
Modeling Pancreatic Cancer with Patient-Derived Organoids Integrating Cancer-Associated Fibroblasts

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Three-Dimensional Organoid Culture System
2.2. Isolation of CAFs from Pancreatic Cancer Tissue
2.3. Co-Culture of Pancreatic Cancer Organoids (PCOs) and CAFs
2.4. KRAS Mutation Analysis
2.5. Histology and Immunohistochemistry
2.6. Immunofluorescence
2.7. Flow Cytometry for Cell Sorting
2.8. Quantitative Reverse Transcription-Polymerase Chain Reaction
2.9. Cell Viability Analysis
2.10. In Vivo Experiments
2.11. Statistical Analysis
3. Results
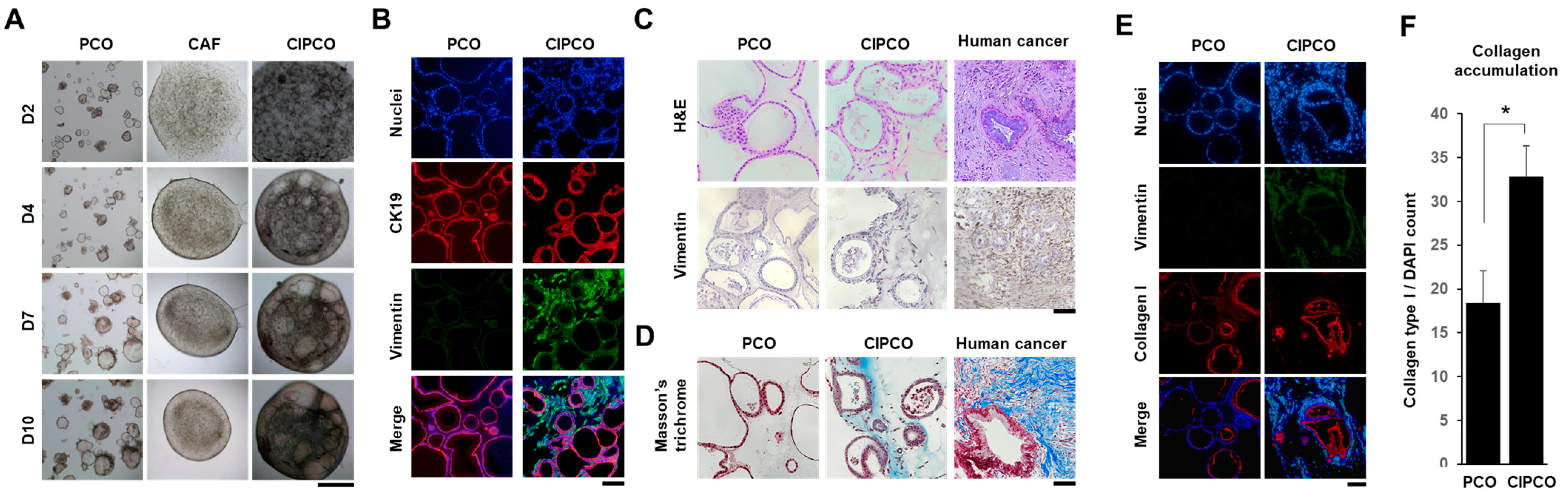
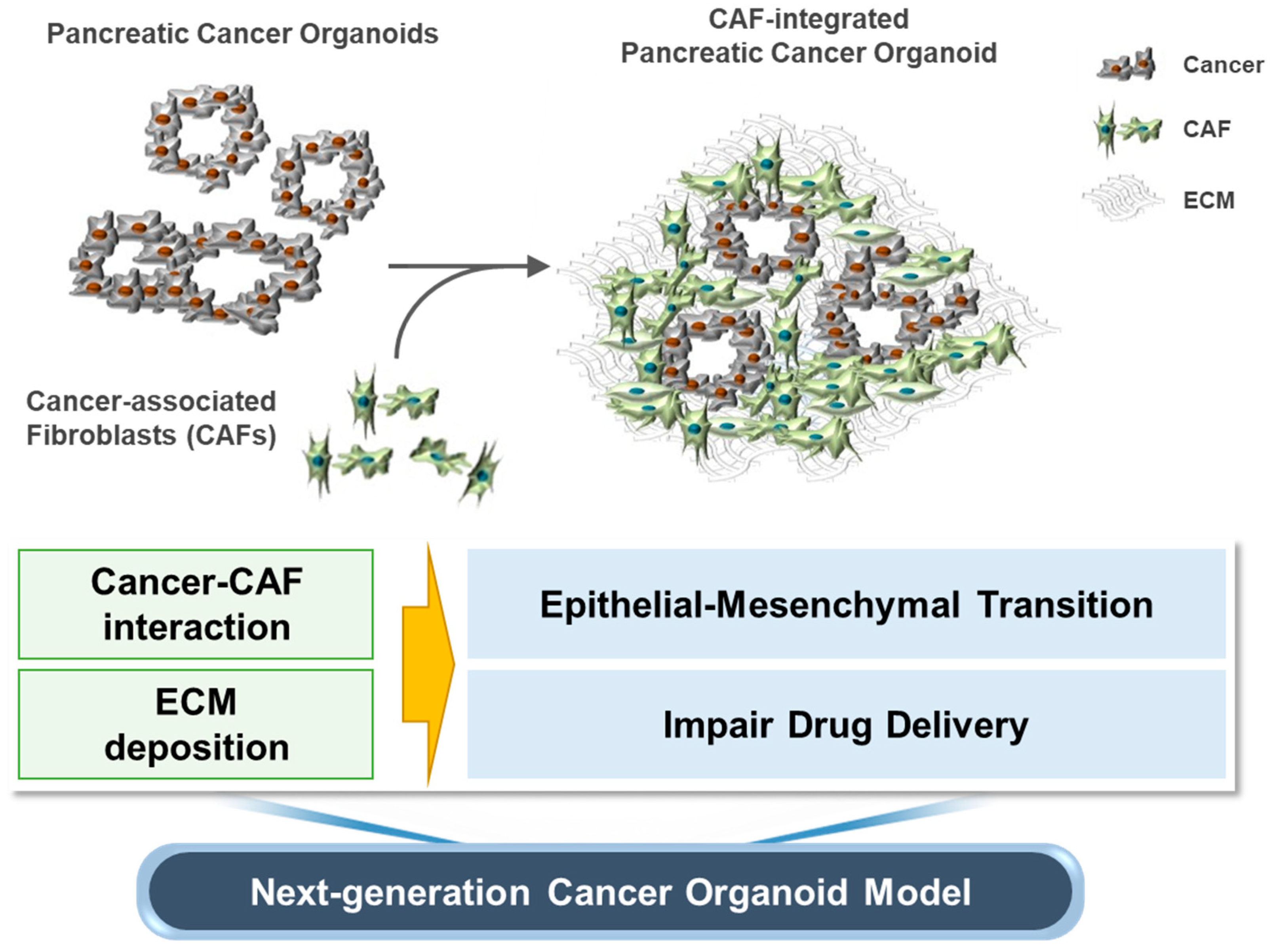
3.1. Establishment of CAF-Integrated Pancreatic Cancer Organoids
3.2. Characterization of CAFs and Cancer Cells in CIPCOs
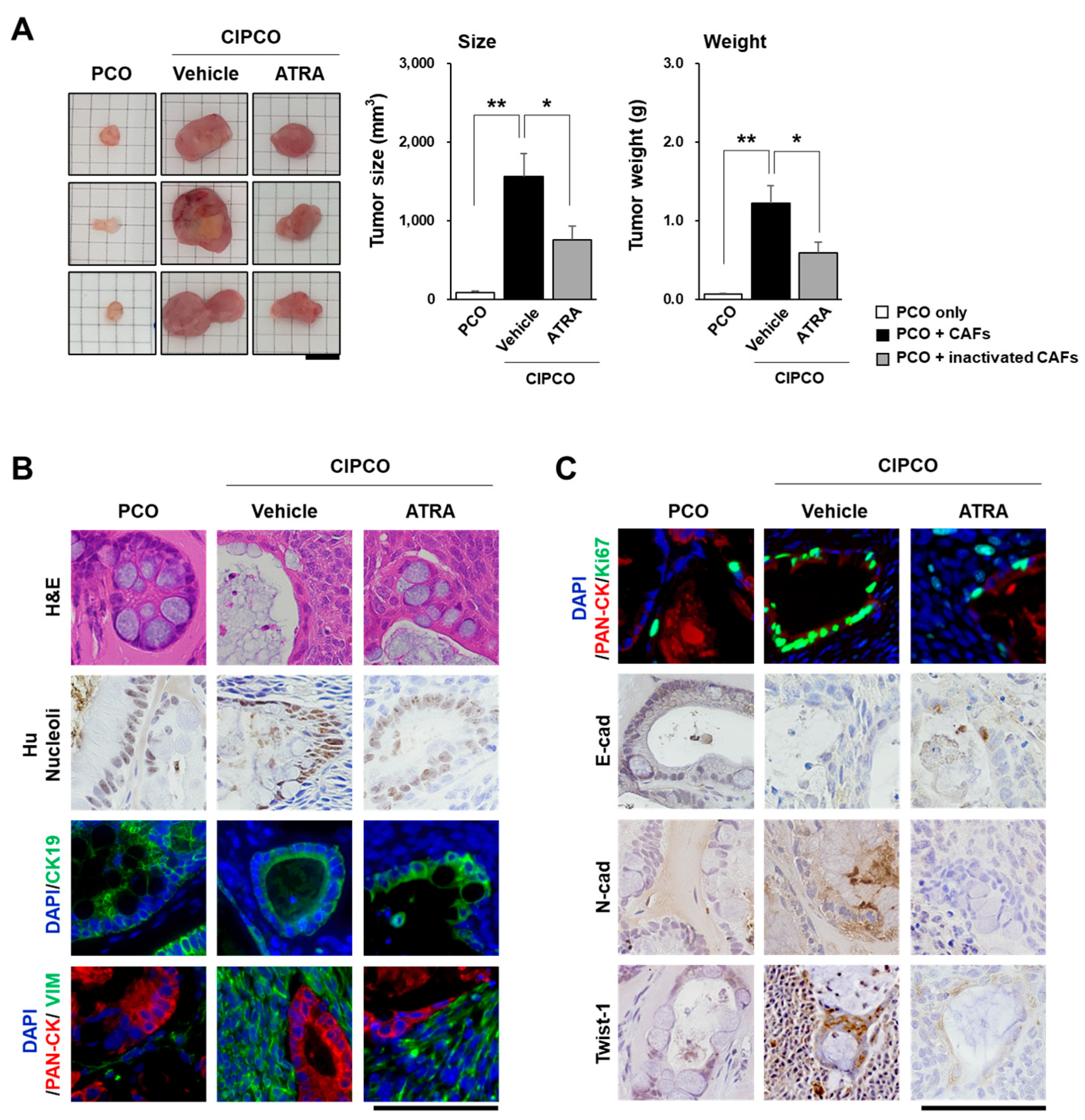
3.3. In Vivo Tumor Formation from CIPCOs
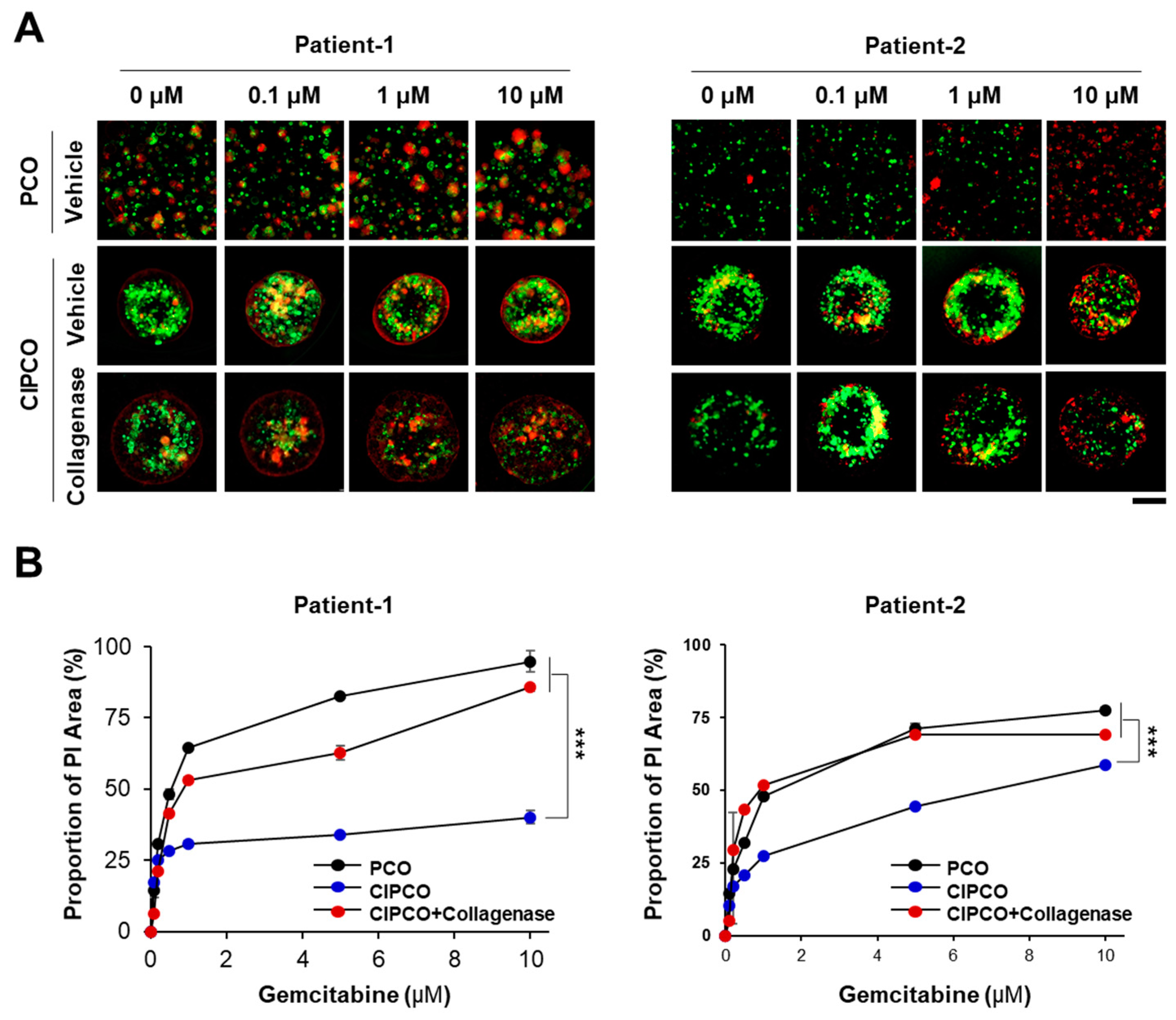
3.4. Differential Sensitivity to Gemcitabine between PCOs and CIPCOs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CAF | cancer-associated fibroblast |
| ECM | extracellular matrix |
| TME | tumor microenvironment |
| PCO | pancreatic cancer organoid |
| CIPCO | CAF-integrated pancreatic cancer organoid |
| ATRA | all-trans retinoic acid |
| EMT | epithelial–mesenchymal transition |
| PI | propidium iodide |
References
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Springfeld, C.; Jäger, D.; Büchler, M.W.; Strobel, O.; Hackert, T.; Palmer, D.H.; Neoptolemos, J.P. Chemotherapy for pancreatic cancer. La Presse Medicale 2019, 48, e159–e174. [Google Scholar] [CrossRef] [PubMed]
- Helms, E.; Onate, M.K.; Sherman, M.H. Fibroblast heterogeneity in the pancreatic tumor microenvironment. Cancer Discov. 2020, 10, 648–656. [Google Scholar] [CrossRef] [Green Version]
- Tomás-Bort, E.; Kieler, M.; Sharma, S.; Candido, J.B.; Loessner, D. 3D approaches to model the tumor microenvironment of pancreatic cancer. Theranostics 2020, 10, 5074. [Google Scholar] [CrossRef]
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T. Targeting hypoxic tumor microenvironment in pancreatic cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and biology of cancer-associated fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.H.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H. Virtual microdissection identifies distinct tumor-and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168. [Google Scholar] [CrossRef]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Ragulan, C.; Cros, J.; Patil, Y.; Martinet, M.; Erkan, M.; Kleeff, J.; Wilson, J.; Apte, M. Inter-and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019, 248, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Koo, B.K.; Knoblich, J.A. Human organoids: Model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020, 21, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Schutgens, F.; Clevers, H. Human organoids: Tools for understanding biology and treating diseases. Annu. Rev. Pathol. 2020, 15, 211–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, T.; Chen, S.; Chen, X.; Lin, W.R.; Li, W.; Ma, J.; Wu, T.; Cui, X.; Ji, H.; Li, Y. Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol. Rep. 2017, 37, 1971–1979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; García, R.; Ghassemi, S.; Fabry, B. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.P.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- La Rosa, S.; Rigoli, E.; Uccella, S.; Novario, R.; Capella, C. Prognostic and biological significance of cytokeratin 19 in pancreatic endocrine tumours. Histopathology 2007, 50, 597–606. [Google Scholar] [CrossRef]
- Maehira, H.; Miyake, T.; Iida, H.; Tokuda, A.; Mori, H.; Yasukawa, D.; Mukaisho, K.; Shimizu, T.; Tani, M. Vimentin Expression in Tumor Microenvironment Predicts Survival in Pancreatic Ductal Adenocarcinoma: Heterogeneity in Fibroblast Population. Ann. Surg. Oncol. 2019, 26, 4791–4804. [Google Scholar] [CrossRef]
- Stylianou, A.; Gkretsi, V.; Louca, M.; Zacharia, L.C.; Stylianopoulos, T. Collagen content and extracellular matrix cause cytoskeletal remodelling in pancreatic fibroblasts. J. R. Soc. Interface 2019, 16, 20190226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beuran, M.; Negoi, I.; Paun, S.; Ion, A.D.; Bleotu, C.; Negoi, R.I.; Hostiuc, S. The epithelial to mesenchymal transition in pancreatic cancer: A systematic review. Pancreatology 2015, 15, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, X.; Zhang, Y.; Zhu, D.; Zhang, L.; Li, Y.; Zhu, Y.; Li, D.; Zhou, J. HIF-2α promotes epithelial-mesenchymal transition through regulating Twist2 binding to the promoter of E-cadherin in pancreatic cancer. J. Exp. Clin. Cancer Res. 2016, 35, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.-M.; Li, A.; Olino, K.; Wolfgang, C.L.; Herman, J.M.; Schulick, R.D.; Iacobuzio-Donahue, C.; Hruban, R.H.; Goggins, M. Loss of E-cadherin expression and outcome among patients with resectable pancreatic adenocarcinomas. Mod. Pathol. 2011, 24, 1237–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piersma, B.; Hayward, M.; Weaver, V.M. Fibrosis and cancer: A strained relationship, Biochimica et Biophysica Acta (BBA). Rev. Cancer 2020, 1873, 188356. [Google Scholar]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [Green Version]
- Sachs, N.; Clevers, H. Organoid cultures for the analysis of cancer phenotypes. Curr. Opin. Genet. Dev. 2014, 24, 68–73. [Google Scholar] [CrossRef]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef]
- Fan, H.; Demirci, U.; Chen, P. Emerging organoid models: Leaping forward in cancer research. J. Hematol. Oncol. 2019, 12, 142. [Google Scholar] [CrossRef] [Green Version]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [Green Version]
- Driehuis, E.; van Hoeck, A.; Moore, K.; Kolders, S.; Francies, H.E.; Gulersonmez, M.C.; Stigter, E.C.A.; Burgering, B.; Geurts, V.; Gracanin, A.; et al. Pancreatic cancer organoids recapitulate disease and allow personalized drug screening. Proc. Natl. Acad. Sci. USA 2019, 116, 26580–26590. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J. Development of primary human pancreatic cancer organoids, matched stromal and immune cells and 3D tumor microenvironment models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef]
- Shan, T.; Chen, S.; Chen, X.; Lin, W.R.; Li, W.; Ma, J.; Wu, T.; Ji, H.; Li, Y.; Cui, X. Prometastatic mechanisms of CAF-mediated EMT regulation in pancreatic cancer cells. Int. J. Oncol. 2017, 50, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.M.; Xu, W.; Du, J.; Zhang, K.S.; Zhang, Q.G.; Wang, X.W.; Liu, Z.G.; Liu, S.Q.; Xie, W.Y.; Liu, H.F. The application of the fibroblast activation protein alpha-targeted immunotherapy strategy. Oncotarget 2016, 7, 33472–33482. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.O.; Mullins, S.R.; Franco-Barraza, J.; Valianou, M.; Cukierman, E.; Cheng, J.D. FAP-overexpressing fibroblasts produce an extracellular matrix that enhances invasive velocity and directionality of pancreatic cancer cells. BMC Cancer 2011, 11, 245. [Google Scholar] [CrossRef] [Green Version]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Torphy, R.J.; Steiger, K.; Hongo, H.; Ritchie, A.J.; Kriegsmann, M.; Horst, D.; Umetsu, S.E.; Joseph, N.M.; McGregor, K. Pancreatic ductal adenocarcinoma progression is restrained by stromal matrix. J. Clin. Investig. 2020, 130, 4704–4709. [Google Scholar] [CrossRef]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic cancer chemoresistance to gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarvepalli, D.; Rashid, M.U.; Rahman, A.U.; Ullah, W.; Hussain, I.; Hasan, B.; Jehanzeb, S.; Khan, A.K.; Jain, A.G.; Khetpal, N. Gemcitabine: A review of chemoresistance in pancreatic cancer. Crit. Rev. Oncog. 2019, 24, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef] [Green Version]
- Muranen, T.; Selfors, L.M.; Worster, D.T.; Iwanicki, M.P.; Song, L.; Morales, F.C.; Gao, S.; Mills, G.B.; Brugge, J.S. Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 2012, 21, 227–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; He, X.; Wei, W.; Wang, J.; Zhang, T.; Shen, X. Tenascin-C induces resistance to apoptosis in pancreatic cancer cell through activation of ERK/NF-kappaB pathway. Apoptosis 2015, 20, 843–857. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Go, Y.-H.; Choi, W.H.; Bae, W.J.; Jung, S.-I.; Cho, C.-H.; Lee, S.A.; Park, J.S.; Ahn, J.M.; Kim, S.W.; Lee, K.J.; et al. Modeling Pancreatic Cancer with Patient-Derived Organoids Integrating Cancer-Associated Fibroblasts. Cancers 2022, 14, 2077. https://doi.org/10.3390/cancers14092077
Go Y-H, Choi WH, Bae WJ, Jung S-I, Cho C-H, Lee SA, Park JS, Ahn JM, Kim SW, Lee KJ, et al. Modeling Pancreatic Cancer with Patient-Derived Organoids Integrating Cancer-Associated Fibroblasts. Cancers. 2022; 14(9):2077. https://doi.org/10.3390/cancers14092077
Chicago/Turabian StyleGo, Yoon-Ha, Woo Hee Choi, Won Jung Bae, Sook-In Jung, Chang-Hoon Cho, Seung Ah Lee, Joon Seong Park, Ji Mi Ahn, Sung Won Kim, Kyung Jin Lee, and et al. 2022. "Modeling Pancreatic Cancer with Patient-Derived Organoids Integrating Cancer-Associated Fibroblasts" Cancers 14, no. 9: 2077. https://doi.org/10.3390/cancers14092077
APA StyleGo, Y.-H., Choi, W. H., Bae, W. J., Jung, S.-I., Cho, C.-H., Lee, S. A., Park, J. S., Ahn, J. M., Kim, S. W., Lee, K. J., Lee, D., & Yoo, J. (2022). Modeling Pancreatic Cancer with Patient-Derived Organoids Integrating Cancer-Associated Fibroblasts. Cancers, 14(9), 2077. https://doi.org/10.3390/cancers14092077