef]
- Gibbons, D.L.; Byers, L.A.; Kurie, J.M. Smoking, P53 Mutation, and Lung Cancer. Mol. Cancer Res. 2014, 12, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Adams, H.P.; Yao, L.; Yaung, S.; Lal, P.; Balasubramanyam, A.; Fuhlbrück, F.; Tikoo, N.; Lovejoy, A.F.; Froehler, S.; et al. Concordance of Genomic Alterations by Next-Generation Sequencing in Tumor Tissue versus Cell-Free DNA in Stage I–IV Non–Small Cell Lung Cancer. J. Mol. Diagn. 2020, 22, 228–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siravegna, G.; Marsoni, S.; Siena, S.; Bardelli, A. Integrating Liquid Biopsies into the Management of Cancer. Nat. Rev. Clin. Oncol. 2017, 14, 531–548. [Google Scholar] [CrossRef]
- Wan, M.J.C.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid Biopsies Come of Age: Towards Implementation of Circulating Tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Quesne, J.L.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic CtDNA Analysis Depicts Early Stage Lung Cancer Evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Fiala, C.; Diamandis, E.P. Utility of Circulating Tumor DNA in Cancer Diagnostics with Emphasis on Early Detection. BMC Med. 2018, 16, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.C.; Maddala, T.; Aravanis, A.; Hubbell, E.; Beausang, J.F.; Filippova, D.; Gross, S.; Jamshidi, A.; Kurtzman, K.; Shen, L.; et al. Breast Cancer Cell-Free DNA (CfDNA) Profiles Reflect Underlying Tumor Biology: The Circulating Cell-Free Genome Atlas (CCGA) Study. J. Clin. Oncol. 2018, 36, 536. [Google Scholar] [CrossRef] [Green Version]
- Gasi Tandefelt, D.; de Bono, J. Circulating Cell-Free DNA: Translating Prostate Cancer Genomics into Clinical Care. Mol. Aspects Med. 2020, 72, 100837. [Google Scholar] [CrossRef]
- Caruso, R.; Parisi, A.; Bonanno, A.; Paparo, D.; Emilia, Q.; Branca, G.; Scardigno, M.; Fedele, F. Histologic Coagulative Tumour Necrosis as a Prognostic Indicator of Aggressiveness in Renal, Lung, Thyroid and Colorectal Carcinomas: A Brief Review. Oncol. Lett. 2012, 3, 16–18. [Google Scholar] [CrossRef] [Green Version]
- Raman, L.; van der Linden, M.; de Vriendt, C.; van den Broeck, B.; Muylle, K.; Deeren, D.; Dedeurwaerdere, F.; Verbeke, S.; Dendooven, A.; de Grove, K.; et al. Shallow-Depth Sequencing of Cell-Free DNA for Hodgkin and Diffuse Large B-Cell Lymphoma (Differential) Diagnosis: A Standardized Approach with Underappreciated Potential. Haematologica 2022, 107, 211–220. [Google Scholar] [CrossRef]
- Vandenberghe, P.; Wlodarska, I.; Tousseyn, T.; Dehaspe, L.; Dierickx, D.; Verheecke, M.; Uyttebroeck, A.; Bechter, O.; Delforge, M.; Vandecaveye, V.; et al. Non-Invasive Detection of Genomic Imbalances in Hodgkin/Reed-Sternberg Cells in Early and Advanced Stage Hodgkin’s Lymphoma by Sequencing of Circulating Cell-Free DNA: A Technical Proof-of-Principle Study. Lancet Haematol. 2015, 2, e55–e65. [Google Scholar] [CrossRef] [Green Version]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Trani, M.D.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating Tumor DNA Reveals Genetics, Clonal Evolution, and Residual Disease in Classical Hodgkin Lymphoma. Blood 2018, 131, 2413–2425. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, D.M.; Scherer, F.; Jin, M.C.; Soo, J.; Craig, A.F.M.; Esfahani, M.S.; Chabon, J.J.; Stehr, H.; Liu, C.L.; Tibshirani, R.; et al. Circulating Tumor DNA Measurements as Early Outcome Predictors in Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2018, 36, 2845–2853. [Google Scholar] [CrossRef]
- Ohara, S.; Suda, K.; Sakai, K.; Nishino, M.; Chiba, M.; Shimoji, M.; Takemoto, T.; Fujino, T.; Koga, T.; Hamada, A.; et al. Prognostic Implications of Preoperative versus Postoperative Circulating Tumor DNA in Surgically Resected Lung Cancer Patients: A Pilot Study. Transl. Lung Cancer Res. 2020, 9, 1915–1923. [Google Scholar] [CrossRef]
- Serrano, M.J.; Garrido-Navas, M.C.; Mochon, J.J.D.; Cristofanilli, M.; Gil-Bazo, I.; Pauwels, P.; Malapelle, U.; Russo, A.; Lorente, J.A.; Ruiz-Rodriguez, A.J.; et al. Precision Prevention and Cancer Interception: The New Challenges of Liquid Biopsy. Cancer Discov. 2020, 10, 1635–1644. [Google Scholar] [CrossRef]
- De Koker, A.; Van Paemel, R.; De Wilde, B.; De Preter, K.; Callewaert, N. A Versatile Method for Circulating Cell-Free DNA Methylome Profiling by Reduced Representation Bisulfite Sequencing. bioRxiv 2019, 663195. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Patients | Controls | ||
|---|---|---|---|---|
| n = 51 | n = 12 | |||
| Age (yrs) | 66 | (36–83) | 64 | (57–74) |
| Smoking | ||||
| Yes | 44 | (86.3%) | 9 | (75.0%) |
| No | 3 | (5.9%) | 2 | (16.7%) |
| NA | 4 | (7.8%) | 1 | (8.3%) |
| Histology | ||||
| LUAD | 32 | (62.7%) | - | - |
| LUSC | 19 | (37.3%) | - | - |
| T stage | ||||
| T1mi | 3 | (5.9%) | - | - |
| T1a | 6 | (11.8%) | - | - |
| T1b | 8 | (15.7%) | - | - |
| T1c | 5 | (9.8%) | - | - |
| T2a | 11 | (21.6%) | - | - |
| T2b | 6 | (11.8%) | - | - |
| T3 | 7 | (13.7%) | - | - |
| T4 | 5 | (9.8%) | - | - |
| Overall stage | ||||
| I | 25 | (49.0%) | - | - |
| II | 12 | (23.5%) | - | - |
| III | 13 | (25.5%) | - | - |
| IV | 1 | (2.0%) | - | - |
| Sample | Gene | Coding Change | Amino Acid Change | AF | FFPET AF | WBC AF | FFPET TC% | Validation Method | CNV Score |
|---|---|---|---|---|---|---|---|---|---|
| C02 | KRAS | c.37G>T | p.Gly13Cys | 2.16 | 40.47 | N/A | 50 | Amplicon-based | N/A |
| C11 | TP53 | c.833C>T | p.Pro278Leu | 11.10 | 14.00 | Absent | 50 | Capture-based | N/A |
| C11 | NFE2L2 | c.77A>T | p.Gln26Leu | 10.75 | 53.30 | N/A | 50 | Amplicon-based | N/A |
| C11 | SLITRK4 | c.1960G>A | p.Glu654Lys | 3.61 | 34.25 | N/A | 50 | Amplicon-based | N/A |
| C13 | BRAF | c.1781A>C | p.Asp594Ala | 29.47 | Absent | N/A | 70 | AVENIO tumor surveillance panel | N/A |
| C25 | KRAS | c.35G>A | p.Gly12Asp | 5.25 | Absent | N/A | 30 | AVENIO tumor surveillance panel | N/A |
| C25 | KEAP1 | c.463G>T | p.Val155Phe | 3.38 | Absent | N/A | 30 | AVENIO tumor surveillance panel | N/A |
| C25 | STK11 | c.725G>T | p.Gly242Val | 2.87 | Absent | N/A | 30 | AVENIO tumor surveillance panel | N/A |
| C32 | KRAS | c.34G>T | p.Gly12Cys | 1.48 | 23.82 | N/A | 32 | Capture-based | N/A |
| C32 | ZNF521 | c.1544G>T | p.Cys515Phe | 3.68 | N/A | N/A | N/A | Not validated | N/A |
| C33 | TP53 | c.722C>G | p.Ser241Cys | 10.91 | Absent | 14.49 | 35 | AVENIO tumor surveillance panel | N/A |
| C38 | TP53 | c.493C>T | p.Gln165* | 19.92 | 52.41 | N/A | 60 | AVENIO tumor surveillance panel | N/A |
| C39 | PDGFRA | c.248C>T | p.Thr83Met | 27.38 | Absent | N/A | 60 | AVENIO tumor surveillance panel | N/A |
| C45 | TP53 | c.747G>T | p.Arg249Ser | 1.19 | N/A | N/A | N/A | Not validated | N/A |
| C45 | CSMD3 | c.3406C>A | p.Leu1136Met | 1.85 | N/A | N/A | N/A | Not validated | N/A |
| C45 | FAM135B | c.1347G>T | p.Met449Ile | 1.25 | N/A | N/A | N/A | Not validated | N/A |
| C46 | TP53 | c.844C>T | p.Arg282Trp | 5.28 | N/A | N/A | N/A | Not validated | N/A |
| C46 | TP53 | c.746G>T | p.Arg249Met | 1.81 | N/A | N/A | N/A | Not validated | N/A |
| C46 | PIK3CA | c.1624G>A | p.Glu542Lys | 3.22 | N/A | N/A | N/A | Not validated | N/A |
| C46 | EGFR | N/A | N/A | N/A | N/A | N/A | N/A | Not validated | 6.47 |
| C46 | POM121L12 | c.325G>T | p.Gly109Trp | 3.95 | N/A | N/A | N/A | Not validated | N/A |
| C47 | TP53 | c.376-1G>A | N/A | 2.29 | N/A | N/A | N/A | Not validated | N/A |
| C48 | NFE2L2 | c.100C>G | p.Arg34Gly | 14.65 | N/A | N/A | N/A | Not validated | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van der Linden, M.; Van Gaever, B.; Raman, L.; Vermaelen, K.; Demedts, I.; Surmont, V.; Himpe, U.; Lievens, Y.; Ferdinande, L.; Dedeurwaerdere, F.; et al. Application of an Ultrasensitive NGS-Based Blood Test for the Diagnosis of Early-Stage Lung Cancer: Sensitivity, a Hurdle Still Difficult to Overcome. Cancers 2022, 14, 2031. https://doi.org/10.3390/cancers14082031
Van der Linden M, Van Gaever B, Raman L, Vermaelen K, Demedts I, Surmont V, Himpe U, Lievens Y, Ferdinande L, Dedeurwaerdere F, et al. Application of an Ultrasensitive NGS-Based Blood Test for the Diagnosis of Early-Stage Lung Cancer: Sensitivity, a Hurdle Still Difficult to Overcome. Cancers. 2022; 14(8):2031. https://doi.org/10.3390/cancers14082031
Chicago/Turabian StyleVan der Linden, Malaïka, Bram Van Gaever, Lennart Raman, Karim Vermaelen, Ingel Demedts, Veerle Surmont, Ulrike Himpe, Yolande Lievens, Liesbeth Ferdinande, Franceska Dedeurwaerdere, and et al. 2022. "Application of an Ultrasensitive NGS-Based Blood Test for the Diagnosis of Early-Stage Lung Cancer: Sensitivity, a Hurdle Still Difficult to Overcome" Cancers 14, no. 8: 2031. https://doi.org/10.3390/cancers14082031
APA StyleVan der Linden, M., Van Gaever, B., Raman, L., Vermaelen, K., Demedts, I., Surmont, V., Himpe, U., Lievens, Y., Ferdinande, L., Dedeurwaerdere, F., Van der Meulen, J., Claes, K., Menten, B., & Van Dorpe, J. (2022). Application of an Ultrasensitive NGS-Based Blood Test for the Diagnosis of Early-Stage Lung Cancer: Sensitivity, a Hurdle Still Difficult to Overcome. Cancers, 14(8), 2031. https://doi.org/10.3390/cancers14082031