Simple Summary
A symbiotic relationship with the host gut microbiome influences the immune system’s development, functions, and activities. In the mucosa, the gut microbiome mediates several immune activities such as the induction of naïve T-cells differentiation, production of cytokines, and myeloid cells activation. The gut-immune interaction and GI cancer development were investigated more recently. Understanding the interaction’s underlying mechanism provides insight to use them as potential anti-cancer targets. Even though multiple reports support the role of gut-immune interactions in targeting cancer-related pathways such as inflammation, apoptosis, and cellular proliferation, efforts are required to assess their interaction and impact on current treatment options.
Abstract
Gastrointestinal cancer (GI) is a global health disease with a huge burden on a patient’s physical and psychological aspects of life and on health care providers. It is associated with multiple disease related challenges which can alter the patient’s quality of life and well-being. GI cancer development is influenced by multiple factors such as diet, infection, environment, and genetics. Although activating immune pathways and components during cancer is critical for the host’s survival, cancerous cells can target those pathways to escape and survive. As the gut microbiome influences the development and function of the immune system, research is conducted to investigate the gut microbiome–immune interactions, the underlying mechanisms, and how they reduce the risk of GI cancer. This review addresses and summarizes the current knowledge on the major immune cells and gut microbiome interactions. Additionally, it highlights the underlying mechanisms of immune dysregulation caused by gut microbiota on four major cancerous pathways, inflammation, cellular proliferation, apoptosis, and metastasis. Overall, gut-immune interactions might be a key to understanding GI cancer development, but further research is needed for more detailed clarification.
1. Introduction
1.1. Gastrointestinal Cancer
Globally, cancers are a significant cause of death and disability [1]. They are characterized by impaired homeostasis and cellular functions [2]. Cancers are classified based on the organ, tissue of origin, or the cancer cell’s molecular characteristics [3] and the development of cancers is influenced by environmental and genetic factors such as obesity, diet, smoking, and infections with pathogenic agents [4]. Gastrointestinal cancers (GI) are considered a major public health problem with challenging economic and medical burdens due to their high prevalence and mortality rate [5]. The symptoms and signs of GI cancers depend on the type of cancer (gastric cancer (GC), colorectal cancer (CRC), esophageal cancer (EC), pancreatic cancer (PC), and hepatocellular carcinoma (HCC)). They might include weight loss, abdominal pain, dysphagia, and anorexia [6] and the progression of GI cancers occurs in a multistage process. They result from uncontrolled cellular proliferation, the loss of apoptotic functions through the intrinsic and extrinsic apoptotic pathways, and the impairment of major pathways such as epithelial–mesenchymal transition (EMT), phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT), and nuclear factor-kappa (NF-κB) signaling pathways [7,8]. Efforts are required to understand GI-cancers’ underlying mechanisms through these specific impaired pathways.
1.2. The Immune System in Cancer Pathogenesis
The human immune system is defined as a group of cells that protect the body from foreign antigens such as toxins, microbes, viruses, and cancer cells [9]. The immune system has two lines of defense that complement each other; innate and adaptive immunity. Imbalance or defects in either line of defense could result in an inappropriate immune response in the body [10].
Cancer and the immune system have been widely discussed for a century [11]. The underlying mechanism between cancer cells and the immune system interaction involves three processes of how the immune system defends and protects the host; (i) the identification of non-self cells, (ii) the production of effector cells to specifically target the cancerous cells, and (iii) the development of immunological memory as a defense mechanism [12]. The role of immune cells in cancer includes both a pro-tumorigenic and an anti-tumorigenic function [11]. Inflammatory immune cells activation in cancer can present in different tumorigenesis stages and can lead to epigenetic modification, the induction of cancerous cellular proliferation, genomic instability, and the enhancement of a cancerous anti-apoptotic pathway, therefore, leading to cancer progression and dissemination [11]. During the pathogenesis of cancer, multiple components and pathways of innate and adaptive immunity are activated to identify cancerous cells and target their genetic and epigenetic alterations and modifications, thus leading to cancer elimination [13]. Such pathways include complement proteins activation aiding in cancer eradication, natural killer (NK) cells, cytotoxic immune cells which recognize and eliminate immunogenic cancerous cells, neutrophil protease activation, anti-tumor macrophages which display a pro-inflammatory like polarization playing a role in the elimination of immunogenic cancerous cells, CD4+ T-cells activation, the production of IL-22 promoting T-cells proliferation, and naïve B cells activation [14,15]. Despite these mechanisms, cancer can manage to overcome immune components as in the case of T-cells, in which cancerous cells can impair the functions of anti-tumor T-cells such as their ability to infiltrate the tumor survival, cytotoxicity, and proliferation abilities [15].
Advances in the development of immuno-oncology have changed the treatment of GI cancer. Multiple ongoing clinical trials evaluate the efficacy and safety of immunotherapy agents such as avelumab (anti-PD-L1) and relatlimab (anti-LAG3) in patients with advanced gastric cancer [16]. Additionally, as for CRC, two immune checkpoint inhibitors target programmed death-ligand 1 (PD-1) in metastatic cancer, namely, KEYNOTE 028 and CheckMate 142, with an objective response rate of 40% and 55%, respectively [17]. More studies are required to identify the common side effects of these treatments, to estimate the impact on patients with immunodeficiency, and to evaluate the role of gut microbiota in treatment utilization.
1.3. Gut Microbiota: Role in GI Cancer Immunity
In the human body, trillions of microorganisms, such as bacteria, viruses, fungi, and protozoan, are known as the microbiota [18]. The microbiota resides mainly on the respiratory and gastrointestinal tract’s mucosal surfaces with different concentrations and relative abundances [19]. Over time, changes in the microbiome composition occur due to internal or external factors such as lifestyle, genetics, geographical locations, and age, leading to significant variations between individuals [20]. The gut microbiome plays a role in protection from infections, vitamin production, and immune cells development and activity [21], but intestinal dysbiosis and the imbalance in the number of microbes and their diversity in the gut is linked to several pathogeneses such as cancer [22].
Studies have reported the impact of microbiota on the development, activities, and function of immune cells [23]. In mucosal sites, where the microbiota prominently resides, early B-lineage cells development occurs under the influence of extracellular signals from the microbiota [24]. Additionally, the gut microbiome promotes the differentiation of naïve T-cells into colonic Treg cells with unique T-cell receptors on their surfaces [25]. During the invasion of pathogenic bacteria, the gut microbiome promotes the activation of myeloid cells leading to cytokines production [23].
In cancer, the gut microbiome influences the anti-tumor immune response through (1) the induction of the T-cells response, (2) the engagement of a pattern recognition receptor that has pro-inflammatory effects, or (3) the mediation of specific metabolites, which can activate T-cell receptors [26]. Efforts are required to investigate and understand the underlying mechanisms between the gut microbiome and the immune system in the context of cancer and how those mechanisms can be utilized as targets for cancer therapy. Figure 1 summarizes the most common pathogens in the GI tract, their relative abundance, and reported immune regulations.
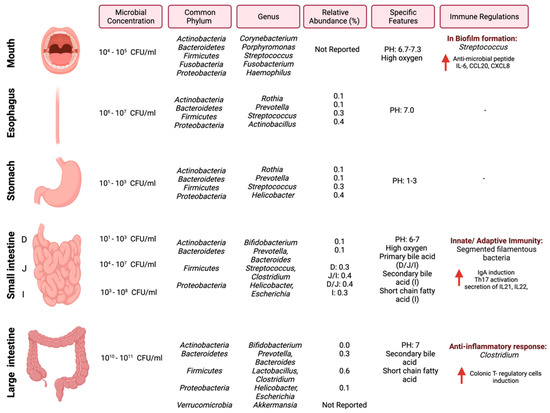
Figure 1.
Schematic illustration of regional diversity of the microbiome along the GI tract. The figure is divided into different regions of the GI tract and highlights the microbial concentration ranges, common phylum and genus, relative abundance [27], and immune regulations specific to each region. “Created with BioRender.com”.
This review has analyzed published studies that report the crosstalk between the gut microbiota and the immune system, assessing the impact of this communication on specific GI cancer pathways. Additionally, it identifies gaps in the current literature.
2. Search Strategy and Selection Criteria
Using the databases “Medline”, “Scopus”, and “PubMed”, papers published from 2001 were searched, using the search terms “Immune cells”, “microbiota”, “Immune cells AND microbiota”, “microbial metabolism”, “Innate immunity AND microbiome”, “Microbiome AND GI cancer”, “gut microbiota enzymes”, “gut microbiome AND immune cells AND GI cancer”, “gut microbiome AND immune cells AND gastric cancer”, and “gut microbiome AND immune cells AND colorectal cancer”. The search yielded around 2000 articles, and in this article, we selected 166 articles and analyzed them in detail. Duplicate studies were excluded, and eligible studies were selected based on inclusion and exclusion criteria. The inclusion criteria included papers that discussed gastrointestinal cancer models or tissues and highlighted gut microbiome interactions.
3. Microbiota–Immune Interactions
The colonization of the gut with microorganisms led to physiological adaptation in the body, as seen with immune cells development, maturation, and interaction [28]. The relationship between the microbiota and the human body is tightly regulated through a controlled immune response to avoid immune activation that might harm the body [29]. This section will discuss three major interactions between the gut microbiome and the immune system: (1) segmented filamentous bacteria (SFB), (2) antimicrobial peptides, and (3) dietary fibers such as short-chain fatty acids (SCFA). Figure 2 highlights these three interactions.
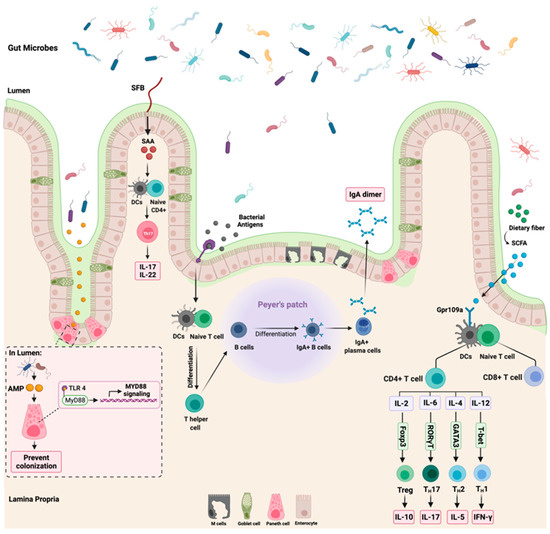
Figure 2.
Summary of the most reported gut microbiome and immune interactions. While the left side of the figure illustrates the influence of segmented filamentous bacteria (SFB) and antimicrobial peptides (AMP) on the host immune system through the activation of Th17 and MyD88 signaling, respectively, the right section of the figure highlights the role of dietary fibers and short chain fatty acids (SCFA) on T cells expression. The role of bacterial antigens on the production of IgA dimer is shown in the middle part of the figure. “Created with BioRender.com”.
3.1. Segmented Filamentous Bacteria
SFB are commensal bacteria found mainly in the small intestine [30]. They are gram-positive bacteria identified by their long and filamentous appearance [31]. A genome sequencing listed SFB as a member of Clostridiales. Additionally, the sequencing results reported a lack of amino acid biosynthetic enzymes in SFB and an expression of typical flagella and spore-forming genes. This suggests that those bacteria depend on the host for essential nutrients [32].
The colonization of SFB in the intestine regulates and influences the immune response in the body [33]. SFB regulates the level of IL-17A and IL-22 expression in the intestine through the modulation of serum amyloid A (SAA) [34]. Additionally, SFB play a role in postnatal maturation of the immune system through the production of IL-17 producing CD4+ T-cells, which is critical in host protection against extracellular pathogens [35]. The observed effects of SFB on the immune system occur due to their ability to adhere to the intestinal epithelium, which is a crucial step to induce Th17 cells differentiation. SFB models lacking the ability to adhere failed to induce intestinal Th17 differentiation [36]. Following the adherence of SFB to the intestinal epithelium is the secretion of SAA, which is essential for cytokines production and secretion [37]. Despite what is known so far about the role of SFB in shaping the intestinal immune response, more research is still required to understand the interaction of SFB with other microbiomes such as viruses and how they all impact the immune system. Additionally, the mechanism in which SFB modulates the immune system requires further understanding as well as how distal organs react to those immune changes modulated by SFB.
3.2. Short Chain Fatty Acids
SCFAs are fermented fatty acids generated by the gut microbiota, such as Faecalibacterium prausnitzii, from the digestion of complex carbohydrates [38]. They are considered the most abundant microbial-derived metabolites in the human gut lumen. They consist mainly of propionate, butyrate, and acetate [39]. SCFAs play a critical role in improving the function of the gut barrier, protecting against microbial invasions, and reducing intestinal inflammation, thus improving the host’s overall health status [40]. Those observed positive effects of SCFAs are due to the activation of G-protein coupled receptors (GPCRs) such as GPR109a or the suppression of histone/histone deacetylases (HDACs) which influence genetic expression [41].
In colonocytes, the sodium-dependent monocarboxylate transporter-1 (SLC5A8) facilitates and mediates the entry of SCFAs (specifically butyrate) from the lumen to the colonic epithelial cells. This leads to the suppression and activation of HDACs and GPCRs, respectively [42]. SCFAs are essential regulators of immune cells’ recruitment, activation, and differentiation, such as dendritic cells (DC), neutrophils, macrophages, and T-lymphocytes. Additionally, SCFAs regulate the expression of pro-inflammatory cytokines such as IL-12 and IL-6 [43]. Moreover, the binding of butyrate to GPR109a receptor on DCs results in an increased expression of IL-10 and a decreased expression of IL-6, which results in increased T-reg cells development, thus inhibiting Th17 cells expansion [44]. This indicates that the GPR109a receptor is vital in anti-inflammatory pathways such as apoptosis, especially in inflammation-induced colon cancer [45]. Figure 2 summarizes the reported interaction between butyrate and GPR109a and the subsequent cytokines production. Despite the observed tumor suppressor effects of GPR109a receptors, some reports highlighted that the activation of this receptor leads to the activation of inflammatory signaling pathways, which suggest that GPR109a could act as a tumor activator and suppressor depending on the affected sites and tissues [46]. Further research is required to investigate the effect of the GPR109a receptor and other GPCRs receptors. Additionally, more efforts are necessary to understand the effect of other SCFAs such as acetate on the host immune system.
3.3. Antimicrobial Peptides
The intestine contains many microorganisms that provide multiple benefits in metabolism, nutrients, and immunity [47]. The symbiotic relationship between the host and the gut microbiota is mediated by chemical and physical gut mucosal barriers, preventing unregulated interaction between the host immune system and the gut microbiota [48]. Antimicrobial peptides (AMPs) are considered a chemical mucosal barrier of basic amino acid-rich proteins with a broad spectrum of antimicrobial properties, such as being cytolytic, microbicidal, and bacteriostatic [49]. Shortly after infection, AMPs are synthesized promptly to rapidly neutralize the invading microbes [50]. Additionally, AMPs include the defensin protein family, such as alpha and beta. They bind to the microbial cell membrane and disrupt the membrane integrity by forming pore-like structures. Deficiency in alpha defensin, a highly expressed protein in Paneth cells, is associated with gut microbiota alteration suggesting that AMPs play a role in gut environment hemostasis [51]. Moreover, patients with inflammatory bowel disease have reported having intestinal barrier dysfunction as the level of AMPs production was reduced [52].
The Toll-like receptor (TLR) family plays a role in enhancing the function of the epithelial barrier and innate immunity [53]. After an infection, TLRs recognize the synthesized bacterial products such as AMPs which activate the cytoplasmic adaptor protein MyD88 [54]. Activated MyD88 is essential in protecting the intestinal epithelial cells from mucus-associated bacteria and opportunistic bacteria. Additionally, a loss of MyD88 signaling activation can disrupt microbiota and host tissue segregation, compromise epithelial barrier function, and alter the balance of the gut microbiota community [55]. Moreover, TLRs are critical for IgA antibody secretion. After producing bacterial antigens, the TLRs sense those antigens, leading to T-cell differentiation and subsequently producing IgA dimers. The production of IgA is essential to distinguish between pathogenic and commensal bacteria where it neutralizes the mobility and adhesions of the pathogenic bacteria [56]. Figure 2 summarizes the role of TLRs in MyD88 signaling activation and IgA production.
4. Microbiota–Immune Interactions: Role in GI Cancer Development
The gut microbiome plays a critical role in the pathogenesis of host diseases such as cancer [57]. As the gut microbiome is influenced by several factors such as diet, genetics, and lifestyle, its dysbiosis, either in the bacterial composition, bacterial bioactivity or diversity, can impair the balance of specific bacterial species and increase the abundance of inflammation-inducing species that can cause several diseases including inflammatory bowel disease and cancer [58]. The gut microbiome influences the host immune response to regulate cancer mechanisms such as progression, genetic instability, and the response to treatment [59]. Animal studies have reported that specific microbes such as Bacteroides fragilis and Escherichia coli can promote cancer development by releasing genotoxins, damaging the host DNA [60]. Additionally, the gut microbiome could impact the efficacy of cancer treatment, as seen in antibiotic-treated mice [61]. This suggests the critical role of intact microbiota in the gut for optimal treatment outcomes.
Additionally, the gut microbiome impacts the function of the mucosal B and T cells, which are essential for immune homeostasis as they inhibit the unregulated response to harmless antigens and preserve the mucosal barrier integrity in the intestine [62]. Disruption of the gut barrier facilitates the interaction between the immune cells and the microorganisms, resulting in cancer development through the induction of immunosuppressive or pro-inflammatory pathways [63]. Gut microbiome dysbiosis can influence cancer pathways by recruiting lymphocytes to the intestine, leading to cellular proliferation by activating the IL-6 pathway [64]. Moreover, TLRs upregulation can activate the nuclear factor (NF)-κB and JAK/STAT3, which are critical for immunosuppression and cellular proliferation [65]. Table 1 summarizes the main findings from the reported studies.

Table 1.
Representative microbial–immune interactions and their underlying anticancer effects.
Table 1.
Representative microbial–immune interactions and their underlying anticancer effects.
| Targeted Cancer Pathway | Type of Cancer (s) | Microbial Species | Targeted Metabolites/ Proteins/ Genes/ Species | Targeted Immune Cells/ Pathways/ Products | Site of Interaction | Mechanism of Action | Methods of Testing | Model Used | References | |
|---|---|---|---|---|---|---|---|---|---|---|
| In Vivo | In Vitro | |||||||||
| Inflammation | Colon cancer | Enterococcus Bacteroidetes Lactobacillus E. coli Segmented filamentous bacteria | Short-chain fatty acids (SCFA) | IL-18 IL-6 IL-22 | Colon Intestine |
| Quantitative reverse transcription PCR Flow cytometry FISH Confocal microscopy |
| [66] | |
| Colon tumorigenesis | Erysipelotrichaceae Prevotellaceae Lachnospiraceae | Not specified | CD8 T cells IFN-γ IL-1β | Colon |
| 16S rRNA sequencing linear discriminant analysis (LDA) Quantitative reverse transcription PCR Antibiotic and antifungal studies Flow cytometry |
| [67] | ||
| Colon cancer | Mix of enteric flora from fecal samples | Compound K | IL-8 | Colon |
| Flow cytometry liquid chromatography quadrupole time-of-flight mass spectrometry ELISA |
| [68] | ||
| Colon cancer | Segmented filamentous bacteria Proteobacteria Firmicutes | FAM3D (cytokine like family) a gut secreted protein | CD3 T cells B220+ B cells CD11b+ myeloid cells | Colon |
| Immunofluorescent staining Real-time PCR Western blot Quantitative reverse transcription PCR FISH | - C57BL/6 mice | [69] | ||
| Colon cancer | Bacteroidetes Prevotellaceae Firmicutes | Gpr109a | IL-17 IL-23 ILC3 | Colon |
| Antibody treatment Quantitative PCR Microbiome sequencing | - C57BL/6 mice | [70] | ||
| Colon cancer | Helicobacter hepaticus Lachnospiraceae | TGF-β | NF-β | Colon |
| DNA/RNA sequencing Multi-omics studies | - Smad3 mice | [71] | ||
| Colon cancer | Bacteroides Firmicutes | IL-23 produced from dendritic cells | IL-1A IL-13 IL-17A CXCL-9 IL-17 | Colon |
| Cell proliferation assays Cell migration and invasion assays ELISA Real-time PCR Ex-vivo studies Immunoblots | - F344 rats |
| [72] | |
| Colon cancer | Prevotellaceae Segmented filamentous Bacteria | LRP5/6-β-catenin-IL-10 signaling axis | TNF-α IL-6 IL-1β | Colon Intestine |
| Antibiotic treatment Fecal microbiota transplant ELISA Cell sorting Flow cytometry Real-time PCR |
| [73] | ||
| Colon cancer | Not specified | TLR-4 | Dual oxidase 2 (DUOX2) NADPH oxidase 1 (NOX1) | Colon |
| Cell viability assays 16s ribosomal RNA polymerase chain reaction 16s ribosomal RNA sequencing |
| [74] | ||
| Colon cancer | Prevotella Escherichia coli Akkermansia Pseudoflavonifractor Ruminococcus Clostridium XlVa | Short chain fatty acids (SCFA) | NOD-like receptor family pyrin domain containing 3 (NLRP3) Tumour necrosis factor-α (TNF-α) Interleukin-1β (IL-1β) | Colon |
| Fecal microbiota transplant Histological studies Immunohistochemistry staining Real-time PCR RNA extraction Western blotting RNA sequencing | - C57BL/6J mice | - Fecal samples | [75] | |
| Cellular Proliferation | Colon cancer | Fusobacterium nucleatum | microRNA-31 | CD3 T cells CD8 T cells CD45RO T cells FOXP3 T cells | Colon |
| Quantitative PCR Ariol image analysis system Microarray Metagenomic analyses | - Colorectal carcinoma tissues from patients | [76] | |
| Colon cancer | Bifidobacterium Prevotellaceae Bacteroides Lachnospiraceae | YYFZBJS (traditional Chinese herbs) | CD4 T cells Foxp3 T-bet ROR-γt | Colon |
| Quantitative PCR Histology Genotyping Antibiotic treatment Fecal microbiota transplantation Flow cytometry Bacterial attachment assay | - ApcMin/+ mice |
| [77] | |
| Colitis-associated colon cancer (CAC) | Not specified | TLR-4 | TNF-a IL-1b | Colon carcinoma |
| Cytokine Quantification Real-Time PCR Flow cytometry | - BALB/c mice | - CT26 cells | [78] | |
| Metastasis | Colon cancer | Not specified | Inflammasome pathway | IL-18 IL-1 Hepatic NK cells | Colon Liver Spleen |
| Quantitative Real-Time PCR Flow cytometry Immunofluorescence staining | - C57BL6/J mice | [79] | |
| Colon cancer | Fusobacterium nucleatum | Fusobacterium nucleatum | CD8 T cells CD33 cells CD163 cells | Colon Liver |
| Immunohistochemical staining DNA extraction Quantitative Real-Time PCR Immunohistochemistry |
| - Colorectal cancer liver metastases cells | [80] | |
| Colon cancer | Firmicutes Proteobacteria | Sodium butyrate | IL-10 IL-17 Hepatic NK cells | Colon Liver |
| Quantitative Real-Time PCR Hematoxylin and eosin stain Flow cytometry | - BALB/c mice | [81] | ||
| Apoptosis | Colon cancer | Erysipelotrichaceae B.fragilis | Follicular helper T (TFH) cells | caspase-3 caspase-7 | Colon |
| Antibiotic treatment Flow cytometry Fecal microbiota transplantation ELISA 16S rRNA gene sequencing Immunohistochemistry staining | - C57BL/6J mice |
| [82] |
| Colon cancer | Bacteroides Firmicutes Prevotellaceae Lactobacillaceae | Fucoidan | β-catenin C-Myc CyclinD1 IL-17 IL-23 Il-4 Il-10 | Colon tissues |
| Flow cytometry Western blotting Immunofluorescence assay 16S rRNA gene sequencing Gas chromatography | - Sprague–Dawley (SD) rats | [83] | ||
| Colon cancer | Not specified | BCL-G (BCL2L14) | IFN-γ TNF-α | Colon |
| Crystal violet staining Microscopy Western blotting Chemokine analysis |
| [84] | ||
4.1. Inflammation
Inflammation is associated with multiple diseases such as diabetes, cardiovascular disease, and multiple stages of cancer [85]. During cancer, acute inflammation is critical in the recruitment and accumulation of neutrophils, the stimulation of antigen presentation, and the maturation of dendritic cells leading to an anti-tumor response. Additionally, and during acute inflammation, the level of C-reactive protein and serum amyloid A protein (SAA), acute phase proteins can increase, with the latter being influenced by segmented filamentous bacteria. On the other hand, chronic inflammation is linked to different stages of cancer development, including transformation, promotion, proliferation, invasion, metastasis, survival, angiogenesis, and treatment resistance, with an accumulation of macrophages, lymphocytes, and plasma cells at the site [86]. In addition, chronic inflammation is considered a risk factor for gastrointestinal cancer development in patients with inflammatory bowel disease, as reports have illustrated a similar inflammatory microenvironment between cancer and inflammatory bowel diseases. Additionally, in both diseases, inflammatory cells produce similar mediators such as IL-6 and IL-12, which suggests the role played by the immune system in both diseases [68]. Damaged tissues in the body caused by either a physical or an ischemic injury, exposure to toxins, or an infection can result in an inflammatory response activation that is necessary to repair the damaged tissues [87]. An inflammatory response can become chronic when the causative agent of the inflammation persists, resulting in cellular proliferation and mutation, thus creating a suitable environment for cancer development [88]. Additionally, and due to chronic inflammation, host leukocytes such as macrophages, dendritic cells, and lymphocytes can be present in tumor areas. They can lead to immunosuppression and cancer growth by producing reactive oxygen species (ROS) that damage the intestinal epithelial cells’ DNA [87].
The gut microbiota in the intestine is usually segregated from the immune cells by a single layer of intestinal epithelial cells joined by tight junctions [89]. Dysbiosis in the gut can alter the permeability of the intestinal barrier, causing a disruption where commensal bacteria and their products can invade the mucosa, thus resulting in low-grade systemic inflammation. Due to that, inflammatory pathways such as Wnt and Notch are activated, affecting the mucosal epithelial cells, thus influencing immune homeostasis and increasing susceptibility to CRC [90]. After activating the myeloid differentiation factor 88 (MyD88), the invading commensal bacteria and their products interact with TLRs on tumor-infiltrating myeloid cells, leading to the production of inflammatory cytokines such as IL-23 activating the production of IL-6, IL-22, and IL-17A [91]. The production of those cytokines can eventually promote the activation of STAT3 and the nuclear factor-kB (NF-kB) signaling pathway [92]. The promoted activation of NF-kB signaling pathway by TLR-4 overexpression can induce COX-2 expression, a CRC biomarker, and an inflammation-associated gene in inflammatory bowel disease [93]. Figure 3 summarizes the interaction between the gut microbiome and the immune cells in GI cancer and its activation of inflammation. Meanwhile, another preclinical study documented that the activation of the inflammatory response significantly correlated with the disturbance of the gut microbiota and changes in the fecal metabolites [94]. The authors found that these changes could be closely related to the occurrence of precancerous lesions of GC. The correlation analysis between inflammatory cytokines and gut microbiota/feces metabolites was evaluated in a N-methyl-N′-nitro-N-nitrosoguanidine multiple factors-induced rat model of GC. The results demonstrated a significant increase in pro-inflammatory serum cytokines such as IL-1β, IL-4, IL-6, IL-10, IFN-γ, TNF-α, and M-CSF.
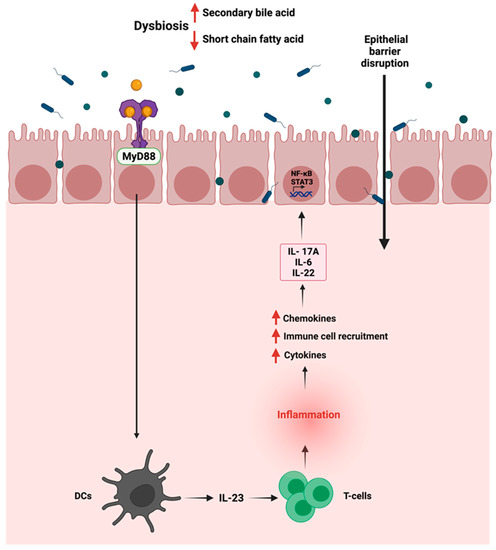
Figure 3.
Schematic representation of the immune—gut interactions during GI cancer and how it influences the inflammatory responses. Due to gut dysbiosis, the low level of short chain fatty acids can lead to the activation of inflammatory pathway, the production of cytokines and chemokines and the activation of STAT3 and NF-kB signaling pathways. “Created with BioRender.com”.
On the other hand, there was a significant decrease in the level of chemokine (C-X-C motif) ligand 1 (CXCL1) in the model group vs. controls. In this regard, the gut microbiota and fecal metabolic phenotype composition in the model group revealed that Lactobacillus and Bifidobacterium significantly increased. At the same time, Turicibacter, Romboutsia, Ruminococcaceae_UCG-014, Ruminococcaceae_UCG-005, and Ruminococcus_1 were significantly decreased compared to the control animals.
4.2. Cellular Proliferation
Cellular proliferation is a fundamental process essential for the development and hemostasis of the organism [95]. It is tightly regulated to ensure a precise and complete genome duplication [96]. Multiple factors, from DNA damage to growth factors, influence the process of DNA replication, especially the entering to the S phase of the cycle [97]. Cancer cells embody multiple characteristics that play a role in their survival and abnormal proliferation [98] and due to epigenetic changes and/or mutations, cancer cells are resistant to cellular proliferation regulators such as growth factors and hormones. Such changes promote the growth and survival of cancerous cells through the stimulation of proliferation pathways and the inhibition of apoptotic pathways [99]. Emerging evidence supports the gut microbiome’s role in influencing cellular proliferation in cancer through contact with immune cells, as seen in the case of Fusobacterium nucleatum, the most studied colon cancer-associated microorganism, which is enriched during cancer [100,101].
F. nucleatum is a commensal opportunistic anaerobic Gram-negative bacillus found mainly in the oral cavity. It is implicated in multiple diseases outside the oral cavity [102]. F. nucleatum plays a role in colon cancer progression and treatment with antibiotics such as metronidazole which reduces their load and cellular proliferation [103]. Additionally, F. nucleatum promotes cellular proliferation in CRC by binding FadA to E-cadherin, which mediates the bacteria’s attachment and invasion. This leads to the activation of β-catenin signaling and the increased expression of Wnt genes, transcription factors, and inflammatory genes, thus impacting T-cells infiltration levels [104,105]. On the other hand, some bacterial strains, such as Holdemanella biformis, are reduced during gut tumorigenesis, which is critical in blocking tumor proliferation [106]. H. biformis impacts cellular proliferation by mediating SCFA such as butyrate, which inhibits histone deacetylase (HDAC) activities by enhancing H3 histone acetylation and reducing the NFATC3 pathway [107].
Efforts are required to identify potential bacteria strains and their role in GI cancer development. Additionally, more research is necessary to assess the feasibility of maybe using specific strains as a treatment option for GI cancer. Figure 4 summarizes the role of the reported bacteria on GI cancer.
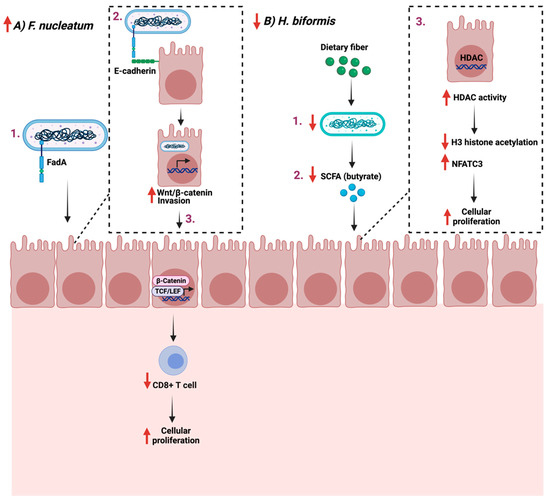
Figure 4.
Schematic illustration of two pathways in which two bacteria Fusobacterium nucleatum and Holdemanella biformis facilitate cancer progression and cellular proliferation through FadA- E-cadherin interaction and short chain fatty acids (SCFA), respectively. (A) represent the proliferative example while (B) the anti-proliferative example. “Created with BioRender.com”.
4.3. Metastasis
Metastasis is defined as the expansion of the primary tumor, leading to secondary tumors distant from the original tumor [108]. Metastasis occurs in a multi-step process that includes the separation from the primary tumor, the invasion through the surrounding tissues, and the entry and survival in the circulation [109]. Understanding the mechanism of metastasis is of great importance to managing and treating cancer. Therefore, assessing the impact of the gut microbiome, a potential therapeutic option, and immune system interaction can provide some insights. F. nucleatum is linked to CRC development and progression [110]. The polymerase chain reaction quantification of F. nucleatum DNA in 181 colorectal cancer liver metastases specimens reported that the presence and the quantity of the bacteria is inversely associated with a lower CD8+ T-cells density. This could suggest the potential involvement of F. nucleatum in cancer metastasis (Table 1) [80]. Mechanistically and in CRC, tissues are overexpressing sugar residues Gal-GalNAc, which is recognized by the F. nucleatum adhesion molecule, Fab2, and which is critical in mediating hemagglutinin and co-aggregation functions. Mechanistically, F. nucleatum could promote metastasis by activating the TLR-4 pathways, upregulating a cytochrome p450 known as CYP2J2. The metabolite of this cytochrome, 12,13-EpOME, then activates EMT, thus promoting CRC metastasis in vitro [111].
Additionally, F. nucleatum can evade anti-cancer immune responses by mediating the recognition and binding of the same Fab2 adhesion molecule to a receptor known as TIGIT, overexpressed on natural killer cells and other lymphocytes. The mediated binding inhibits the functions of lymphocytes and natural killer cells, therefore, protecting F. nucleatum and promoting a pro-tumorigenic environment [112]. Figure 5 highlights the reported mechanisms in which F. nucleatum promotes GI cancer metastasis.
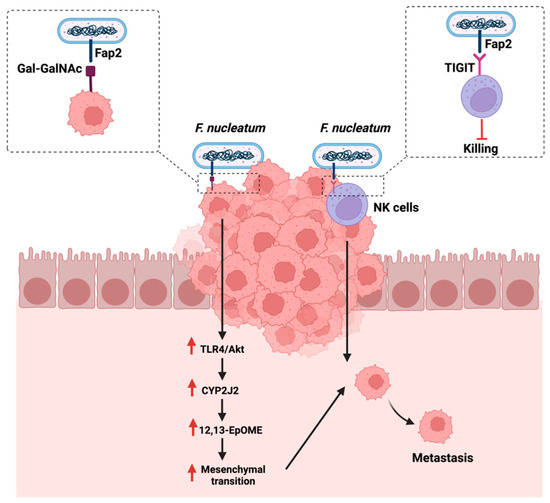
Figure 5.
Summarizes the influence of Fusobacterium nucleatum on cancer metastasis either by targeting sugar residues Gal-GalNAc on cancerous cells or targeting receptors that are overexpressed on natural killer cells (NK). “Created with BioRender.com”.
4.4. Apoptosis
Apoptosis is a basic cellular mechanism that is essential in the development and homeostasis of the organism [113]. Distinct morphological changes characterize it, controlled by intracellular and extracellular signals regulated by the cell environment [114]. Intrinsic and extrinsic pathways are the two major apoptotic pathways where they process the stress signal and execute the death signal in the cell [115]. Both exogenous and endogenous agents such as physical trauma, infectious agents, radiation, and chemotherapeutic drugs can trigger apoptosis [116]. In cancer, downregulation of apoptosis by pro-survival proteins is necessary to maintain the phenotypic properties. Such alteration is observed in the anti-apoptotic Bcl-2 family, which is overexpressed frequently in solid tumors [117]. On the other hand, a study analyzed the expression of human BCL-G, a member of the BCL-2 family in gastrointestinal conditions, and they reported that both variants were highly expressed in a healthy gut. At the same time, their m-RNA level was decreased in colorectal cancer and inflammatory bowel disease conditions [84]. Additionally, the study reported that the depletion of BCL-G affected the secretion of chemokines such as CCL5 thus illustrating a non-apoptotic function of the BCL-2 family. More studies are required to assess the role of the BCL-2 family in shaping the immune system, apoptosis, and maybe the regulation of chemokines (Table 1).
The gut microbiome is a critical mediator of the host’s health by producing certain metabolites essential for immune system regulations [118]. Gut dysbiosis can reduce the beneficial bacteria responsible for producing SCFA, such as butyrate [119]. Butyrate plays a role in maintaining the intestinal barrier function and reducing inflammation in the colon, as they supply colonocytes with 70% of their required energy [120]. Additionally, the butyrate induces IL-18 expression in the colon, which is essential in suppressing colonic inflammation [121]. The administration of butyrate reduces cellular proliferation and pro-inflammatory cytokines production, such as IL-6, while promoting apoptosis [122]. Gut analysis of patients with colon cancer and ulcerative colitis showed a significant reduction in butyrate levels and the number of butyrate-producing bacteria in the colon [123]. During cancer, and when the gut is in dysbiosis, butyrate production is reduced, impacting the butyrate receptor’s activity, GPR109a, found in the colon. This reduces IL-18 and IL-22 production, reducing the mucosal tissue repair capabilities, thus impacting cellular apoptosis [124,125]. Another study described the significant role of moxibustion, a traditional Chinese medicine, in inducing apoptosis of rat GC cells in vivo by regulating intestinal flora [126]. The authors summarized that moxibustion delayed the GC metastasis possibly by lowering the abundance of Ruminococcaceae and Prevotellaceae bacteria (bacteria producing short-chain fatty acids in the gut) and enhancing the occurrence of probiotic Akkermansia in the rat intestine.
Additionally, butyrate induces apoptosis in CRC through the mitochondrial pathway and caspase 3 [127]. When the butyrate level is reduced, the expression of Bcl-2 anti-apoptotic family is enhanced, while the expression of Bax/Bak, cytochrome c is reduced [120]. Figure 6 summarizes the role of gut dysbiosis and butyrate production on cellular apoptosis during cancer.
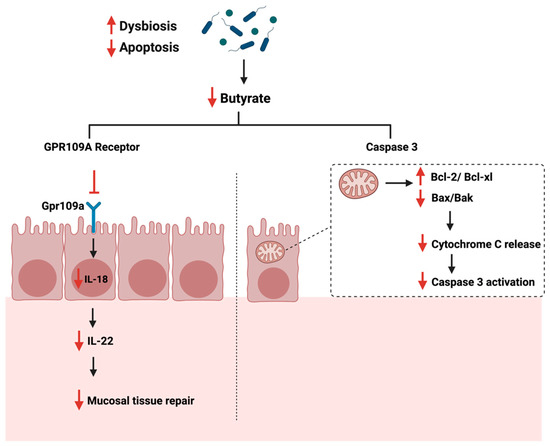
Figure 6.
Illustrations of the role of short-chain fatty acids specifical butyrate on cellular apoptosis. The figure highlights the Gpr109a receptor and the pathways that lead to the reduction of mucosal tissue repair and Caspase 3 activation. “Created with BioRender.com”.
5. Discussion
5.1. Influence of Gut Microbiome on Immunotherapy
Current cancer treatments, including chemotherapy, surgery, endocrine therapy, and radiotherapy, are usually non-specific approaches. They frequently reach a refractory period, leading to treatment failure and disease recurrence [128,129]. Targeting the immune system and enhancing the patient’s immune system to attack the tumor can potentially be therapeutic [130]. Cancer immunotherapy is an alternative approach that utilizes specific components of a patient’s immune system to selectively target and eliminate tumor cells, thus mitigating the side effects of the currently used treatments [131]. Depending on the mechanism by which the therapy activates the immune response, immunotherapy can be passive, such as cell-based therapy and chimeric antigen receptor T cell therapy (CAR-T cell) or active, such as vaccination, immunostimulatory cytokines, and immune checkpoint inhibitors [132,133]. Immune checkpoint inhibitors are used as a treatment option to induce a T-cells mediated response against cancerous cells to selectively block the inhibitory checkpoint receptors manipulated by the tumor cell [134]. Types of inhibitory checkpoint receptors include programmed cell death protein 1 (PD-1), cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), T cell immunoglobulin and mucin protein 3 (TIM-3), and programmed cell death 1 ligand 1 (PD-L1) [135]. To treat CRC, immunomodulatory therapy such as CTLA4, PD-1, and PD-L1 is currently used to target selective checkpoint molecules and inhibit T-cell activation [136]. Despite this, 19 patients with unselected CRC did not demonstrate positive clinical responses when using Nivolumab, a monoclonal antibody that binds to PD-1 receptor [137].
The gut microbiome plays a role in stimulating and influencing immunotherapy against cancer [138]. The intestinal microbiota is an essential factor in providing an optimal CpG-oligonucleotide immunotherapy response which activates innate immune cells [139]. Moreover, the microbiome influences immunotherapy as a community, but specific microbes such as Bacteroides fragilis can enhance PD-1/PD-L1 and CTLA-4 immunotherapy as they activate Th1 cells [140]. Figure 7 summarizes the interaction of B. fragilis with immunotherapy. Additionally, in 74 advanced gastrointestinal cancer patients, the ratio of Prevotella/Bacteroides was elevated with an enhanced anti-PD-1/PD-L1 treatment response [141]. The analysis of DNA sequencing of stool samples collected before the administration of checkpoint inhibitors illustrated a distinct bacterial taxa composition [142], and that microbial species capable of producing SCFA were reported to have better anti-PD-1/PD-L1 positive responses [141]. A mice model study showed that Prevotella CAG:485 and Akkermansia might influence the efficacy of PD-1 immunotherapy through the modulation of glycerophospholipid metabolism, which can affect the expression of cytokines such as IL-2 and IFN-γ [143]. More clinical and experimental trials are necessary to investigate how the gut microbiome impacts immunotherapy.
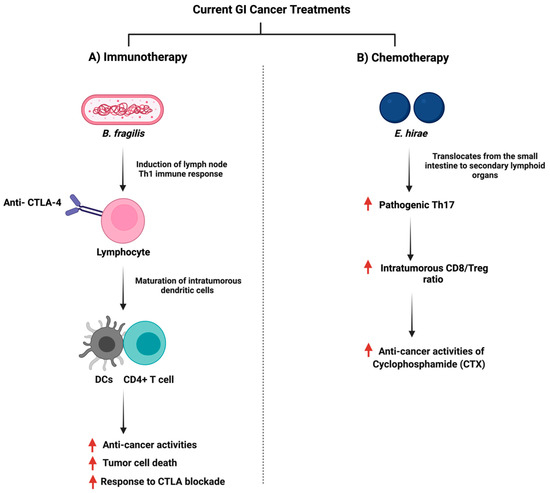
Figure 7.
Illustrations of the gut bacteria and their role in modulating the efficacy of the currently used anti-cancer drugs. The figure summarizes the influence of the reported bacteria on immunotherapy and chemotherapy treatments. “Created with BioRender.com”.
5.2. Chemotherapy Treatment and Immune–Gut Interactions
Chemotherapy is used as a treatment option for cancer, with platinum and fluorouracil being the commonly used drugs [144,145]. Regularly, cancer patients receiving chemotherapy have signs of depression, fatigue, anxiety, and cognitive impairment [146]. Chemotherapy treatment is often accompanied by multiple complications caused by the cytotoxic effect, linked to a bidirectional interaction between the drug and the gut microbiome [147]. Preclinical model studies demonstrated chemotherapy-induced changes in the gut microbiome with a decrease in the total number and diversity of the gut microbiome [148]. Additionally, and depending on the drug used, the overall impact on the gut profile reported a reduction in Lactobacillus and Bifidobacterium, and an increase in Escherichia coli (E. coli) and Staphylococcus. The reported gut microbiome composition disruption was associated with activating inflammatory pathways, thus enhancing the vulnerability to pathogenic infections [149].
On the other hand, the efficacy of chemotherapy can be affected by the gut microbiome. Such a mechanism includes when specific oral or injected drugs, such as CPT-11 (Irinotecan) depend on the gut microbiome to be converted to the active form and the treatment can exert anti-cancer properties [150]. Moreover, the gut microbiome can facilitate the anti-cancer effects of chemotherapy through the induction of enzymatic expression responsible for ROS production, which can induce cellular apoptosis [151]. Additionally, the gut microbiome can impact the ROS pathway through a toll-like receptor agonist, which can downstream the expression of MyD88 and induces inflammatory cytokines such as IL-6 [152,153]. Chemotherapy treatment and the gut microbiome can influence the immune system and changes in the gut microbiome due to chemotherapy can impact innate immunity by reducing the production of inflammatory cytokines and antigen-presenting cells [154]. For example, both Enterococcus hirae and Lactobacillus johnsonii were essential for the anti-cancer activities of Cyclophosphamide (CTX) where they promoted splenic Th1 memory and a Th17 response [155]. Figure 7 summarizes the interaction of E. hirae with CTX treatment.
Although multiple reports illustrate the role of the gut microbiome in chemotherapy, some studies highlight microbiota-induced chemoresistance. The gut of patients with CRC is enriched with F. nucleatum, which was discussed in the above sections along with how it can promote metastasis [156]. This phylum can induce chemoresistance in which the inflammatory pathway is stimulated by the mediated binding of FadA and E-cadherin, which can then increase tumor growth [157]. Additionally, the gut microbiome can inactivate the used chemotherapy drug, inducing chemoresistance as seen with Gammaproteobacteria, which can convert the gemcitabine drug to its inactive metabolite, thus contributing to drug resistance [147]. All data indicate that efforts are required to investigate the bidirectional interaction between the gut microbiota and chemotherapy and the possibility of using this interaction to improve the treatment outcome further and reduce chemoresistance development.
5.3. Challenges with Studying the Field
The area of the gut microbiome and immune interaction research is growing as scientists understand more about microbial communities, their behaviors, core microbial species, their produced metabolites, and their influence on the host immune system in health and disease as in the case of GI cancer. Despite this, the field faces multiple challenges, including protocol standardization, experimental models, and interpretation tools. Additionally, the gut is influenced by several factors such as diet, geographical location, genetic diversity, and medications, thus requiring a systematic and extensive data analysis. Moreover, investigating the mechanistic pathways in which the gut microbiome influences the immune response during cancer is critical as those interactions might provide potential therapeutic targets. Collective efforts from microbiologists, ecologists, bioinformaticians, immunologists, and geneticists are fundamental to improving the field further.
5.4. Future of GI Cancer Treatment?
As discussed in the previous sections, the gut microbiome can interfere directly or indirectly with current treatments such as chemotherapy and immunotherapy, which might impact a treatment’s outcome. Manipulating the gut microbiome composition using fecal microbiota transplantation or phytochemicals might improve therapeutic outcomes [158]. Fecal microbiota transplantation (FMT) is known as the transplantation of microbes from the gut of a healthy donor to a recipient either through the upper or lower gastrointestinal tract [159]. It was first documented in clinical use in 1958 to treat Clostridium difficile infection as it helped treat 80% of the affected patients [160]. The advantages of using FMT include its safety and its ability to restore intestinal microbial diversity [161]. Limited studies are available in the literature that investigates the role and the application of FMT in the context of GI cancer treatment. We found a study that reported the effectiveness of FMT in mice receiving intestinal microbiota from wild mice, as the results showed better resistance to CRC [162]. Additionally, and on a different approach, the usage of phytochemicals for GI cancer treatment has recently gained attention. The bioactive plant-derived compounds generally have lower oral bioavailability due to poor aqueous solubility, and therefore, the gut microbiome is essential for the metabolism and absorption of bioactive compounds [163]. Several data support the role of 13 bioactive secondary compounds on GI cancer [164]. For example, lutein, an abundant fat-soluble bioactive compound found primarily in green leaved vegetables, was reported to significantly reduce aberrant crypt foci (ACF) in the colon of mice, thus reducing cellular proliferation [165]. Despite those reports that support potential treatments, research is much needed to investigate the potential synergetic effects between the currently used treatments and FMT or phytochemicals. Additionally, attention should be given to the required concentration and the appropriate delivery mode of FMT and phytochemicals to avoid toxicity and possible side effects. Moreover, looking at the role of gut enzymes in the metabolism and the utilization of those natural bioactive compounds, research is needed to investigate the underlying mechanisms played by those enzymes that might affect the treatment outcome, as we have shown in our recently published paper [166].
6. Conclusions
The gut microbiome plays an essential role in mediating the immune response, impacting its activities, development, and function. Generally, and during cancer, signature microbes in the gut influence the anti-tumor activities by producing specific metabolites or inducing T-cell responses. On the other hand, some reported bacterial species enhance cellular proliferation and metastasis during cancer and understanding those interactions in the context of cancer may provide potential therapeutic targets. Despite the advances in the field, more research is needed to understand the underlying mechanisms, investigate the impact on current treatments, and identify specific microbes and immune cells that might lead to this interaction. Additionally, clinical trials are essential to assess the influence of immune–gut interaction on immunotherapy treatment in clinical settings.
Author Contributions
Conceptualization, R.K.A.-I. and D.B.; literature review and resources, R.K.A.-I., L.K. and P.K.; writing—original draft preparation, R.K.A.-I.; writing—review and editing, L.K., P.K. and D.B.; figure preparation and editing, R.K.A.-I. and D.B.; visualization, R.K.A.-I. and D.B.; supervision, D.B.; project administration, D.B.; funding acquisition, D.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by two National Priorities Research Program grants (NPRP11S-1214-170101 and NPRP14S-0311-210033); awarded to Professor Dr. Dietrich Büsselberg from the Qatar National Research Fund (QNRF, a member of Qatar Foundation). The statements made herein are solely the responsibility of the authors.
Acknowledgments
The publication costs of this article were funded by the Weill Cornell Medicine—Qatar Distributed eLibrary.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| GI | gastrointestinal |
| CRC | colorectal cancer |
| GC | gastric cancer |
| EC | esophageal cancer |
| HCC | hepatocellular carcinoma |
| NF-κB | nuclear factor kappa |
| EMT | epithelial-mesenchymal transition |
| FISH | Fluorescence in situ hybridization |
| PD-1 | Programmed death ligand 1 |
| PD-1 | Programmed cell death protein 1 |
| SFB | Segmented filamentous bacteria |
| SAA | Serum amyloid A |
| DC | Dendritic cell |
| AMP | Antimicrobial peptide |
| TLR | Toll-like receptor |
| SCFA | Short chain fatty acid |
| GPCRS | G-protein coupled receptors |
| HDAC | Histone deacetylase |
| ROS | Reactive oxygen species |
| F. nucleatum | Fusobacterium nucleatum |
| CTLA-4 | cytotoxic T lymphocyte-associated antigen 4 |
| TIM-3 | T cell immunoglobulin and mucin protein 3 |
| FMT | Fecal microbiota transplant |
| ACF | Aberrant crypt foci |
References
- Hassanzade, J.; Molavi, E.V.H.; Farahmand, M.; Rajaiifard, A.R. Incidence and Mortality Rate of Common Gastrointestinal Cancers in South of Iran, a Population Based Study. Iran. J. Cancer Prev. 2011, 4, 163–169. [Google Scholar] [PubMed]
- Machlowska, J.; Baj, J.; Sitarz, M.; Maciejewski, R.; Sitarz, R. Gastric Cancer: Epidemiology, Risk Factors, Classification, Genomic Characteristics and Treatment Strategies. Int. J. Mol. Sci. 2020, 21, 4012. [Google Scholar] [CrossRef] [PubMed]
- Krieghoff-Henning, E.; Folkerts, J.; Penzkofer, A.; Weg-Remers, S. Cancer-an overview. Med. Mon. Pharm. 2017, 40, 48–54. [Google Scholar]
- Zali, H.; Rezaei-Tavirani, M.; Azodi, M. Gastric cancer: Prevention, risk factors and treatment. Gastroenterol. Hepatol. Bed Bench 2011, 4, 175–185. [Google Scholar] [PubMed]
- Rozen, P. Cancer of the gastrointestinal tract: Early detection or early prevention? Eur. J. Cancer Prev. 2020, 13, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Correa, P. Gastric cancer: Overview. Gastroenterol. Clin. N. Am. 2020, 42, 211–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igney, F.H.; Krammer, P.H. Death and anti-death: Tumour resistance to apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Qiao, L.; Wong, B.C. Targeting apoptosis as an approach for gastrointestinal cancer therapy. Drug Resist. Updates 2009, 12, 55–64. [Google Scholar] [CrossRef]
- Turvey, S.E.; Broide, D.H. Innate immunity. J. Allergy Clin. Immunol. 2010, 125, S24–S32. [Google Scholar] [CrossRef]
- Marshall, J.S.; Warrington, R.; Watson, W.; Kim, H.L. An introduction to immunology and immunopathology. Allergy Asthma Clin. Immunol. 2018, 14, 49. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.L.; Smothers, J.; Srinivasan, R.; Hoos, A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discov. 2015, 14, 603–622. [Google Scholar] [CrossRef] [PubMed]
- Pandya, P.H.; Murray, M.E.; Pollok, K.E.; Renbarger, J.L. The Immune System in Cancer Pathogenesis: Potential Therapeutic Approaches. J. Immunol. Res. 2016, 2016, 4273943. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, T.J.; Drake, C.G. Primer on tumor immunology and cancer immunotherapy. J. Immunother. Cancer 2013, 1, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, A.D.; Houghton, A.M. Tumor-associated neutrophils: New targets for cancer therapy. Cancer Res. 2011, 71, 2411–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, Y.; Seo, A.N.; Lee, H.E.; Lee, H.S. Tumor immune response and immunotherapy in gastric cancer. J. Pathol. Transl. Med. 2020, 54, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Golshani, G.; Zhang, Y. Advances in immunotherapy for colorectal cancer: A review. Ther. Adv. Gastroenterol. 2020, 13, 1756284820917527. [Google Scholar] [CrossRef] [PubMed]
- Garrett, W.S. Cancer and the microbiota. Science 2015, 348, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Nasr, R.; Shamseddine, A.; Mukherji, D.; Nassar, F.; Temraz, S. The Crosstalk between Microbiome and Immune Response in Gastric Cancer. Int. J. Mol. Sci. 2020, 21, 6586. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Zmora, N.; Levy, M.; Elinav, E. The microbiome and innate immunity. Nature 2016, 535, 65–74. [Google Scholar] [CrossRef]
- Meng, C.; Bai, C.; Brown, T.D.; Hood, L.E.; Tian, Q. Human Gut Microbiota and Gastrointestinal Cancer. Genomics Proteomics Bioinformatics 2018, 16, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Pham, F.; Moinard-Butot, F.; Coutzac, C.; Chaput, N. Cancer and immunotherapy: A role for microbiota composition. Eur. J. Cancer 2021, 155, 145–154. [Google Scholar] [CrossRef]
- Yu, Q.; Jia, A.; Li, Y.; Bi, Y.; Liu, G. Microbiota regulate the development and function of the immune cells. Int. Rev. Immunol. 2018, 37, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Wesemann, D.R.; Portuguese, A.J.; Meyers, R.M.; Gallagher, M.P.; Cluff-Jones, K.; Magee, J.M.; Alt, F.W. Microbial colonization influences early B-lineage development in the gut lamina propria. Nature 2013, 501, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Lathrop, S.K.; Bloom, S.M.; Rao, S.M.; Nutsch, K.; Lio, C.W.; Santacruz, N.; Hsieh, C.S. Peripheral education of the immune system by colonic commensal microbiota. Nature 2011, 478, 250–254. [Google Scholar] [CrossRef]
- Zitvogel, L.; Ma, Y.; Raoult, D.; Kroemer, G.; Gajewski, T.F. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 2018, 359, 1366–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Guryn, K.; Leone, V.; Chang, E.B. Regional Diversity of the Gastrointestinal Microbiome. Cell Host Microbe 2019, 26, 314–324. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Chen, H.; Shu, X.; Yin, Y.; Li, J.; Qin, J.; Xiang, C. Presence of Segmented Filamentous Bacteria in Human Children and Its Potential Role in the Modulation of Human Gut Immunity. Front. Microbiol. 2018, 9, 1403. [Google Scholar] [CrossRef]
- Hedblom, G.A.; Reiland, H.A.; Sylte, M.J.; Johnson, T.J.; Baumler, D.J. Segmented Filamentous Bacteria-Metabolism Meets Immunity. Front. Microbiol. 2018, 9, 1991. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.; Oshima, K.; Morita, H.; Fukuda, S.; Imaoka, A.; Kumar, N. Complete genome sequences of rat and mouse segmented filamentous bacteria, a potent inducer of th17 cell differentiation. Cell Host Microbe 2011, 10, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudino, S.J.; Beaupre, M.; Lin, X. IL-22 receptor signaling in Paneth cells is critical for their maturation, microbiota colonization, Th17-related immune responses, and anti-Salmonella immunity. Mucosal Immunol. 2021, 14, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Flannigan, K.L.; Ngo, V.L.; Geem, D.; Harusato, A.; Hirota, S.A.; Parkos, C.A. IL-17Amediated neutrophil recruitment limits expansion of segmented filamentous bacteria. Mucosal Immunol. 2017, 10, 673–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, V.F.; Cox, J.; Kljavin, N.M.; Dengler, H.S.; Reichelt, M.; Kumar, P. Homeostatic IL23 receptor signaling limits Th17 response through IL-22-mediated containment of commensal microbiota. Proc. Natl. Acad. Sci. USA 2014, 111, 13942–13947. [Google Scholar] [CrossRef] [Green Version]
- Flannigan, K.L.; Denning, T.L. Segmented filamentous bacteria-induced immune responses: A balancing act between host protection and autoimmunity. Immunology 2018, 154, 537–546. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Littman, D.R. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Sivaprakasam, S.; Bhutia, Y.D.; Yang, S.; Ganapathy, V. Short-Chain Fatty Acid Transporters: Role in Colonic Homeostasis. Compr. Physiol. 2017, 8, 299–314. [Google Scholar]
- Blaak, E.E.; Canfora, E.E.; Theis, S.; Frost, G.; Groen, A.K.; Mithieux, G.; Verbeke, K. Short chain fatty acids in human gut and metabolic health. Benef. Microbes 2020, 11, 411–455. [Google Scholar] [CrossRef]
- Chambers, E.S.; Preston, T.; Frost, G.; Morrison, D.J. Role of Gut Microbiota-Generated Short-Chain Fatty Acids in Metabolic and Cardiovascular Health. Curr. Nutr. Rep. 2018, 7, 198–206. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.Y.; Groer, M.; Dutra, S.V.O.; Sarkar, A.; McSkimming, D.I. Gut Microbiota and Immune System Interactions. Microorganisms 2020, 8, 1587. [Google Scholar] [CrossRef] [PubMed]
- Vinolo, M.A.; Rodrigues, H.G.; Nachbar, R.T.; Curi, R. Regulation of inflammation by short chain fatty acids. Nutrients 2011, 3, 858–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Nabhani, Z.; Dulauroy, S.; Marques, R.; Cousu, C.; Al Bounny, S.; Déjardin, F.; Sparwasser, T.; Bérard, M.; Cerf-Bensussan, N.; Eberl, G. A weaning reaction to microbiota is required for resistance to immunopathologies in the adult. Immunity 2019, 50, 1276–1288.e1275. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A. GPR109A is a G-protein–coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; He, Z.; Chen, W.; Holzman, I.R.; Lin, J. Effects of butyrate on intestinal barrier function in a Caco-2 cell monolayer model of intestinal barrier. Pediatr. Res. 2007, 61, 37–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Okumura, R.; Takeda, K. Maintenance of intestinal homeostasis by mucosal barriers. Inflamm. Regen. 2018, 38, 5. [Google Scholar] [CrossRef]
- Pasupuleti, M.; Schmidtchen, A.; Malmsten, M. Antimicrobial peptides: Key components of the innate immune system. Crit. Rev. Biotechnol. 2012, 32, 143–171. [Google Scholar] [CrossRef] [Green Version]
- Ostaff, M.J.; Stange, E.F.; Wehkamp, J. Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol. Med. 2013, 5, 1465–1483. [Google Scholar] [CrossRef]
- Salzman, N.H.; Hung, K.; Haribhai, D.; Chu, H.; Karlsson-Sjoberg, J.; Amir, E.; Teggatz, P.; Barman, M.; Hayward, M.; Eastwood, D. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 2010, 11, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Grondin, J.A.; Kwon, Y.H.; Far, P.M.; Haq, S.; Khan, W.I. Mucins in intestinal mucosal defense and inflammation: Learning from clinical and experimental studies. Front. Immunol. 2020, 11, 2054. [Google Scholar] [CrossRef] [PubMed]
- Cario, E. Heads up! How the intestinal epithelium safeguards mucosal barrier immunity through the infl ammasome and beyond. Curr. Opin. Gastroenterol. 2010, 26, 583–590. [Google Scholar] [CrossRef]
- Jobin, C. MyD88 signaling in the intestine: Dr Jekyll and Mr Hyde? Gastroenterology 2010, 139, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Frantz, A.L.; Rogier, E.W.; Weber, C.R.; Shen, L.; Cohen, D.A.; Fenton, L.A.; Kaetzel, C.S. Targeted deletion of MyD88 in intestinal epithelial cells results in compromised antibacterial immunity associated with downregulation of polymeric immunoglobulin receptor, mucin-2, and antibacterial peptides. Mucosal. Immunol. 2012, 5, 501–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tezuka, H.; Ohteki, T. Regulation of IgA Production by Intestinal Dendritic Cells and Related Cells. Front. Immunol. 2019, 10, 1891. [Google Scholar] [CrossRef]
- Cenit, M.C.; Sanz, Y.; Codoner-Franch, P. Influence of gut microbiota on neuropsychiatric disorders. World J. Gastroenterol. 2017, 23, 5486–5498. [Google Scholar] [CrossRef]
- Tajik, N.; Frech, M.; Schulz, O.; Schalter, F.; Lucas, S.; Azizov, V.; Durholz, K.; Steffen, F.; Omata, Y.; Rings, A. Targeting zonulin and intestinal epithelial barrier function to prevent onset of arthritis. Nat. Commun. 2020, 11, 1995. [Google Scholar] [CrossRef] [Green Version]
- Panebianco, C.; Andriulli, A.; Pazienza, V. Pharmacomicrobiomics: Exploiting the drug-microbiota interactions in anticancer therapies. Microbiome 2018, 6, 92. [Google Scholar] [CrossRef]
- Dzutsev, A.; Badger, J.H.; Perez-Chanona, E.; Roy, S.; Salcedo, R.; Smith, C.K.; Trinchieri, G. Microbes and Cancer. Annu. Rev. Immunol. 2017, 35, 199–228. [Google Scholar] [CrossRef]
- Iida, N.; Dzutsev, A.; Stewart, C.A.; Smith, L.; Bouladoux, N.; Weingarten, R.A.; Molina, D.A.; Salcedo, R.; Back, T.; Cramer, S. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science 2013, 342, 967–970. [Google Scholar] [CrossRef]
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nature 2016, 535, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Tsuei, J.; Chau, T.; Mills, D.; Wan, Y.J. Bile acid dysregulation, gut dysbiosis, and gastrointestinal cancer. Exp. Biol. Med. 2014, 239, 1489–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Wang, X.; Guo, Y. Gut microbiota influence tumor development and Alter interactions with the human immune system. J. Exp. Clin. Cancer Res. 2021, 40, 42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, F.; Zhao, C.; Xu, Q.; Liang, C.; Yang, Y.; Wang, H.; Shang, Y.; Wang, Y.; Mu, X. Dysbiosis of the gut microbiome is associated with thyroid cancer and thyroid nodules and correlated with clinical index of thyroid function. Endocrine 2019, 64, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Friesen, L.; Park, J.; Kim, H.M.; Kim, C.H. Microbial metabolites, short-chain fatty acids, restrain tissue bacterial load, chronic inflammation, and associated cancer in the colon of mice. Eur. J. Immunol. 2018, 48, 1235–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, A.I.; Zhao, L.; Eaton, K.A.; Ho, S.; Chen, J.; Poe, S.; Chen, G.Y. Gut Microbiota Modulate CD8 T Cell Responses to Influence Colitis-Associated Tumorigenesis. Cell Rep. 2020, 31, 107471. [Google Scholar] [CrossRef]
- Yao, H.; Wan, J.Y.; Zeng, J.; Huang, W.H.; Sava-Segal, C.; Li, L.; Yuan, C.S. Effects of compound K, an enteric microbiome metabolite of ginseng, in the treatment of inflammation associated colon cancer. Oncol. Lett. 2018, 15, 8339–8348. [Google Scholar] [CrossRef]
- Liang, W.; Peng, X.; Li, Q.; Wang, P.; Lv, P.; Song, Q.; Wang, Y. FAM3D is essential for colon homeostasis and host defense against inflammation associated carcinogenesis. Nat. Commun. 2020, 11, 5912. [Google Scholar] [CrossRef]
- Bhatt, B.; Zeng, P.; Zhu, H.; Sivaprakasam, S.; Li, S.; Xiao, H.; Singh, N. Gpr109a Limits Microbiota-Induced IL-23 Production To Constrain ILC3-Mediated Colonic Inflammation. J. Immunol. 2018, 200, 2905–2914. [Google Scholar] [CrossRef] [Green Version]
- Daniel, S.G.; Ball, C.L.; Besselsen, D.G.; Doetschman, T.; Hurwitz, B.L.; Chia, N. Functional Changes in the Gut Microbiome Contribute to Transforming Growth Factor β -Deficient Colon Cancer. mSystems 2017, 2, e00065-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panneerselvam, J.; Madka, V.; Rai, R.; Morris, K.T.; Houchen, C.W.; Chandrakesan, P.; Rao, C.V. Inflammatory Mediators and Gut Microbial Toxins Drive Colon Tumorigenesis by IL-23 Dependent Mechanism. Cancers 2021, 13, 5159. [Google Scholar] [CrossRef] [PubMed]
- Swafford, D.; Shanmugam, A.; Ranganathan, P.; Manoharan, I.; Hussein, M.S.; Patel, N.; Manicassamy, S. The Wnt-beta-Catenin-IL-10 Signaling Axis in Intestinal APCs Protects Mice from Colitis-Associated Colon Cancer in Response to Gut Microbiota. J. Immunol. 2020, 205, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Burgueno, J.F.; Fritsch, J.; Gonzalez, E.E.; Landau, K.S.; Santander, A.M.; Fernandez, I.; Abreu, M.T. Epithelial TLR4 Signaling Activates DUOX2 to Induce Microbiota-Driven Tumorigenesis. Gastroenterology 2021, 160, 797–808.e796. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, X.; Zhong, W.; Yang, M.; Xu, M.; Sun, Y.; Cao, H. Gut microbiota from colorectal cancer patients enhances the progression of intestinal adenoma in Apc(min/+) mice. EBioMedicine 2019, 48, 301–315. [Google Scholar] [CrossRef] [Green Version]
- Nosho, K.; Sukawa, Y.; Adachi, Y.; Ito, M.; Mitsuhashi, K.; Kurihara, H.; Shinomura, Y. Association of Fusobacterium nucleatum with immunity and molecular alterations in colorectal cancer. World J. Gastroenterol. 2016, 22, 557–566. [Google Scholar] [CrossRef]
- Sui, H.; Zhang, L.; Gu, K.; Chai, N.; Ji, Q.; Zhou, L.; Li, Q. YYFZBJS ameliorates colorectal cancer progression in Apc(Min/+) mice by remodeling gut microbiota and inhibiting regulatory T-cell generation. Cell Commun. Signal. 2020, 18, 113. [Google Scholar] [CrossRef]
- Pastille, E.; Fassnacht, T.; Adamczyk, A.; Ngo Thi Phuong, N.; Buer, J.; Westendorf, A.M. Inhibition of TLR4 Signaling Impedes Tumor Growth in Colitis-Associated Colon Cancer. Front. Immunol. 2021, 12, 669747. [Google Scholar] [CrossRef]
- Dupaul-Chicoine, J.; Arabzadeh, A.; Dagenais, M.; Douglas, T.; Champagne, C.; Morizot, A.; Saleh, M. The Nlrp3 Inflammasome Suppresses Colorectal Cancer Metastatic Growth in the Liver by Promoting Natural Killer Cell Tumoricidal Activity. Immunity 2015, 43, 751–763. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, Y.; Mima, K.; Ishimoto, T.; Ogata, Y.; Imai, K.; Miyamoto, Y.; Baba, H. Relationship between Fusobacterium nucleatum and anti-tumor immunity in colorectal cancer liver metastasis. Cancer Sci. 2021, 112, 4470–4477. [Google Scholar] [CrossRef]
- Ma, X.; Zhou, Z.; Zhang, X.; Fan, M.; Hong, Y.; Feng, Y.; Wang, G. Sodium butyrate modulates gut microbiota and immune response in colorectal cancer liver metastatic mice. Cell Biol. Toxicol. 2020, 36, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Roberti, M.P.; Yonekura, S.; Duong, C.P.M.; Picard, M.; Ferrere, G.; Tidjani Alou, M.; Zitvogel, L. Chemotherapy-induced ileal crypt apoptosis and the ileal microbiome shape immunosurveillance and prognosis of proximal colon cancer. Nat. Med. 2020, 26, 919–931. [Google Scholar] [CrossRef]
- Xue, M.; Liang, H.; Ji, X.; Zhou, Z.; Liu, Y.; Sun, T.; Zhang, L. Effects of fucoidan on gut flora and tumor prevention in 1,2-dimethylhydrazine-induced colorectal carcinogenesis. J. Nutr. Biochem. 2020, 82, 108396. [Google Scholar] [CrossRef] [PubMed]
- Woznicki, J.A.; Flood, P.; Bustamante-Garrido, M.; Stamou, P.; Moloney, G.; Fanning, A.; Nally, K. Human BCL-G regulates secretion of inflammatory chemokines but is dispensable for induction of apoptosis by IFN-gamma and TNF-alpha in intestinal epithelial cells. Cell Death Dis. 2020, 11, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, B.B. Nuclear factor-kappaB: The enemy within. Cancer Cell 2004, 6, 203–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A. Cancer: Inflammation by remote control. Nature 2005, 435, 752–753. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Baby, D.; Rajguru, J.P.; Patil, P.B.; Thakkannavar, S.S.; Pujari, V.B. Inflammation and cancer. Ann. Afr. Med. 2019, 18, 121–126. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 36–44. [Google Scholar] [CrossRef]
- Lee, T.C.; Huang, Y.C.; Lu, Y.Z.; Yeh, Y.C.; Yu, L.C. Hypoxia-induced intestinal barrier changes in balloon-assisted enteroscopy. J. Physiol. 2018, 596, 3411–3424. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Kohno, H.; Suzuki, R.; Hata, K.; Sugie, S.; Niho, N. Dextran sodium sulfate strongly promotes colorectal carcinogenesis in Apc (Min/+) mice: Inflammatory stimuli by dextran sodium sulfate results in development of multiple colonic neoplasms. Int. J. Cancer 2006, 118, 25–34. [Google Scholar] [CrossRef]
- Song, X.; Gao, H.; Lin, Y.; Yao, Y.; Zhu, S.; Wang, J. Alterations in the Microbiota Drive Interleukin-17C Production from Intestinal Epithelial Cells to Promote Tumorigenesis. Immunity 2014, 40, 140–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Ling, Z.; Li, L. The Intestinal Microbiota and Colorectal Cancer. Front. Immunol. 2020, 11, 615056. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Fang, L.; Lee, M.H. Dysbiosis of gut microbiota in promoting the development of colorectal cancer. Gastroenterol. Rep. 2018, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Chu, F.; Li, Y.; Meng, X.; Li, Y.; Li, T.; Zhai, M.; Ding, X. Gut Microbial Dysbiosis and Changes in Fecal Metabolic Phenotype in Precancerous Lesions of Gastric Cancer Induced With N-Methyl-N’-Nitro-N-Nitrosoguanidine, Sodium Salicylate, Ranitidine, and Irregular Diet. Front. Physiol. 2021, 12, 733979. [Google Scholar] [CrossRef]
- Matson, J.P.; Cook, J.G. Cell cycle proliferation decisions: The impact of single cell analyses. FEBS J. 2017, 284, 362–375. [Google Scholar] [CrossRef] [Green Version]
- Hallstrom, T.C.; Nevins, J.R. Balancing the decision of cell proliferation and cell fate. Cell Cycle 2009, 8, 532–535. [Google Scholar] [CrossRef] [Green Version]
- Duronio, R.J.; Xiong, Y. Signaling pathways that control cell proliferation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008904. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Nowsheen, S. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef]
- von Frieling, J.; Fink, C.; Hamm, J.; Klischies, K.; Forster, M.; Bosch, T.C.G.; Sommer, F. Grow with the Challenge-Microbial Effects on Epithelial Proliferation, Carcinogenesis, and Cancer Therapy. Front. Microbiol. 2018, 9, 2020. [Google Scholar] [CrossRef]
- McAllister, F.; Housseau, F.; Sears, C.L. Microbiota and immune responses in colon cancer: More to learn. Cancer J. 2014, 20, 232–236. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Li, Q.; Fu, X. Fusobacterium nucleatum Contributes to the Carcinogenesis of Colorectal Cancer by Inducing Inflammation and Suppressing Host Immunity. Transl. Oncol. 2019, 12, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.W. Fusobacterium nucleatum: A commensal-turned pathogen. Curr. Opin. Microbiol. 2015, 23, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Meyerson, M. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anti-cancer immune response. J. Hematol. Oncol. 2017, 10, 101. [Google Scholar] [CrossRef] [Green Version]
- Zagato, E.; Pozzi, C.; Bertocchi, A.; Schioppa, T.; Saccheri, F.; Guglietta, S.; Rescigno, M. Endogenous murine microbiota member Faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nat. Microbiol. 2020, 5, 511–524. [Google Scholar] [CrossRef]
- Hanus, M.; Parada-Venegas, D.; Landskron, G.; Wielandt, A.M.; Hurtado, C.; Alvarez, K.; De la Fuente, M. Immune System, Microbiota, and Microbial Metabolites: The Unresolved Triad in Colorectal Cancer Microenvironment. Front. Immunol. 2021, 12, 612826. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: A hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Hunter, K.W.; Crawford, N.P.; Alsarraj, J. Mechanisms of metastasis. Breast Cancer Res. 2008, 10, S2. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.N.; Araujo-Perez, F.; Azcarate-Peril, A.; Yeh, J.J.; Sandler, R.S.; Keku, T.O. Fusobacterium is associated with colorectal adenomas. PLoS ONE 2013, 8, e53653. [Google Scholar] [CrossRef]
- Wang, S.; Liu, Y.; Li, J.; Zhao, L.; Yan, W.; Lin, B.; Wei, Y. Fusobacterium nucleatum Acts as a Pro-carcinogenic Bacterium in Colorectal Cancer: From Association to Causality. Front. Cell Dev. Biol. 2021, 9, 710165. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.A.; Garrett, W.S. Fusobacterium nucleatum-symbiont, opportunist and oncobacterium. Nat. Rev. Microbiol. 2019, 17, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Kaczanowski, S. Apoptosis: Its origin, history, maintenance and the medical implications for cancer and aging. Phys. Biol. 2016, 13, 031001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Jan, R.; Chaudhry, G.E. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull 2019, 9, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Yu, J. Role of apoptosis in colon cancer biology, therapy, and prevention. Curr. Colorectal. Cancer Rep. 2013, 9, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Liu, J.; Ma, Y.; Wei, Y.; Liu, J.; Wang, H. Impairment of Intestinal Barrier Function Induced by Early Weaning via Autophagy and Apoptosis Associated with Gut Microbiome and Metabolites. Front. Immunol. 2021, 12, 804870. [Google Scholar] [CrossRef]
- Waldecker, M.; Kautenburger, T.; Daumann, H.; Busch, C.; Schrenk, D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 2008, 19, 587–593. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, K.N.; Vitetta, L. Effects of Intestinal Microbial(-)Elaborated Butyrate on Oncogenic Signaling Pathways. Nutrients 2019, 11, 1026. [Google Scholar] [CrossRef] [Green Version]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; Gonzalez, M.J.; Quera, R.; Dijkstra, G.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Y.; Xu, Q.; Sun, L.; Ye, Y.; Ji, G. Short-chain fatty acids administration is protective in colitis-associated colorectal cancer development. J. Nutr. Biochem. 2018, 57, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012, 6, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salcedo, R.; Worschech, A.; Cardone, M.; Jones, Y.; Gyulai, Z.; Dai, R.-M.; Wang, E.; Ma, W.; Haines, D.; O’Huigin, C. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: Role of interleukin 18. J. Exp. Med. 2010, 207, 1625–1636. [Google Scholar] [CrossRef]
- Tye, H.; Yu, C.H.; Simms, L.A.; de Zoete, M.R.; Kim, M.L.; Zakrzewski, M.; Masters, S.L. NLRP1 restricts butyrate producing commensals to exacerbate inflammatory bowel disease. Nat. Commun. 2018, 9, 3728. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.J.; Ma, S.Y.; Wen, J.; Zhang, X.Q.; Xing, H.J.; Jia, C.S. Direct contact moxibustion promotes apoptosis of gastric cancer cells in rats by regulating intestinal flora. J. Tradit. Chin. Med. 2021, 41, 943–952. [Google Scholar] [PubMed]
- Zhang, Y.; Zhou, L.; Bao, Y.L.; Wu, Y.; Yu, C.L.; Huang, Y.X.; Sun, Y.; Zheng, L.H.; Li, Y.X. Butyrate induces cell apoptosis through activation of JNK MAP kinase pathway in human colon cancer RKO cells. Chem. Biol. Interact. 2010, 185, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [Green Version]
- Urruticoechea, A.; Alemany, R.; Balart, J.; Villanueva, A.; Vinals, F.; Capella, G. Recent advances in cancer therapy: An overview. Curr. Pharm. Des. 2010, 16, 3–10. [Google Scholar] [CrossRef]
- Seager, R.J.; Hajal, C.; Spill, F.; Kamm, R.D.; Zaman, M.H. Dynamic interplay between tumour, stroma and immune system can drive or prevent tumour progression. Converg. Sci. Phys. Oncol. 2017, 3, 034002. [Google Scholar] [CrossRef]
- Inthagard, J.; Edwards, J.; Roseweir, A.K. Immunotherapy: Enhancing the efficacy of this promising therapeutic in multiple cancers. Clin. Sci. 2019, 133, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? BMC Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vacchelli, E.; Bravo-San Pedro, J.M.; Buque, A.; Senovilla, L.; Baracco, E.E.; Kroemer, G. Classification of current anti-cancer immunotherapies. Oncotarget 2014, 5, 12472–12508. [Google Scholar] [CrossRef] [Green Version]
- Dine, J.; Gordon, R.; Shames, Y.; Kasler, M.K.; Barton-Burke, M. Immune Checkpoint Inhibitors: An Innovation in Immunotherapy for the Treatment and Management of Patients with Cancer. Asia Pac. J. Oncol. Nurs. 2017, 4, 127–135. [Google Scholar] [CrossRef]
- Koury, J.; Lucero, M.; Cato, C.; Chang, L.; Geiger, J.; Henry, D.; Tran, A. Immunotherapies: Exploiting the Immune System for Cancer Treatment. J. Immunol. Res. 2018, 2018, 9585614. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Sznol, M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Manz, S.M.; Losa, M.; Fritsch, R.; Scharl, M. Efficacy and side effects of immune checkpoint inhibitors in the treatment of colorectal cancer. Ther. Adv. Gastroenterol. 2021, 14, 17562848211002018. [Google Scholar] [CrossRef] [PubMed]
- Vivarelli, S.; Salemi, R.; Candido, S.; Falzone, L.; Santagati, M.; Stefani, S.; Libra, M. Gut Microbiota and Cancer: From Pathogenesis to Therapy. Cancers 2019, 11, 38. [Google Scholar] [CrossRef] [Green Version]
- Shui, L.; Yang, X.; Li, J.; Yi, C.; Sun, Q.; Zhu, H. Gut Microbiome as a Potential Factor for Modulating Resistance to Cancer Immunotherapy. Front. Immunol. 2019, 10, 2989. [Google Scholar] [CrossRef] [Green Version]
- Vetizou, M.; Pitt, J.M.; Daillere, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Zitvogel, L. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Cheng, S.; Kou, Y.; Wang, Z.; Jin, R.; Hu, H.; Shen, L. The Gut Microbiome Is Associated with Clinical Response to Anti-PD-1/PD-L1 Immunotherapy in Gastrointestinal Cancer. Cancer Immunol. Res. 2020, 8, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Fessler, J.; Matson, V.; Gajewski, T.F. Exploring the emerging role of the microbiome in cancer immunotherapy. J. Immunother. Cancer 2019, 7, 108. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Lv, J.; Guo, F.; Li, J.; Jia, Y.; Jiang, D.; Li, Z. Gut Microbiome Influences the Efficacy of PD-1 Antibody Immunotherapy on MSS-Type Colorectal Cancer via Metabolic Pathway. Front. Microbiol. 2020, 11, 814. [Google Scholar] [CrossRef]
- Lin, C.; Cai, X.; Zhang, J.; Wang, W.; Sheng, Q.; Hua, H.; Zhou, X. Role of Gut Microbiota in the Development and Treatment of Colorectal Cancer. Digestion 2019, 100, 72–78. [Google Scholar] [CrossRef]
- Goubet, A.G.; Daillere, R.; Routy, B.; Derosa, L.; Roberti, P.M.; Zitvogel, L. The impact of the intestinal microbiota in therapeutic responses against cancer. C R Biol. 2018, 341, 284–289. [Google Scholar] [CrossRef]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Editorial: Adverse Effects of Cancer Chemotherapy: Anything New to Improve Tolerance and Reduce Sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef]
- Li, W.; Deng, X.; Chen, T. Exploring the Modulatory Effects of Gut Microbiota in Anti-Cancer Therapy. Front. Oncol. 2021, 11, 644454. [Google Scholar] [CrossRef]
- Lin, X.B.; Dieleman, L.A.; Ketabi, A.; Bibova, I.; Sawyer, M.B.; Xue, H. Irinotecan (CPT-11) chemotherapy alters intestinal microbiota in tumour bearing rats. PLoS ONE 2012, 7, e39764. [Google Scholar] [CrossRef]
- Secombe, K.R.; Coller, J.K.; Gibson, R.J.; Wardill, H.R.; Bowen, J.M. The bidirectional interaction of the gut microbiome and the innate immune system: Implications for chemotherapy-induced gastrointestinal toxicity. Int. J. Cancer 2019, 144, 2365–2376. [Google Scholar] [CrossRef]
- Klaassen, C.D.; Cui, J.Y. Review: Mechanisms of How the Intestinal Microbiota Alters the Effects of Drugs and Bile Acids. Drug Metab. Dispos. 2015, 43, 1505–1521. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Yu, K.; Zhang, X.; Yu, S. Dual functional roles of the MyD88 signaling in colorectal cancer development. Biomed. Pharmacother. 2018, 107, 177–184. [Google Scholar] [CrossRef]
- Chang, C.W.; Lee, H.C.; Li, L.H.; Chiang Chiau, J.S.; Wang, T.E.; Chuang, W.H. Fecal Microbiota Transplantation Prevents Intestinal Injury, Upregulation of Toll-Like Receptors, and 5-Fluorouracil/Oxaliplatin-Induced Toxicity in Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirmer, M.; Smeekens, S.P.; Vlamakis, H.; Jaeger, M.; Oosting, M.; Franzosa, E.A. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 2016, 167, 1125–1136.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viaud, S.; Saccheri, F.; Mignot, G.; Yamazaki, T.; Daillère, R.; Hannani, D. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 2013, 342, 971–976. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Fang, J.Y. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563.e516. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.; Brunner, V.; Tschurtschenthaler, M. Microbiota and Colorectal Cancer: From Gut to Bedside. Front. Pharmacol. 2021, 12, 760280. [Google Scholar] [CrossRef]
- Chen, D.; Wu, J.; Jin, D.; Wang, B.; Cao, H. Fecal microbiota transplantation in cancer management: Current status and perspectives. Int. J. Cancer 2019, 145, 2021–2031. [Google Scholar] [CrossRef] [Green Version]
- Van Nood, E.; Vrieze, A.; Nieuwdorp, M. Duodenal infusion of donor feces for recurrent Clostridium difficile. N. Engl. J. Med. 2013, 368, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.O.; Gluck, M. Fecal Microbiota Transplantation: An Update on Clinical Practice. Clin. Endosc. 2019, 52, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Rosshart, S.P.; Vassallo, B.G.; Angeletti, D. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell 2017, 171, 1015–1028.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassidy, A.; Minihane, A.M. The role of metabolism (and the microbiome) in defining the clinical efficacy of dietary flavonoids. Am. J. Clin. Nutr. 2017, 105, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ishaq, R.K.; Overy, A.J.; Busselberg, D. Phytochemicals and Gastrointestinal Cancer: Cellular Mechanisms and Effects to Change Cancer Progression. Biomolecules 2020, 10, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gali-Muhtasib, H.U.; Younes, I.H.; Karchesy, J.J.; el-Sabban, M.E. Plant tannins inhibit the induction of aberrant crypt foci and colonic tumors by 1,2-dimethylhydrazine in mice. Nutr. Cancer 2001, 39, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Al-Ishaq, R.K.; Liskova, A.; Kubatka, P.; Busselberg, D. Enzymatic Metabolism of Flavonoids by Gut Microbiota and Its Impact on Gastrointestinal Cancer. Cancers 2021, 13, 3934. [Google Scholar] [CrossRef]
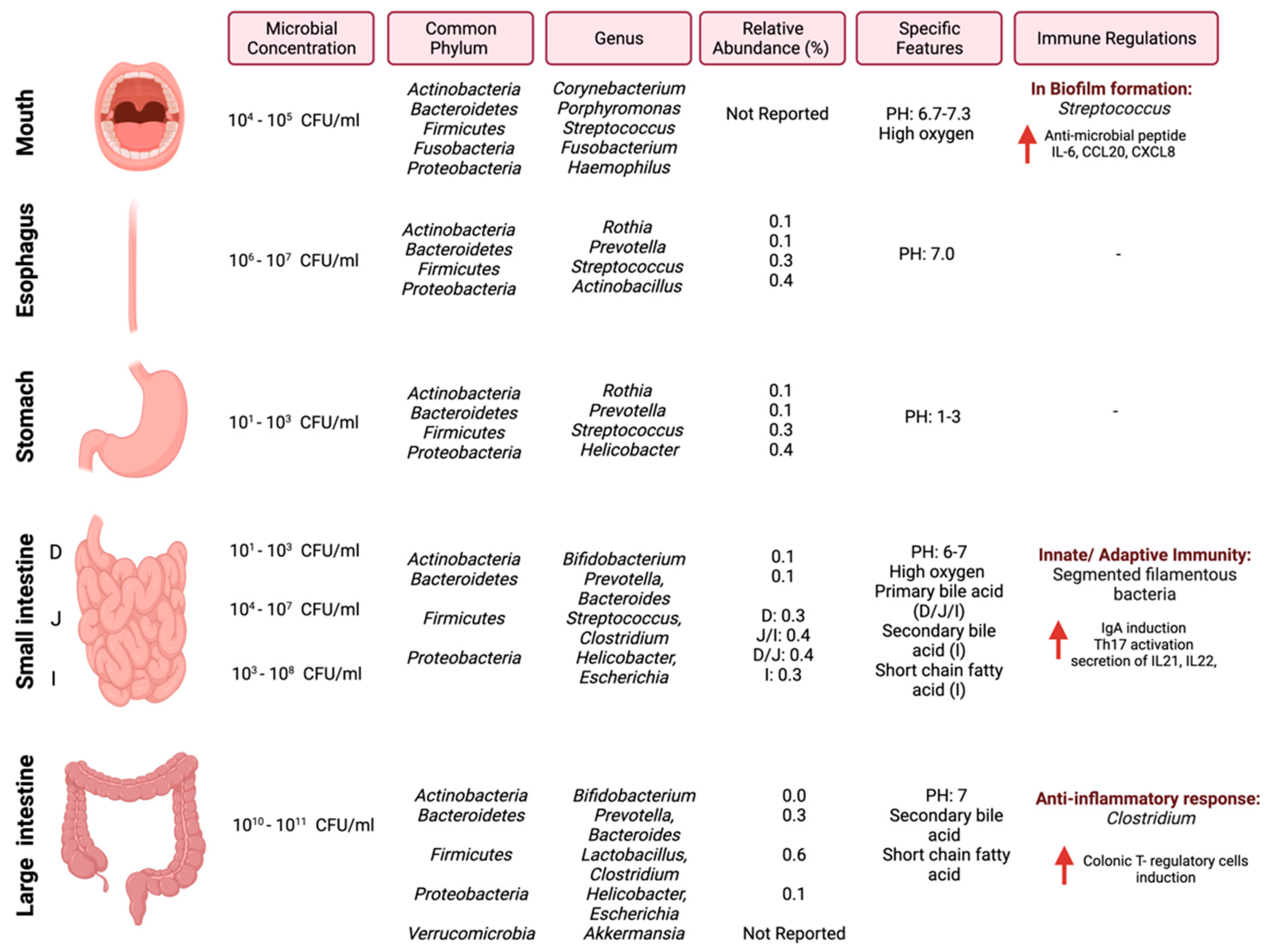
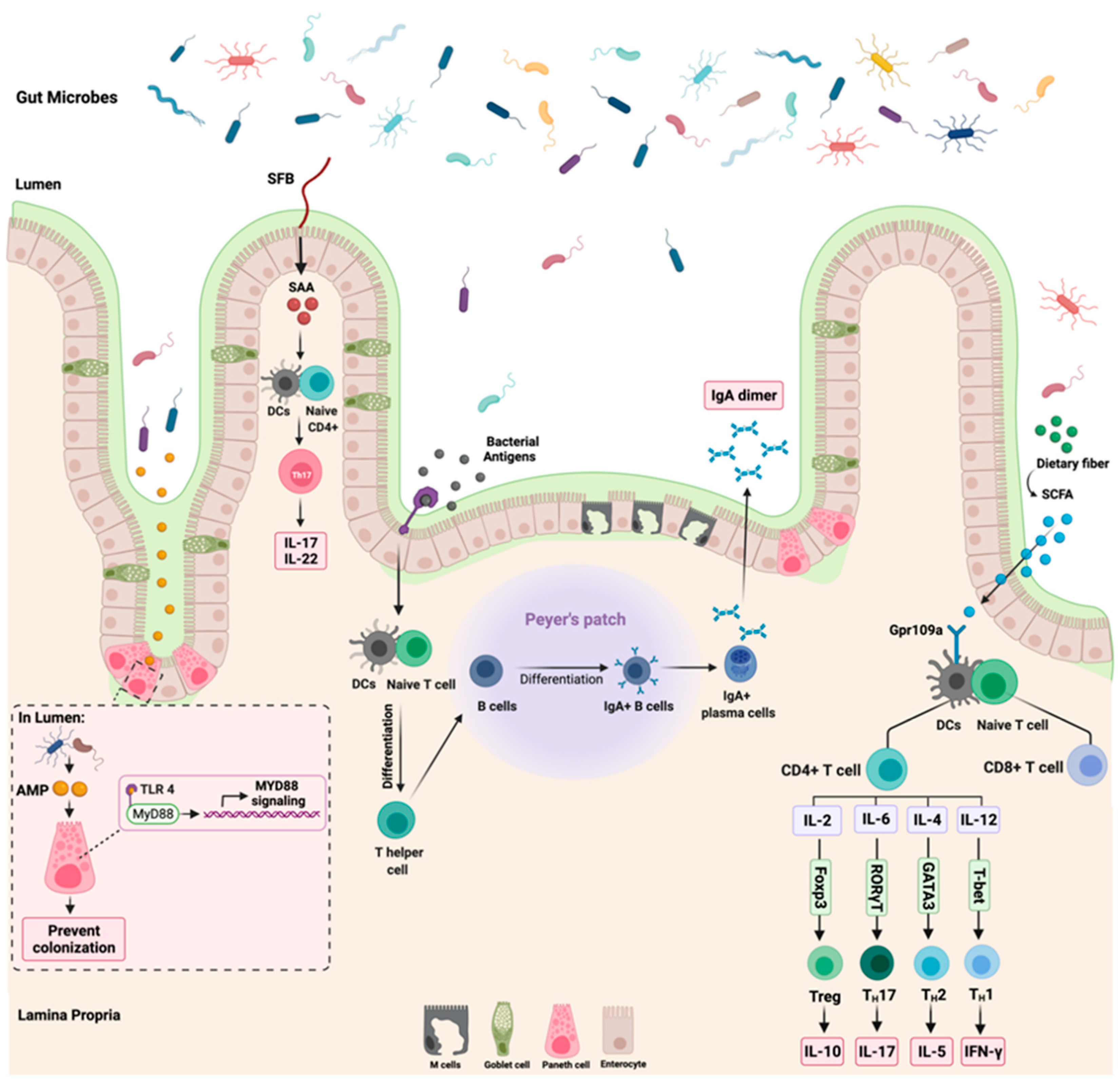
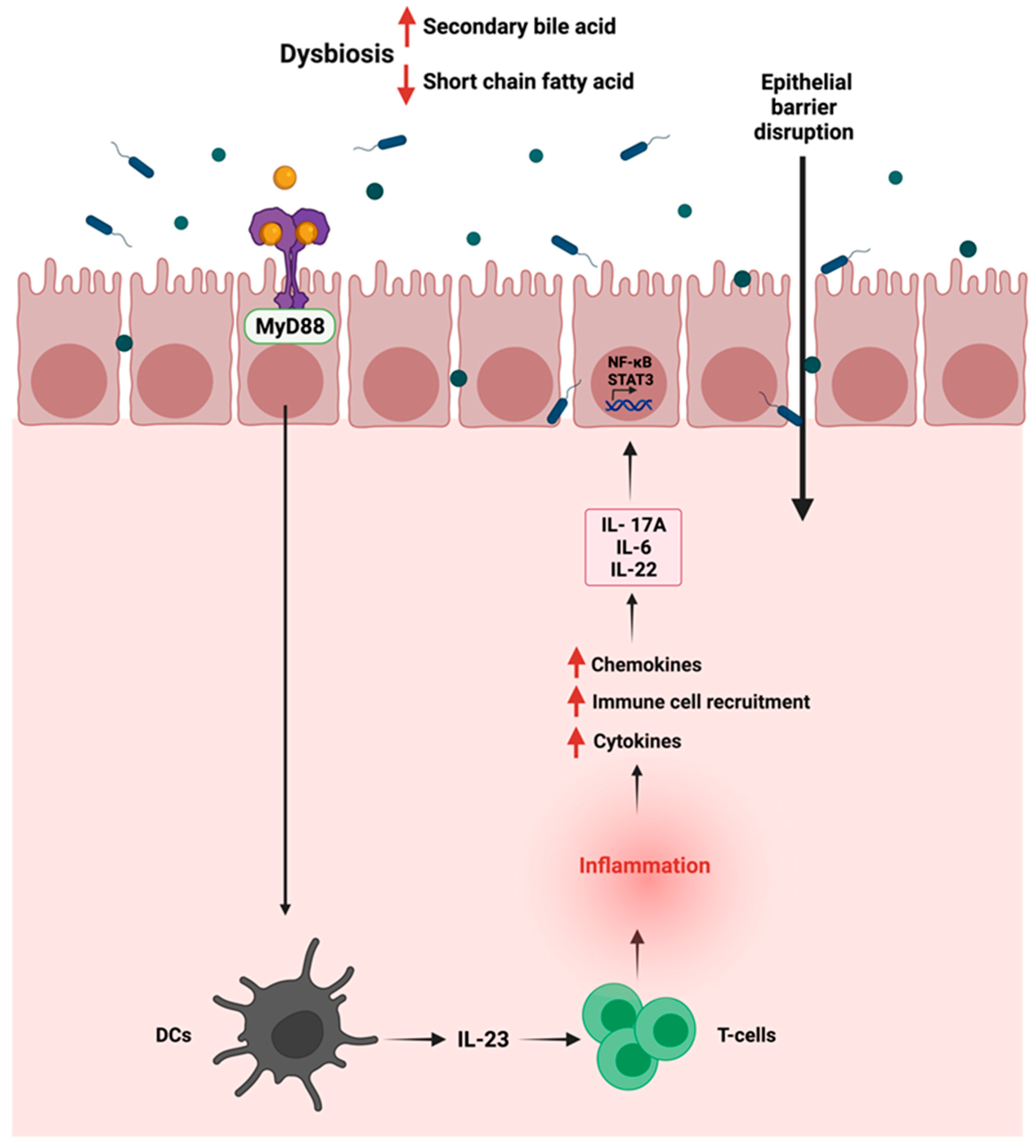
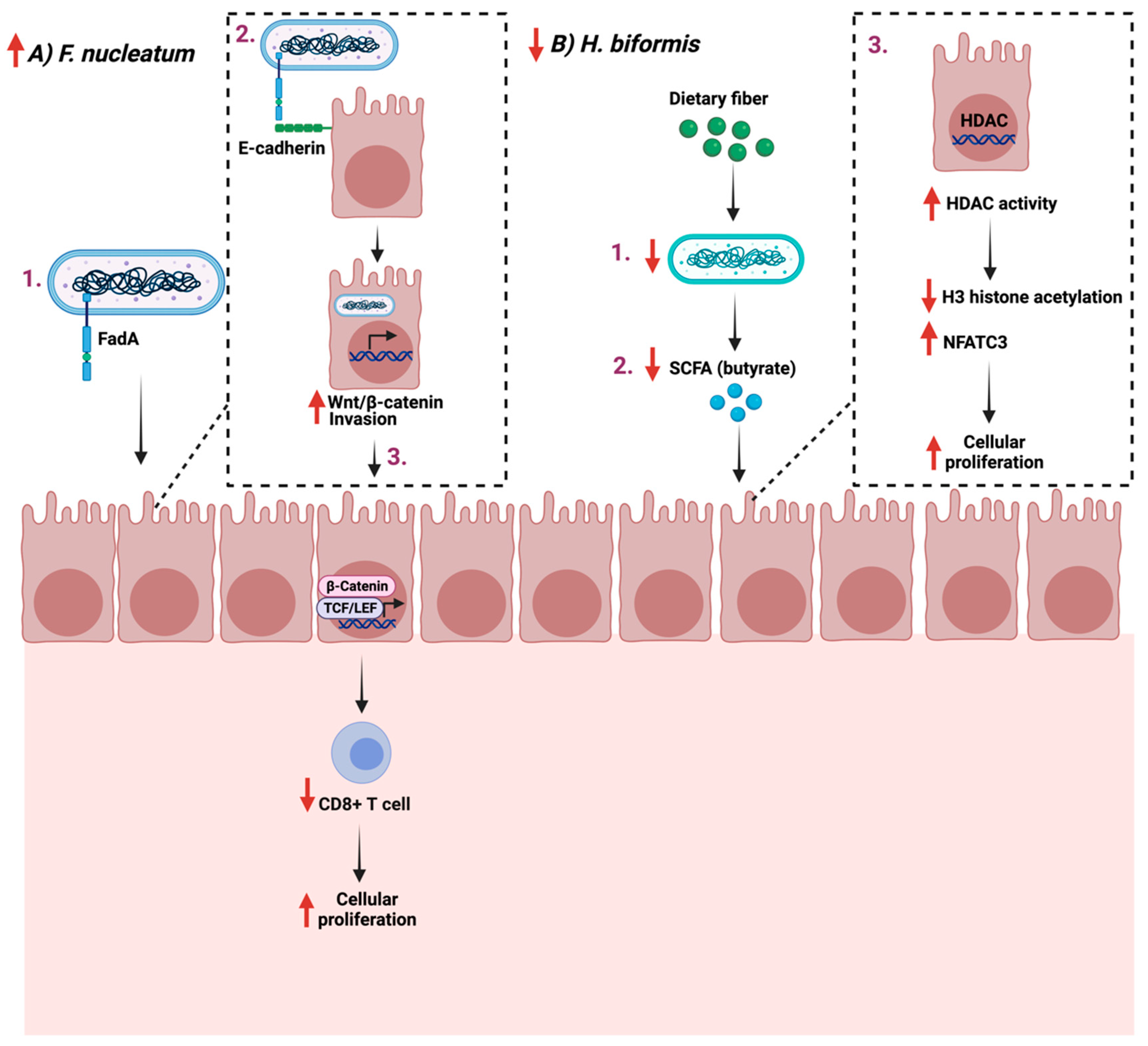
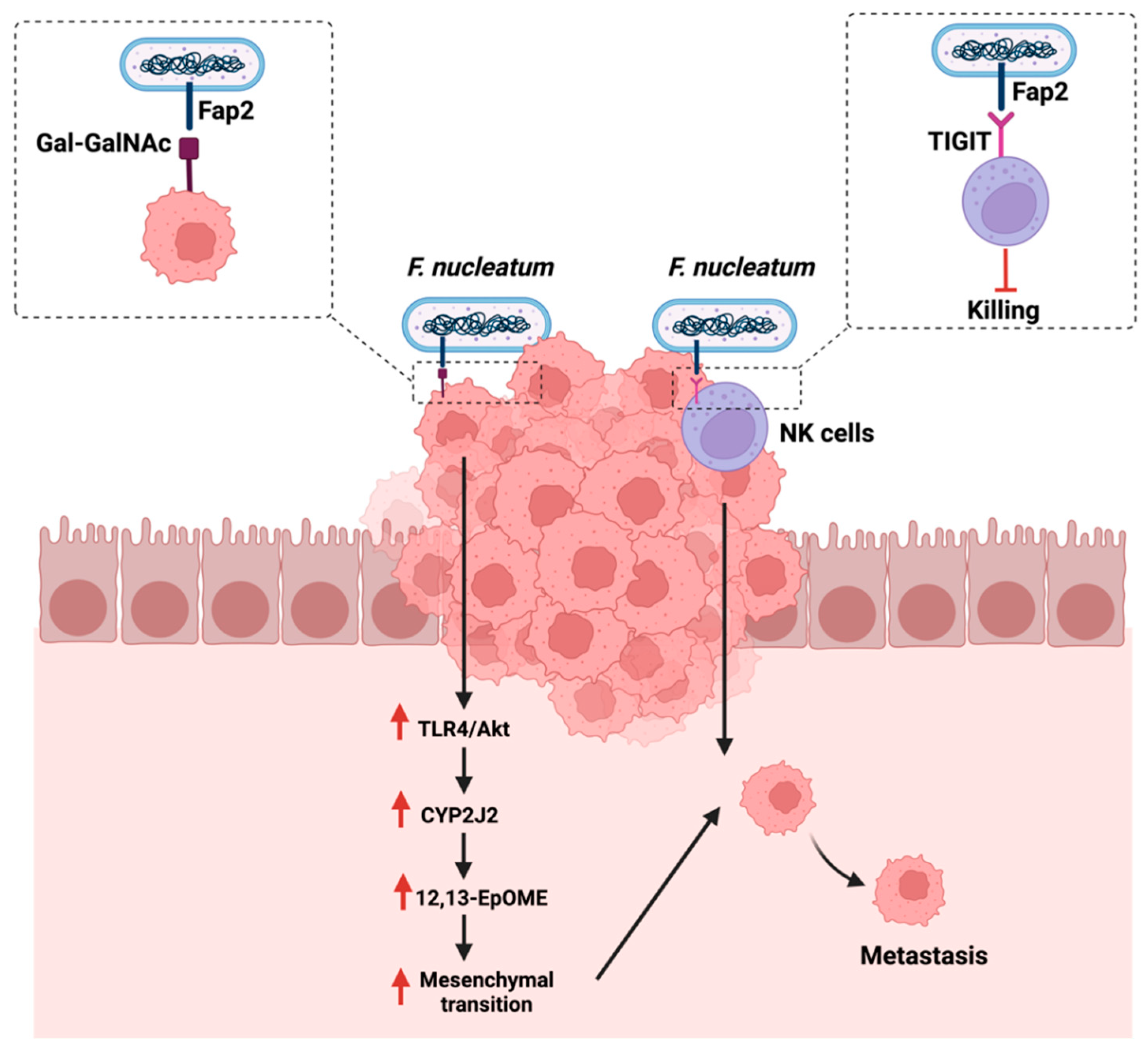
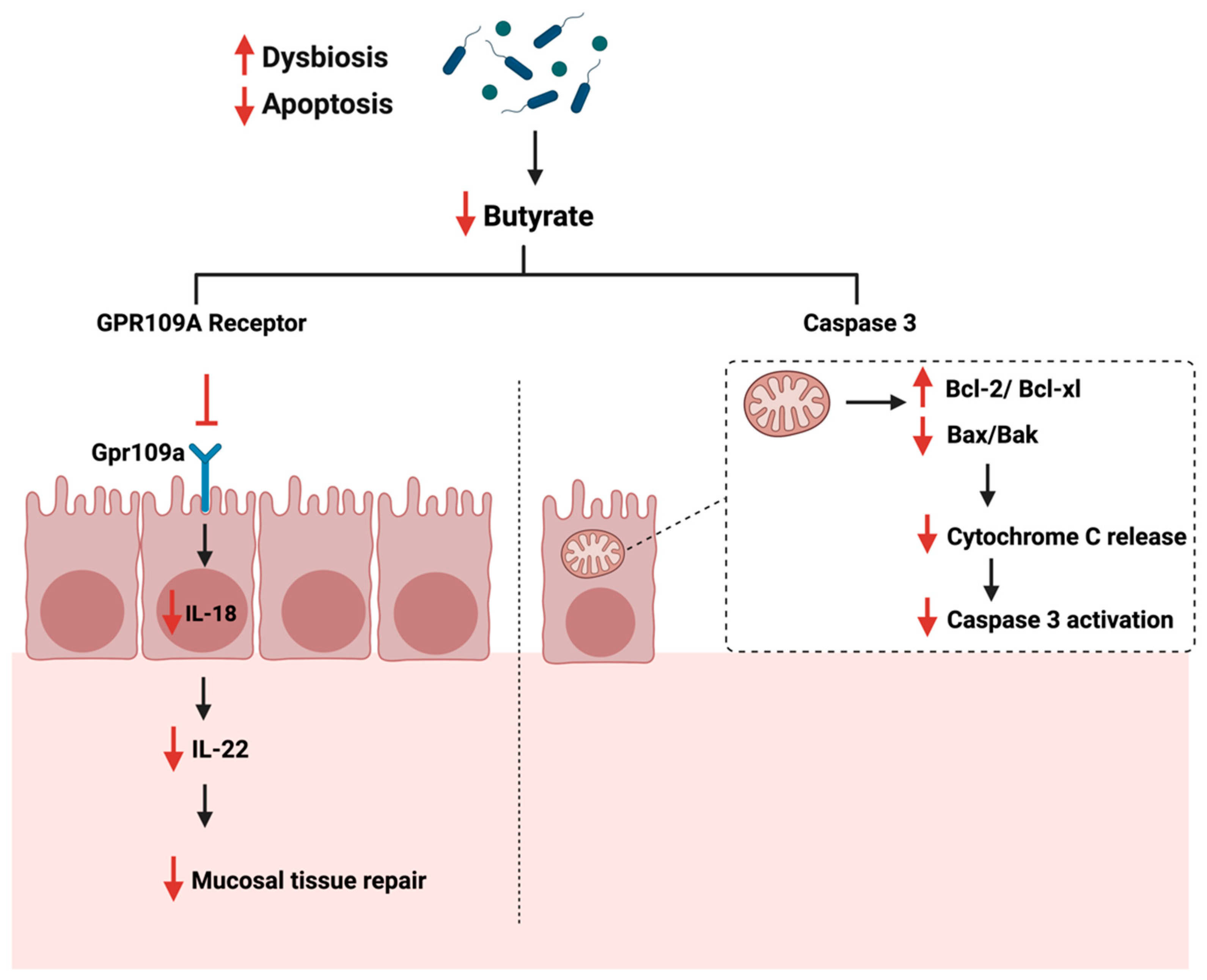
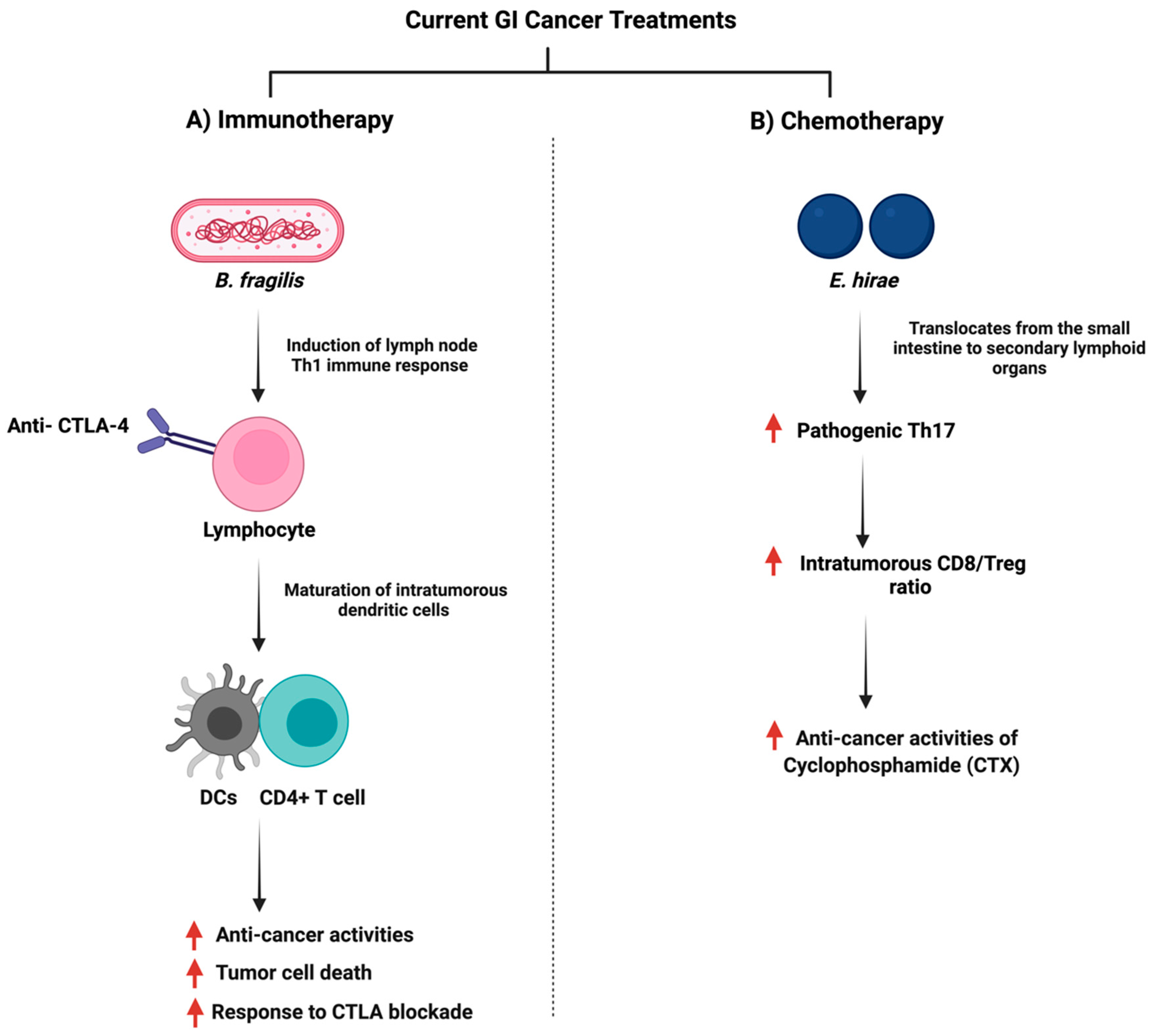
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).