
Targeting Sphingolipid Metabolism as a Therapeutic Strategy in Cancer Treatment



Abstract
:Simple Summary
Abstract
1. Introduction
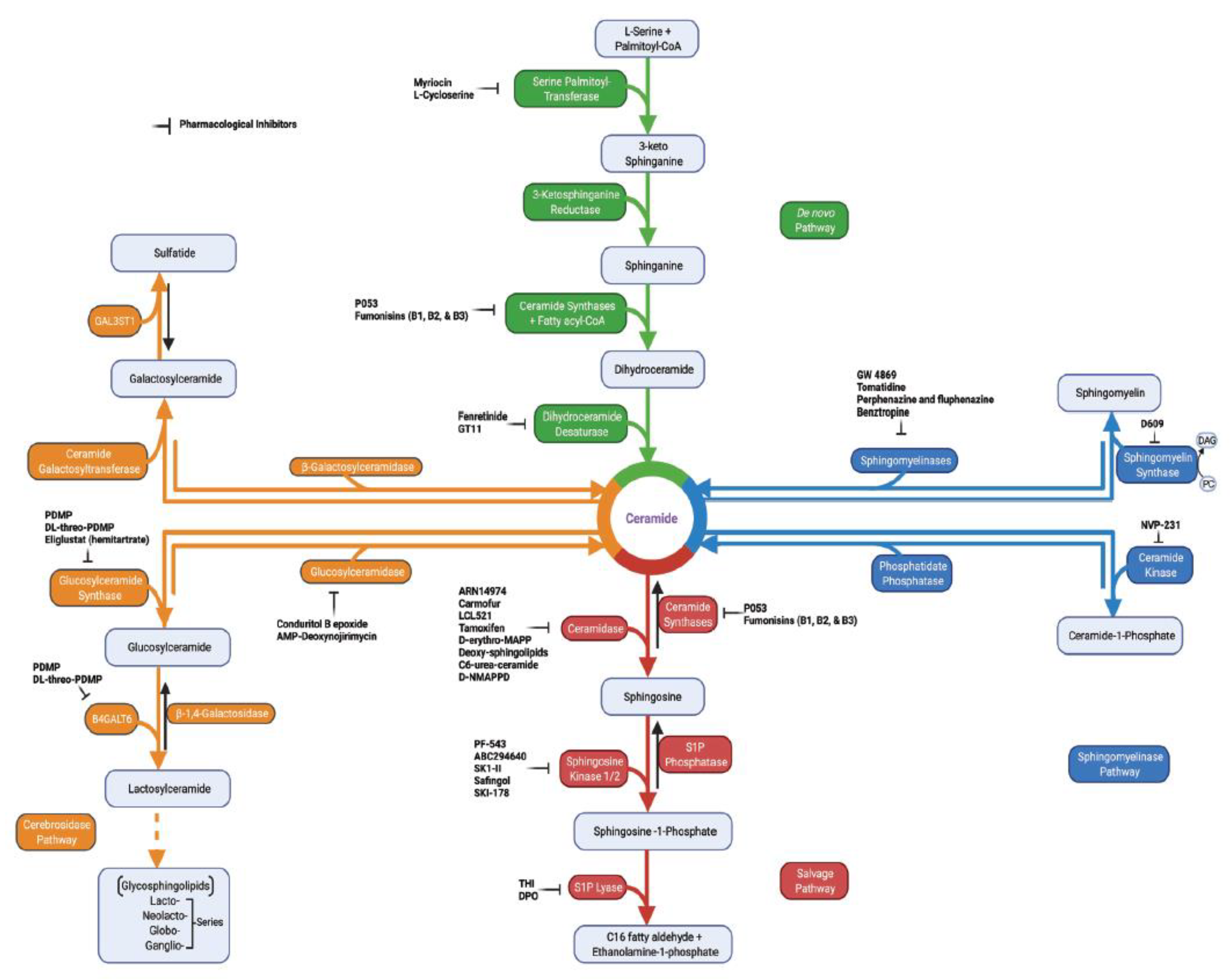
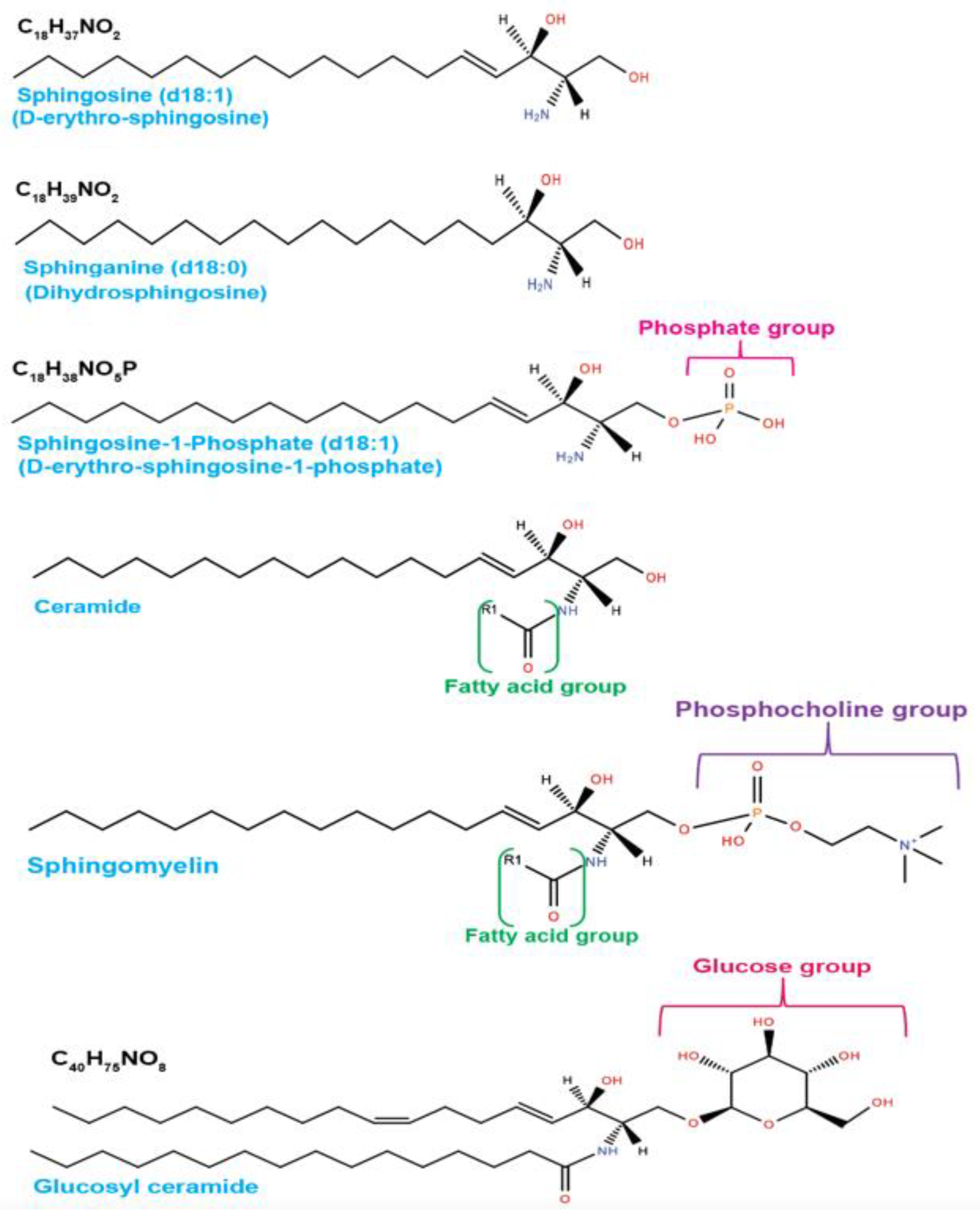
2. Sphingolipid Metabolism in Tumor Pathogenesis
3. Key Sphingolipid Enzymes and Their Roles in Cancer Progression
3.1. Sphingosine Kinases (SPHKs)
3.2. Sphingosine-1-Phosphate Lyase 1 (SGPL1)
3.3. Ceramide Kinase (CERK)
3.4. Ceramidases (CDases)
3.5. Ceramide Synthases 1–6 (CerS1–6)
3.6. Sphingomyelinases (SMases)
3.7. Sphingomyelin Synthase (SMS)
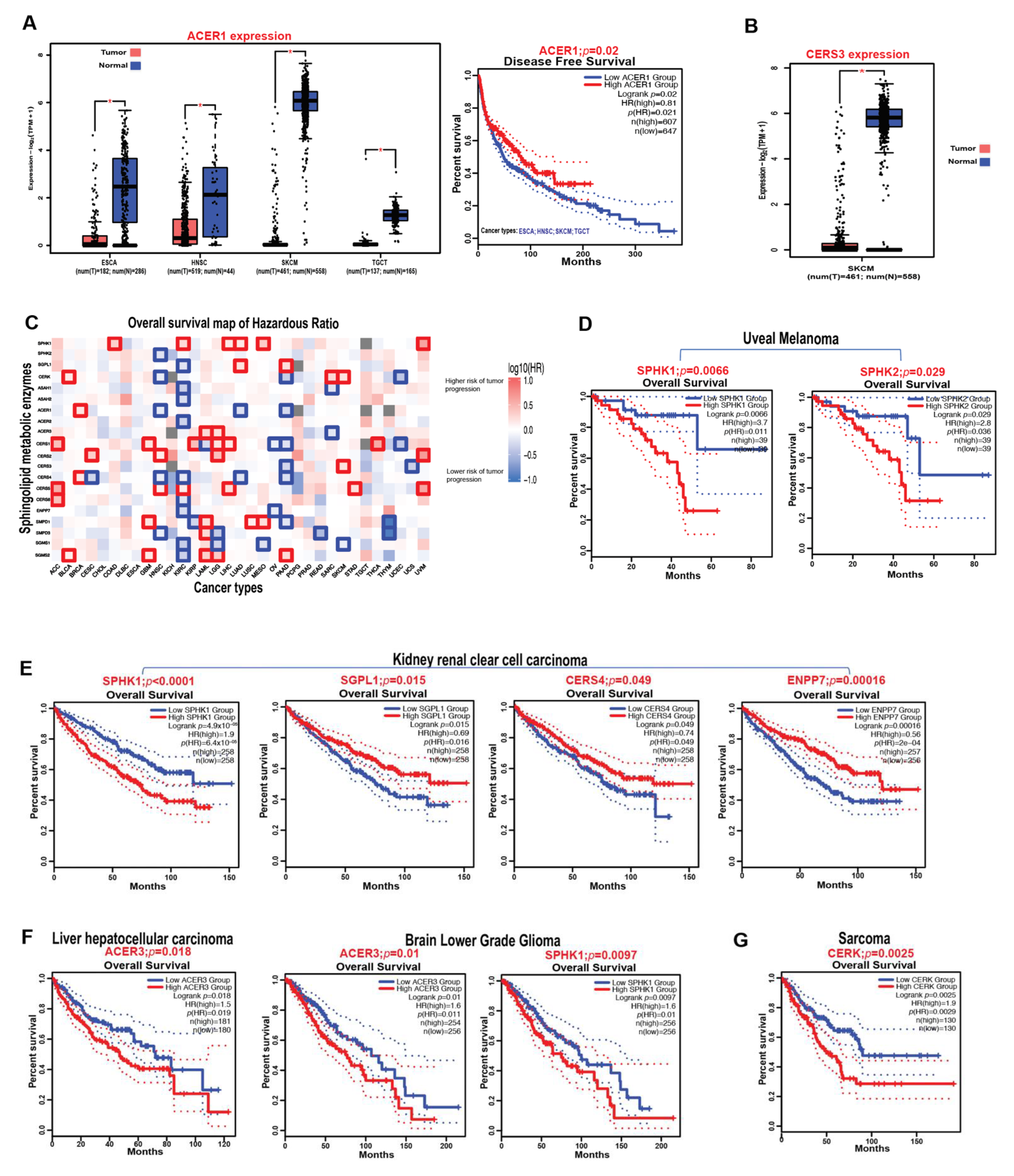
3.8. Prognostic Impact of Sphingolipid Metabolic Enzymes on the Survival of Cancer Patients
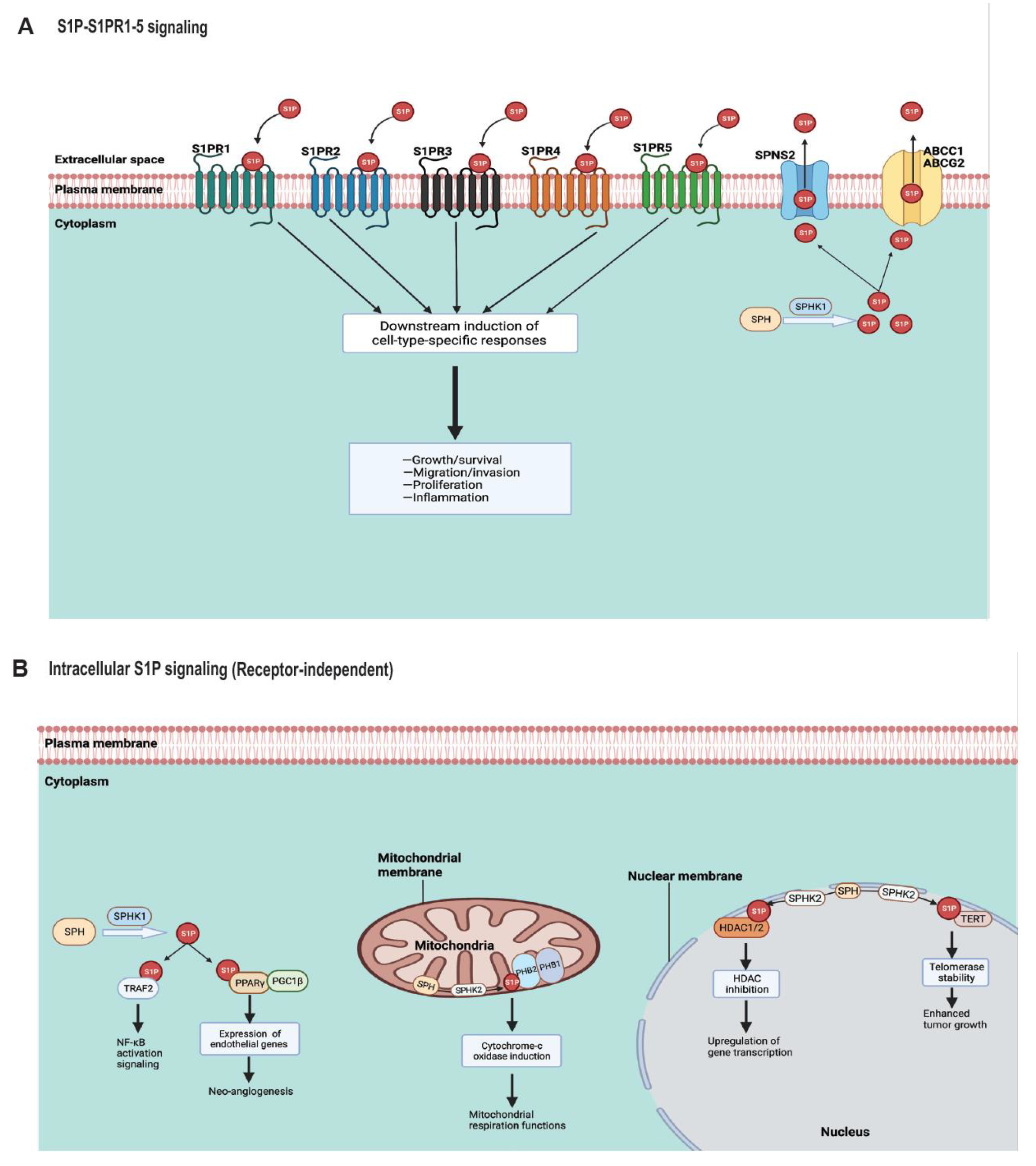
4. S1P Signaling in Cancer
4.1. S1P Transporters
4.2. S1P Receptors (S1PR1–5)
4.3. Endogenous S1P Signaling Targets
5. Sphingolipid Therapeutics in Cancer
5.1. Chemotherapy, Radiotherapy, and Immunotherapy
5.2. Anticancer Drugs Targeting Sphingolipids
5.2.1. ABC294640 (Yeliva, Opaganib)
5.2.2. Fingolimod (FTY720)
5.2.3. Ceramide Nanoliposomes (CNLs)
5.2.4. Sonepcizumab
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weigert, A.; Olesch, C.; Brüne, B. Sphingosine-1-phosphate and macrophage biology—How the sphinx tames the big eater. Front. Immunol. 2019, 10, 1706. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: An enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol. 2003, 4, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Hartung, H.-P. Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol. 2010, 33, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Presa, N.; Gomez-Larrauri, A.; Dominguez-Herrera, A.; Trueba, M.; Gomez-Munoz, A. Novel signaling aspects of ceramide 1-phosphate. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2020, 1865, 158630. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366, eaar5551. [Google Scholar] [CrossRef] [PubMed]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Liu, Y.; Gulbins, E.; Grassmé, H. The Anti-Infectious Role of Sphingosine in Microbial Diseases. Cells 2021, 10, 1105. [Google Scholar] [CrossRef]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2018, 18, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Pyne, S.; Adams, D.R.; Pyne, N.J. Sphingosine 1-phosphate and sphingosine kinases in health and disease: Recent advances. Prog. Lipid Res. 2016, 62, 93–106. [Google Scholar] [CrossRef] [Green Version]
- Janneh, A.H.; Kassir, M.F.; Dwyer, C.J.; Chakraborty, P.; Pierce, J.S.; Flume, P.A.; Li, H.; Nadig, S.N.; Mehrotra, S.; Ogretmen, B. Alterations of lipid metabolism provide serologic biomarkers for the detection of asymptomatic versus symptomatic COVID-19 patients. Sci. Rep. 2021, 11, 14232. [Google Scholar] [CrossRef]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Mayo, L.; Trauger, S.A.; Blain, M.; Nadeau, M.; Patel, B.; Alvarez, J.I.; Mascanfroni, I.D.; Yeste, A.; Kivisäkk, P.; Kallas, K.; et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat. Med. 2014, 20, 1147–1156. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Li, J.; Qiu, Y.; Sun, H. 1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP) facilitates curcumin-induced melanoma cell apoptosis by enhancing ceramide accumulation, JNK activation, and inhibiting PI3K/AKT activation. Mol. Cell. Biochem. 2012, 361, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Cannavo, A.; Liccardo, D.; Komici, K.; Corbi, G.; de Lucia, C.; Femminella, G.D.; Elia, A.; Bencivenga, L.; Ferrara, N.; Koch, W.J.; et al. Sphingosine kinases and sphingosine 1-phosphate receptors: Signaling and actions in the cardiovascular system. Front. Pharmacol. 2017, 8, 556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siow, D.; Wattenberg, B. The compartmentalization and translocation of the sphingosine kinases: Mechanisms and functions in cell signaling and sphingolipid metabolism. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; de Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef]
- Chakraborty, P.; Vaena, S.G.; Thyagarajan, K.; Chatterjee, S.; Al-Khami, A.; Selvam, S.P.; Nguyen, H.; Kang, I.; Wyatt, M.W.; Baliga, U.; et al. Pro-survival lipid sphingosine-1-phosphate metabolically programs T cells to limit anti-tumor activity. Cell Rep. 2019, 28, 1879–1893. [Google Scholar] [CrossRef] [Green Version]
- Dai, L.; Wang, C.; Song, K.; Wang, W.; Di, W. Activation of SphK1 by adipocytes mediates epithelial ovarian cancer cell proliferation. J. Ovarian Res. 2021, 14, 62. [Google Scholar] [CrossRef]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.-C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-H.; Shi, W.-N.; Wu, S.-H.; Miao, R.-R.; Sun, S.-Y.; Luo, D.-D.; Wan, S.-B.; Guo, Z.-K.; Wang, W.-Y.; Yu, X.-F.; et al. SphK2 confers 5-fluorouracil resistance to colorectal cancer via upregulating H3K56ac-mediated DPD expression. Oncogene 2020, 39, 5214–5227. [Google Scholar] [CrossRef]
- Song, K.; Dai, L.; Long, X.; Wang, W.; Di, W. Follicle-stimulating hormone promotes the proliferation of epithelial ovarian cancer cells by activating sphingosine kinase. Sci. Rep. 2020, 10, 13834. [Google Scholar] [CrossRef]
- Adamus, A.; Engel, N.; Seitz, G. SGPL1 321 mutation: One main trigger for invasiveness of pediatric alveolar rhabdomyosarcoma. Cancer Gene Ther. 2020, 27, 571–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovric, S.; Goncalves, S.; Gee, H.Y.; Oskouian, B.; Srinivas, H.; Choi, W.-I.; Shril, S.; Ashraf, S.; Tan, W.; Rao, J.; et al. Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J. Clin. Investig. 2017, 127, 912–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwiebs, A.; San Juan, M.H.; Schmidt, K.G.; Wiercinska, E.; Anlauf, M.; Ottenlinger, F.; Thomas, D.; Elwakeel, E.; Weigert, A.; Farin, H.F.; et al. Cancer-induced inflammation and inflammation-induced cancer in colon: A role for S1P lyase. Oncogene 2019, 38, 4788–4803. [Google Scholar] [CrossRef]
- Faqar-Uz-Zaman, W.F.; Schmidt, K.G.; Thomas, D.; Pfeilschifter, J.M.; Radeke, H.H.; Schwiebs, A. S1P Lyase siRNA Dampens Malignancy of DLD-1 Colorectal Cancer Cells. Lipids 2021, 56, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Al-Rashed, F.; Ahmad, Z.; Snider, A.J.; Thomas, R.; Kochumon, S.; Melhem, M.; Sindhu, S.; Obeid, L.M.; Al-Mulla, F.; Hannun, Y.A.; et al. Ceramide kinase regulates TNF-α-induced immune responses in human monocytic cells. Sci. Rep. 2021, 11, 8259. [Google Scholar] [CrossRef] [PubMed]
- Bajjalieh, S.; Martin, T.; Floor, E. Synaptic vesicle ceramide kinase: A calcium-stimulated lipid kinase that co-purifies with brain synaptic vesicles. J. Biol. Chem. 1989, 264, 14354–14360. [Google Scholar] [CrossRef]
- Mitsutake, S.; Kim, T.-J.; Inagaki, Y.; Kato, M.; Yamashita, T.; Igarashi, Y. Ceramide kinase is a mediator of calcium-dependent degranulation in mast cells. J. Biol. Chem. 2004, 279, 17570–17577. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.; Xu, Y.; Wang, L.; Liao, S.; Wang, Y.; Shi, M.; Tu, Y.; Zhou, Y.; Wei, W. Ceramide kinase mediates intrinsic resistance and inferior response to chemotherapy in triple-negative breast cancer by upregulating Ras/ERK and PI3K/Akt pathways. Cancer Cell Int. 2021, 21, 42. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, S.; Erhardt, M.; Römer, I.; Pfeilschifter, J.; Zangemeister-Wittke, U.; Huwiler, A. Ceramide kinase is upregulated in metastatic breast cancer cells and contributes to migration and invasion by activation of PI 3-kinase and Akt. Int. J. Mol. Sci. 2020, 21, 1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastukhov, O.; Schwalm, S.; Zangemeister-Wittke, U.; Fabbro, D.; Bornancin, F.; Japtok, L.; Kleuser, B.; Pfeilschifter, J.; Huwiler, A. The ceramide kinase inhibitor NVP-231 inhibits breast and lung cancer cell proliferation by inducing M phase arrest and subsequent cell death. Br. J. Pharmacol. 2014, 171, 5829–5844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, A.W.; Pant, D.K.; Pan, T.-C.; Chodosh, L.A. Ceramide kinase promotes tumor cell survival and mammary tumor recurrence. Cancer Res. 2014, 74, 6352–6363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, C.; Obeid, L.M. Ceramidases: Regulators of cellular responses mediated by ceramide, sphingosine, and sphingosine-1-phosphate. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2008, 1781, 424–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govindarajah, N.; Clifford, R.; Bowden, D.; Sutton, P.; Parsons, J.; Vimalachandran, D. Sphingolipids and acid ceramidase as therapeutic targets in cancer therapy. Crit. Rev. Oncol. Hematol. 2019, 138, 104–111. [Google Scholar] [CrossRef]
- Mahdy, A.E.; Cheng, J.C.; Li, J.; Elojeimy, S.; Meacham, W.D.; Turner, L.S.; Bai, A.; Gault, C.R.; McPherson, A.S.; Garcia, N.; et al. Acid ceramidase upregulation in prostate cancer cells confers resistance to radiation: AC inhibition, a potential radiosensitizer. Mol. Ther. 2009, 17, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Leclerc, J.; Garandeau, D.; Pandiani, C.; Gaudel, C.; Bille, K.; Nottet, N.; Garcia, V.; Colosetti, P.; Pagnotta, S.; Bahadoran, P.; et al. Lysosomal acid ceramidase ASAH1 controls the transition between invasive and proliferative phenotype in melanoma cells. Oncogene 2019, 38, 1282–1295. [Google Scholar] [CrossRef]
- Clifford, R.E.; Govindarajah, N.; Bowden, D.; Sutton, P.; Glenn, M.; Darvish-Damavandi, M.; Buczacki, S.; McDermott, U.; Szulc, Z.; Ogretmen, B.; et al. Targeting Acid Ceramidase to Improve the Radiosensitivity of Rectal Cancer. Cells 2020, 9, 2693. [Google Scholar] [CrossRef]
- Lai, M.; Amato, R.; La Rocca, V.; Bilgin, M.; Freer, G.; Spezia, P.; Quaranta, P.; Piomelli, D.; Pistello, M. Acid ceramidase controls apoptosis and increases autophagy in human melanoma cells treated with doxorubicin. Sci. Rep. 2021, 11, 11221. [Google Scholar] [CrossRef]
- Cheng, J.C.; Bai, A.; Beckham, T.H.; Marrison, S.T.; Yount, C.L.; Young, K.; Lu, P.; Bartlett, A.M.; Wu, B.X.; Keane, B.J.; et al. Radiation-induced acid ceramidase confers prostate cancer resistance and tumor relapse. J. Clin. Investig. 2013, 123, 4344–4358. [Google Scholar] [CrossRef] [Green Version]
- Bedia, C.; Casas, J.; Andrieu-Abadie, N.; Fabriàs, G.; Levade, T. Acid ceramidase expression modulates the sensitivity of A375 melanoma cells to dacarbazine. J. Biol. Chem. 2011, 286, 28200–28209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coant, N.; Hannun, Y.A. Neutral ceramidase: Advances in mechanisms, cell regulation, and roles in cancer. Adv. Biol. Regul. 2019, 71, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Coant, N.; García-Barros, M.; Zhang, Q.; Obeid, L.M.; Hannun, Y.A. AKT as a key target for growth promoting functions of neutral ceramidase in colon cancer cells. Oncogene 2018, 37, 3852–3863. [Google Scholar] [CrossRef]
- García-Barros, M.; Coant, N.; Kawamori, T.; Wada, M.; Snider, A.J.; Truman, J.P.; Wu, B.X.; Furuya, H.; Clarke, C.J.; Bialkowska, A.B.; et al. Role of neutral ceramidase in colon cancer. FASEB J. 2016, 30, 4159–4171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, R.; Boasiako, P.A.; Mao, C. Alkaline ceramidase family: The first two decades. Cell. Signal. 2021, 78, 109860. [Google Scholar] [CrossRef]
- Liakath-Ali, K.; Vancollie, V.E.; Lelliott, C.J.; Speak, A.O.; Lafont, D.; Protheroe, H.J.; Ingvorsen, C.; Galli, A.; Green, A.; Gleeson, D.; et al. Alkaline ceramidase 1 is essential for mammalian skin homeostasis and regulating whole-body energy expenditure. J. Pathol. 2016, 239, 374–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Xu, R.; Hu, W.; Jin, J.; Crellin, H.A.; Bielawski, J.; Szulc, Z.M.; Thiers, B.H.; Obeid, L.M.; Mao, C. Upregulation of the human alkaline ceramidase 1 and acid ceramidase mediates calcium-induced differentiation of epidermal keratinocytes. J. Investig. Dermatol. 2008, 128, 389–397. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.X.; Fukunaga-Kalabis, M.; Herlyn, M. Crosstalk in skin: Melanocytes, keratinocytes, stem cells, and melanoma. J. Cell Commun. Signal. 2016, 10, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Jin, J.; Hu, W.; Sun, W.; Bielawski, J.; Szulc, Z.; Taha, T.; Obeid, L.M.; Mao, C. Golgi alkaline ceramidase regulates cell proliferation and survival by controlling levels of sphingosine and S1P. FASEB J. 2006, 20, 1813–1825. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Jin, J.; Xu, R.; Hu, W.; Szulc, Z.M.; Bielawski, J.; Obeid, L.M.; Mao, C. Substrate specificity, membrane topology, and activity regulation of human alkaline ceramidase 2 (ACER2). J. Biol. Chem. 2010, 285, 8995–9007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Xiao, J.; Dong, M.; Qiu, Z.; Jin, J. Human alkaline ceramidase 2 promotes the growth, invasion, and migration of hepatocellular carcinoma cells via sphingomyelin phosphodiesterase acid-like 3B. Cancer Sci. 2020, 111, 2259. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wang, K.; Mileva, I.; Hannun, Y.A.; Obeid, L.M.; Mao, C. Alkaline ceramidase 2 and its bioactive product sphingosine are novel regulators of the DNA damage response. Oncotarget 2016, 7, 18440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, C.; Jin, Y.; He, Q.; Liu, Z.; Ai, Q.; Lei, Y.; Li, Y.; Song, F.; Bu, Y.; et al. Alkaline ceramidase 2 is a novel direct target of p53 and induces autophagy and apoptosis through ROS generation. Sci. Rep. 2017, 7, 44573. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Xu, R.; Sun, W.; Szulc, Z.M.; Bielawski, J.; Obeid, L.M.; Mao, C. Alkaline ceramidase 3 (ACER3) hydrolyzes unsaturated long-chain ceramides, and its down-regulation inhibits both cell proliferation and apoptosis. J. Biol. Chem. 2010, 285, 7964–7976. [Google Scholar] [CrossRef] [Green Version]
- Vasiliauskaité-Brooks, I.; Healey, R.D.; Rochaix, P.; Saint-Paul, J.; Sounier, R.; Grison, C.; Waltrich-Augusto, T.; Fortier, M.; Hoh, F.; Saied, E.M.; et al. Structure of a human intramembrane ceramidase explains enzymatic dysfunction found in leukodystrophy. Nat. Commun. 2018, 9, 5437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Xu, M.; Gao, J.; Li, M. Alkaline ceramidase 3 promotes growth of hepatocellular carcinoma cells via regulating S1P/S1PR2/PI3K/AKT signaling. Pathol.-Res. Pract. 2018, 214, 1381–1387. [Google Scholar] [CrossRef]
- Chen, C.; Yin, Y.; Li, C.; Chen, J.; Xie, J.; Lu, Z.; Li, M.; Wang, Y.; Zhang, C.C. ACER3 supports development of acute myeloid leukemia. Biochem. Biophys. Res. Commun. 2016, 478, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Xu, R.; Snider, A.; Schrandt, J.; Li, Y.; Bialkowska, A.; Li, M.; Zhou, J.; Hannun, Y.; Obeid, L.; et al. Alkaline ceramidase 3 deficiency aggravates colitis and colitis-associated tumorigenesis in mice by hyperactivating the innate immune system. Cell Death Dis. 2016, 7, e2124. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Many ceramides. J. Biol. Chem. 2011, 286, 27855–27862. [Google Scholar] [CrossRef] [Green Version]
- Pewzner-Jung, Y.; Ben-Dor, S.; Futerman, A.H. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: Insights into the regulation of ceramide synthesis. J. Biol. Chem. 2006, 281, 25001–25005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senkal, C.E.; Salama, M.F.; Snider, A.J.; Allopenna, J.J.; Rana, N.A.; Koller, A.; Hannun, Y.A.; Obeid, L.M. Ceramide is metabolized to acylceramide and stored in lipid droplets. Cell Metab. 2017, 25, 686–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sentelle, R.D.; Senkal, C.E.; Jiang, W.; Ponnusamy, S.; Gencer, S.; Selvam, S.P.; Ramshesh, V.K.; Peterson, Y.K.; Lemasters, J.J.; Szulc, Z.M.; et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat. Chem. Biol. 2012, 8, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Obeid, L.M.; Linardic, C.M.; Karolak, L.A.; Hannun, Y.A. Programmed cell death induced by ceramide. Science 1993, 259, 1769–1771. [Google Scholar] [CrossRef]
- Bose, R.; Verheij, M.; Haimovitz-Friedman, A.; Scotto, K.; Fuks, Z.; Kolesnick, R. Ceramide synthase mediates daunorubicin-induced apoptosis: An alternative mechanism for generating death signals. Cell 1995, 82, 405–414. [Google Scholar] [CrossRef] [Green Version]
- Mullen, T.D.; Hannun, Y.A.; Obeid, L.M. Ceramide synthases at the centre of sphingolipid metabolism and biology. Biochem. J. 2012, 441, 789–802. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, K.; Riebeling, C.; Bodennec, J.; Riezman, H.; Allegood, J.C.; Sullards, M.C.; Merrill, A.H.; Futerman, A.H. Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast Longevity Assurance Gene 1 (LAG1), regulatesN-Stearoyl-sphinganine (C18-(Dihydro) ceramide) synthesis in a fumonisin B1-independent manner in mammalian cells. J. Biol. Chem. 2002, 277, 35642–35649. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.; Futerman, A.H. Mammalian ceramide synthases. IUBMB Life 2010, 62, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wu, C.; Chen, Y.; Guo, Y.; Qiu, L.; Liu, Z.; Sun, H.; Chen, S.; An, Z.; Zhang, Z.; et al. Downregulation of ceramide synthase 1 promotes oral cancer through endoplasmic reticulum stress. Int. J. Oral Sci. 2021, 13, 10. [Google Scholar] [CrossRef]
- Meyers-Needham, M.; Ponnusamy, S.; Gencer, S.; Jiang, W.; Thomas, R.J.; Senkal, C.E.; Ogretmen, B. Concerted functions of HDAC1 and microRNA-574-5p repress alternatively spliced ceramide synthase 1 expression in human cancer cells. EMBO Mol. Med. 2012, 4, 78–92. [Google Scholar] [CrossRef]
- Senkal, C.E.; Ponnusamy, S.; Bielawski, J.; Hannun, Y.A.; Ogretmen, B. Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J. 2010, 24, 296–308. [Google Scholar] [CrossRef] [Green Version]
- Thomas, R.J.; Oleinik, N.; Panneer Selvam, S.; Vaena, S.G.; Dany, M.; Nganga, R.N.; Depalma, R.; Baron, K.D.; Kim, J.; Szulc, Z.M.; et al. HPV/E7 induces chemotherapy-mediated tumor suppression by ceramide-dependent mitophagy. EMBO Mol. Med. 2017, 9, 1030–1051. [Google Scholar] [CrossRef] [PubMed]
- Koybasi, S.; Senkal, C.E.; Sundararaj, K.; Spassieva, S.; Bielawski, J.; Osta, W.; Day, T.A.; Jiang, J.C.; Jazwinski, S.M.; Hannun, Y.A.; et al. Defects in cell growth regulation by C18: 0-ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J. Biol. Chem. 2004, 279, 44311–44319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dany, M.; Gencer, S.; Nganga, R.; Thomas, R.J.; Oleinik, N.; Baron, K.D.; Szulc, Z.M.; Ruvolo, P.; Kornblau, S.; Andreeff, M.; et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood J. Am. Soc. Hematol. 2016, 128, 1944–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Sakamoto, W.; Canals, D.; Ishibashi, M.; Matsuda, M.; Nishida, K.; Toyoshima, M.; Shigeta, S.; Taniguchi, M.; Senkal, C.E.; et al. Ceramide synthase 2-C24: 1-ceramide axis limits the metastatic potential of ovarian cancer cells. FASEB J. 2021, 35, e21287. [Google Scholar] [PubMed]
- Pani, T.; Rajput, K.; Kar, A.; Sharma, H.; Basak, R.; Medatwal, N.; Saha, S.; Dev, G.; Kumar, S.; Gupta, S.; et al. Alternative splicing of ceramide synthase 2 alters levels of specific ceramides and modulates cancer cell proliferation and migration in Luminal B breast cancer subtype. Cell Death Dis. 2021, 12, 171. [Google Scholar] [CrossRef]
- Mesicek, J.; Lee, H.; Feldman, T.; Jiang, X.; Skobeleva, A.; Berdyshev, E.V.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Ceramide synthases 2, 5, and 6 confer distinct roles in radiation-induced apoptosis in HeLa cells. Cell. Signal. 2010, 22, 1300–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennemann, R.; Rabionet, M.; Gorgas, K.; Epstein, S.; Dalpke, A.; Rothermel, U.; Bayerle, A.; van der Hoeven, F.; Imgrund, S.; Kirsch, J.; et al. Loss of ceramide synthase 3 causes lethal skin barrier disruption. Hum. Mol. Genet. 2012, 21, 586–608. [Google Scholar] [CrossRef] [Green Version]
- Gencer, S.; Oleinik, N.; Kim, J.; Panneer Selvam, S.; De Palma, R.; Dany, M.; Nganga, R.; Thomas, R.J.; Senkal, C.E.; Howe, P.H.; et al. TGF-β receptor I/II trafficking and signaling at primary cilia are inhibited by ceramide to attenuate cell migration and tumor metastasis. Sci. Signal. 2017, 10, eaam7464. [Google Scholar] [CrossRef] [Green Version]
- Hammerschmidt, P.; Ostkotte, D.; Nolte, H.; Gerl, M.J.; Jais, A.; Brunner, H.L.; Sprenger, H.-G.; Awazawa, M.; Nicholls, H.T.; Turpin-Nolan, S.M.; et al. CerS6-derived sphingolipids interact with Mff and promote mitochondrial fragmentation in obesity. Cell 2019, 177, 1536–1552. [Google Scholar] [CrossRef]
- Vaena, S.; Chakraborty, P.; Lee, H.G.; Janneh, A.H.; Kassir, M.F.; Beeson, G.; Hedley, Z.; Yalcinkaya, A.; Sofi, M.H.; Li, H.; et al. Aging-dependent mitochondrial dysfunction mediated by ceramide signaling inhibits antitumor T cell response. Cell Rep. 2021, 35, 109076. [Google Scholar] [CrossRef] [PubMed]
- El-Hindi, K.; Brachtendorf, S.; Hartel, J.C.; Oertel, S.; Birod, K.; Trautmann, S.; Thomas, D.; Ulshöfer, T.; Weigert, A.; Utermöhlen, O.; et al. Ceramide synthase 5 deficiency aggravates dextran sodium sulfate-induced colitis and colon carcinogenesis and impairs T-cell activation. Cancers 2020, 12, 1753. [Google Scholar] [CrossRef] [PubMed]
- Qi, D.; Song, X.; Xue, C.; Yao, W.; Shen, P.; Yu, H.; Zhang, Z. AKT1/FOXP3 axis-mediated expression of CerS6 promotes p53 mutant pancreatic tumorigenesis. Cancer Lett. 2021, 522, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Senkal, C.E.; Ponnusamy, S.; Manevich, Y.; Meyers-Needham, M.; Saddoughi, S.A.; Mukhopadyay, A.; Dent, P.; Bielawski, J.; Ogretmen, B. Alteration of ceramide synthase 6/C16-ceramide induces activating transcription factor 6-mediated endoplasmic reticulum (ER) stress and apoptosis via perturbation of cellular Ca2+ and ER/Golgi membrane network. J. Biol. Chem. 2011, 286, 42446–42458. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; White-Gilbertson, S.; Beeson, G.; Beeson, C.; Ogretmen, B.; Norris, J.; Voelkel-Johnson, C. Ceramide Synthase 6 Maximizes p53 Function to Prevent Progeny Formation from Polyploid Giant Cancer Cells. Cancers 2021, 13, 2212. [Google Scholar] [CrossRef] [PubMed]
- Pavoine, C.; Pecker, F. Sphingomyelinases: Their regulation and roles in cardiovascular pathophysiology. Cardiovasc. Res. 2009, 82, 175–183. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, P.; Xu, S.-C.; Yang, L.; Voss, U.; Ekblad, E.; Wu, Y.; Min, Y.; Hertervig, E.; Nilsson, Å.; et al. Enhanced colonic tumorigenesis in alkaline sphingomyelinase (NPP7) knockout mice. Mol. Cancer Ther. 2015, 14, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Mauhin, W.; Levade, T.; Vanier, M.T.; Froissart, R.; Lidove, O. Prevalence of Cancer in Acid Sphingomyelinase Deficiency. J. Clin. Med. 2021, 10, 5029. [Google Scholar] [CrossRef]
- Romiti, E.; Vasta, V.; Meacci, E.; Farnararo, M.; Linke, T.; Ferlinz, K.; Sandhoff, K.; Bruni, P. Characterization of sphingomyelinase activity released by thrombin-stimulated platelets. Mol. Cell. Biochem. 2000, 205, 75–81. [Google Scholar] [CrossRef]
- Carpinteiro, A.; Becker, K.A.; Japtok, L.; Hessler, G.; Keitsch, S.; Požgajovà, M.; Schmid, K.W.; Adams, C.; Müller, S.; Kleuser, B.; et al. Regulation of hematogenous tumor metastasis by acid sphingomyelinase. EMBO Mol. Med. 2015, 7, 714–734. [Google Scholar] [CrossRef] [PubMed]
- Hannun, Y.A.; Newcomb, B. A new twist to the emerging functions of ceramides in cancer: Novel role for platelet acid sphingomyelinase in cancer metastasis. EMBO Mol. Med. 2015, 7, 692–694. [Google Scholar] [CrossRef] [PubMed]
- Montfort, A.; Bertrand, F.; Rochotte, J.; Gilhodes, J.; Filleron, T.; Milhès, J.; Dufau, C.; Imbert, C.; Riond, J.; Tosolini, M.; et al. Neutral Sphingomyelinase 2 Heightens Anti-Melanoma Immune Responses and Anti–PD-1 Therapy Efficacy. Cancer Immunol. Res. 2021, 9, 568–582. [Google Scholar] [CrossRef]
- Jabalee, J.; Towle, R.; Lawson, J.; Dickman, C.; Garnis, C. Sphingomyelin phosphodiesterase 3 methylation and silencing in oral squamous cell carcinoma results in increased migration and invasion and altered stress response. Oncotarget 2020, 11, 523. [Google Scholar] [CrossRef] [PubMed]
- Huitema, K.; van den Dikkenberg, J.; Brouwers, J.F.; Holthuis, J.C. Identification of a family of animal sphingomyelin synthases. EMBO J. 2004, 23, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.-C.; Li, Z.; Yazdanyar, A. Sphingolipids and HDL Metabolism. In The HDL Handbook; Elsevier: Amsterdam, The Netherlands, 2014; pp. 133–158. [Google Scholar]
- Zheng, K.; Chen, Z.; Feng, H.; Chen, Y.; Zhang, C.; Yu, J.; Luo, Y.; Zhao, L.; Jiang, X.; Shi, F. Sphingomyelin synthase 2 promotes an aggressive breast cancer phenotype by disrupting the homoeostasis of ceramide and sphingomyelin. Cell Death Dis. 2019, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- Fernández-García, P.; Rosselló, C.A.; Rodríguez-Lorca, R.; Beteta-Göbel, R.; Fernández-Díaz, J.; Lladó, V.; Busquets, X.; Escribá, P.V. The opposing contribution of SMS1 and SMS2 to glioma progression and their value in the therapeutic response to 2OHOA. Cancers 2019, 11, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, S.; Pan, Z.; Liu, S.; Yuan, S.; Yan, N. Sphingomyelin synthase 2 overexpression promotes cisplatin-induced apoptosis of HepG2 cells. Oncol. Lett. 2018, 15, 483–488. [Google Scholar] [CrossRef] [Green Version]
- Jing, F.; Jing, C.; Dai, X.; Zhou, G.; Di, S.; Bi, X.; Dai, T.; Qin, T.; Hong, L. Sphingomyelin synthase 2 but not sphingomyelin synthase 1 is upregulated in ovarian cancer and involved in migration, growth and survival via different mechanisms. Am. J. Transl. Res. 2021, 13, 4412. [Google Scholar] [PubMed]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Wang, P.; Yuan, Y.; Lin, W.; Zhong, H.; Xu, K.; Qi, X. Roles of sphingosine-1-phosphate signaling in cancer. Cancer Cell Int. 2019, 19, 295. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Yamada, A.; Katsuta, E.; Aoyagi, T.; Huang, W.-C.; Terracina, K.P.; Hait, N.C.; Allegood, J.C.; Tsuchida, J.; Yuza, K.; et al. Targeting the SphK1/S1P/S1PR1 axis that links obesity, chronic inflammation, and breast cancer metastasis. Cancer Res. 2018, 78, 1713–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takabe, K.; Spiegel, S. Export of sphingosine-1-phosphate and cancer progression. J. Lipid Res. 2014, 55, 1839–1846. [Google Scholar] [CrossRef] [Green Version]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strub, G.M.; Paillard, M.; Liang, J.; Gomez, L.; Allegood, J.C.; Hait, N.C.; Maceyka, M.; Price, M.M.; Chen, Q.; Simpson, D.C.; et al. Sphingosine-1-phosphate produced by sphingosine kinase 2 in mitochondria interacts with prohibitin 2 to regulate complex IV assembly and respiration. FASEB J. 2011, 25, 600–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panneer Selvam, S.; De Palma, R.M.; Oaks, J.J.; Oleinik, N.; Peterson, Y.K.; Stahelin, R.V.; Skordalakes, E.; Ponnusamy, S.; Garrett-Mayer, E.; Smith, C.D.; et al. Binding of the sphingolipid S1P to hTERT stabilizes telomerase at the nuclear periphery by allosterically mimicking protein phosphorylation. Sci. Signal. 2015, 8, ra58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Hou, J.; Cao, Y.; Shan, J.-J.; Zhao, J. Spinster homolog 2 in cancers, its functions and mechanisms. Cell. Signal. 2021, 77, 109821. [Google Scholar] [CrossRef]
- Kawahara, A.; Nishi, T.; Hisano, Y.; Fukui, H.; Yamaguchi, A.; Mochizuki, N. The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science 2009, 323, 524–527. [Google Scholar] [CrossRef] [Green Version]
- Osborne, N.; Brand-Arzamendi, K.; Ober, E.A.; Jin, S.-W.; Verkade, H.; Holtzman, N.G.; Yelon, D.; Stainier, D.Y. The spinster homolog, two of hearts, is required for sphingosine 1-phosphate signaling in zebrafish. Curr. Biol. 2008, 18, 1882–1888. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zheng, Y.; Wang, F.; Zhong, J.; Zhao, T.; Xie, Q.; Zhu, T.; Ma, F.; Tang, Q.; Zhou, B.; et al. Mfsd2a and Spns2 are essential for sphingosine-1-phosphate transport in the formation and maintenance of the blood-brain barrier. Sci. Adv. 2020, 6, eaay8627. [Google Scholar] [CrossRef]
- Spiegel, S.; Maczis, M.A.; Maceyka, M.; Milstien, S. New insights into functions of the sphingosine-1-phosphate transporter SPNS2. J. Lipid Res. 2019, 60, 484–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, T.M.; Ishizu, A.-N.; Foo, J.C.; Toh, X.R.; Zhang, F.; Whee, D.M.; Torta, F.; Cazenave-Gassiot, A.; Matsumura, T.; Kim, S.; et al. Mfsd2b is essential for the sphingosine-1-phosphate export in erythrocytes and platelets. Nature 2017, 550, 524–528. [Google Scholar] [CrossRef]
- Fukuhara, S.; Simmons, S.; Kawamura, S.; Inoue, A.; Orba, Y.; Tokudome, T.; Sunden, Y.; Arai, Y.; Moriwaki, K.; Ishida, J.; et al. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J. Clin. Investig. 2012, 122, 1416–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hisano, Y.; Kobayashi, N.; Yamaguchi, A.; Nishi, T. Mouse SPNS2 functions as a sphingosine-1-phosphate transporter in vascular endothelial cells. PLoS ONE 2012, 7, e38941. [Google Scholar]
- Chandrakanthan, M.; Nguyen, T.Q.; Hasan, Z.; Muralidharan, S.; Vu, T.M.; Li, A.W.L.; Le, U.T.N.; Thi Thuy Ha, H.; Baik, S.-H.; Tan, S.H.; et al. Deletion of Mfsd2b impairs thrombotic functions of platelets. Nat. Commun. 2021, 12, 2286. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.Q.; Vu, T.M.; Tukijan, F.; Muralidharan, S.; Foo, J.C.; Chin, J.F.L.; Hasan, Z.; Torta, F.; Nguyen, L.N. Erythrocytes efficiently utilize exogenous sphingosines for S1P synthesis and export via Mfsd2b. J. Biol. Chem. 2021, 296, 100201. [Google Scholar] [CrossRef]
- van der Weyden, L.; Arends, M.J.; Campbell, A.D.; Bald, T.; Wardle-Jones, H.; Griggs, N.; Velasco-Herrera, M.D.C.; Tüting, T.; Sansom, O.J.; Karp, N.A. Genome-wide in vivo screen identifies novel host regulators of metastatic colonization. Nature 2017, 541, 233–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adada, M.M.; Canals, D.; Jeong, N.; Kelkar, A.D.; Hernandez-Corbacho, M.; Pulkoski-Gross, M.J.; Donaldson, J.C.; Hannun, Y.A.; Obeid, L.M. Intracellular sphingosine kinase 2-derived sphingosine-1-phosphate mediates epidermal growth factor-induced ezrin-radixin-moesin phosphorylation and cancer cell invasion. FASEB J. 2015, 29, 4654–4669. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.; Yi, Q.; Yan, Y.; Chao, F.; Li, M. SPNS2 downregulation induces EMT and promotes colorectal cancer metastasis via activating AKT signaling pathway. Front. Oncol. 2021, 11, 1790. [Google Scholar] [CrossRef]
- Cole, S.; Bhardwaj, G.; Gerlach, J.; Mackie, J.; Grant, C.; Almquist, K.; Stewart, A.; Kurz, E.; Duncan, A.; Deeley, R.G. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science 1992, 258, 1650–1654. [Google Scholar] [CrossRef]
- Stride, B.D.; Grant, C.E.; Loe, D.W.; Hipfner, D.R.; Cole, S.P.; Deeley, R.G. Pharmacological characterization of the murine and human orthologs of multidrug-resistance protein in transfected human embryonic kidney cells. Mol. Pharmacol. 1997, 52, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Takabe, K.; Kim, R.H.; Allegood, J.C.; Mitra, P.; Ramachandran, S.; Nagahashi, M.; Harikumar, K.B.; Hait, N.C.; Milstien, S.; Spiegel, S. Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J. Biol. Chem. 2010, 285, 10477–10486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Jeldres, T.; Alvarez-Lobos, M.; Rivera-Nieves, J. Targeting Sphingosine-1-Phosphate Signaling in Immune-Mediated Diseases: Beyond Multiple Sclerosis. Drugs 2021, 81, 985–1002. [Google Scholar] [CrossRef] [PubMed]
- Cantalupo, A.; Di Lorenzo, A. S1P signaling and de novo biosynthesis in blood pressure homeostasis. J. Pharmacol. Exp. Ther. 2016, 358, 359–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkerson, B.A.; Grass, G.D.; Wing, S.B.; Argraves, W.S.; Argraves, K.M. Sphingosine 1-phosphate (S1P) carrier-dependent regulation of endothelial barrier: High density lipoprotein (HDL)-S1P prolongs endothelial barrier enhancement as compared with albumin-S1P via effects on levels, trafficking, and signaling of S1P1. J. Biol. Chem. 2012, 287, 44645–44653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, B.-S.; Yang, D.; Swendeman, S.L.; Christoffersen, C.; Nielsen, L.B.; Friedman, S.L.; Powell, C.A.; Hla, T.; Cao, Z. Aging suppresses sphingosine-1-phosphate chaperone ApoM in circulation resulting in maladaptive organ repair. Dev. Cell 2020, 53, 677–690. [Google Scholar] [CrossRef]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Deng, J.; Wang, L.; Lee, H.; Armstrong, B.; Scuto, A.; Kowolik, C.; Weiss, L.M.; Forman, S.; Yu, H. S1PR1 is an effective target to block STAT3 signaling in activated B cell–like diffuse large B-cell lymphoma. Blood J. Am. Soc. Hematol. 2012, 120, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, S.; Selvam, S.P.; Mehrotra, S.; Kawamori, T.; Snider, A.J.; Obeid, L.M.; Shao, Y.; Sabbadini, R.; Ogretmen, B. Communication between host organism and cancer cells is transduced by systemic sphingosine kinase 1/sphingosine 1-phosphate signalling to regulate tumour metastasis. EMBO Mol. Med. 2012, 4, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Stelling, A.; Hashwah, H.; Bertram, K.; Manz, M.G.; Tzankov, A.; Müller, A. The tumor suppressive TGF-β/SMAD1/S1PR2 signaling axis is recurrently inactivated in diffuse large B-cell lymphoma. Blood J. Am. Soc. Hematol. 2018, 131, 2235–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flori, M.; Schmid, C.A.; Sumrall, E.T.; Tzankov, A.; Law, C.W.; Robinson, M.D.; Müller, A. The hematopoietic oncoprotein FOXP1 promotes tumor cell survival in diffuse large B-cell lymphoma by repressing S1PR2 signaling. Blood J. Am. Soc. Hematol. 2016, 127, 1438–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartier, A.; Leigh, T.; Liu, C.H.; Hla, T. Endothelial sphingosine 1-phosphate receptors promote vascular normalization and antitumor therapy. Proc. Natl. Acad. Sci. USA 2020, 117, 3157–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, J.A.; Lewis, A.C.; Zhu, W.; Toubia, J.; Pitman, M.R.; Wallington-Beddoe, C.T.; Moretti, P.A.; Iarossi, D.; Samaraweera, S.E.; Cummings, N.; et al. Targeting sphingosine kinase 1 induces MCL1-dependent cell death in acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2017, 129, 771–782. [Google Scholar] [CrossRef] [Green Version]
- Hirata, N.; Yamada, S.; Shoda, T.; Kurihara, M.; Sekino, Y.; Kanda, Y. Sphingosine-1-phosphate promotes expansion of cancer stem cells via S1PR3 by a ligand-independent Notch activation. Nat. Commun. 2014, 5, 4806. [Google Scholar] [CrossRef]
- Wang, S.; Liang, Y.; Chang, W.; Hu, B.; Zhang, Y. Triple negative breast cancer depends on sphingosine kinase 1 (SphK1)/sphingosine-1-phosphate (S1P)/sphingosine 1-phosphate receptor 3 (S1PR3)/notch signaling for metastasis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 1912. [Google Scholar] [CrossRef]
- Shen, Y.; Zhao, S.; Wang, S.; Pan, X.; Zhang, Y.; Xu, J.; Jiang, Y.; Li, H.; Zhang, Q.; Gao, J.; et al. S1P/S1PR3 axis promotes aerobic glycolysis by YAP/c-MYC/PGAM1 axis in osteosarcoma. EBioMedicine 2019, 40, 210–223. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Liu, J.; Lee, J.-F.; Zhang, W.; Kandouz, M.; VanHecke, G.C.; Chen, S.; Ahn, Y.-H.; Lonardo, F.; Lee, M.-J. TGF-β/SMAD3 Pathway stimulates sphingosine-1 phosphate receptor 3 expression implication of sphingosine-1 phosphate receptor 3 in lung adenocarcinoma progression. J. Biol. Chem. 2016, 291, 27343–27353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesch, C.; Sirait-Fischer, E.; Berkefeld, M.; Fink, A.F.; Susen, R.M.; Ritter, B.; Michels, B.E.; Steinhilber, D.; Greten, F.R.; Savai, R.; et al. S1PR4 ablation reduces tumor growth and improves chemotherapy via CD8+ T cell expansion. J. Clin. Investig. 2020, 130, 5461–5476. [Google Scholar] [CrossRef]
- Burkard, T.; Dreis, C.; Herrero San Juan, M.; Huhn, M.; Weigert, A.; Pfeilschifter, J.M.; Radeke, H.H. Enhanced CXCR4 Expression of Human CD8Low T Lymphocytes Is Driven by S1P4. Front. Immunol. 2021, 12, 3435. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-F.; Dang, A.; Hernandez, E.; Pong, R.-C.; Chen, B.; Sonavane, R.; Raj, G.; Kapur, P.; Lin, H.-Y.; Wu, S.-R.; et al. Activation of sphingosine kinase by lipopolysaccharide promotes prostate cancer cell invasion and metastasis via SphK1/S1PR4/matriptase. Oncogene 2019, 38, 5580–5598. [Google Scholar]
- Evrard, M.; Wynne-Jones, E.; Peng, C.; Kato, Y.; Christo, S.N.; Fonseca, R.; Park, S.L.; Burn, T.N.; Osman, M.; Devi, S.; et al. Sphingosine 1-phosphate receptor 5 (S1PR5) regulates the peripheral retention of tissue-resident lymphocytes. J. Exp. Med. 2022, 219, e20210116. [Google Scholar]
- Andrieu, G.; Ledoux, A.; Branka, S.; Bocquet, M.; Gilhodes, J.; Walzer, T.; Kasahara, K.; Inagaki, M.; Sabbadini, R.A.; Cuvillier, O.; et al. Sphingosine 1-phosphate signaling through its receptor S1P5 promotes chromosome segregation and mitotic progression. Sci. Signal. 2017, 10, eaah4007. [Google Scholar] [PubMed]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.-S.; Choi, S.; Shin, B.; Yu, J.; Yu, J.; Hwang, J.-M.; Yun, H.; Chung, Y.-H.; Choi, J.-S.; Choi, Y.; et al. Tumor necrosis factor (TNF) receptor-associated factor (TRAF)-interacting protein (TRIP) negatively regulates the TRAF2 ubiquitin-dependent pathway by suppressing the TRAF2-sphingosine 1-phosphate (S1P) interaction. J. Biol. Chem. 2015, 290, 9660–9673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Lee, H.J.; Mariko, B.; Lu, Y.-C.; Dannenberg, A.J.; Haka, A.S.; Maxfield, F.R.; Camerer, E.; Proia, R.L.; Hla, T. Sphingosine kinases are not required for inflammatory responses in macrophages. J. Biol. Chem. 2013, 288, 32563–32573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etemadi, N.; Chopin, M.; Anderton, H.; Tanzer, M.C.; Rickard, J.A.; Abeysekera, W.; Hall, C.; Spall, S.K.; Wang, B.; Xiong, Y.; et al. TRAF2 regulates TNF and NF-κB signalling to suppress apoptosis and skin inflammation independently of Sphingosine kinase 1. eLife 2015, 4, e10592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parham, K.A.; Zebol, J.R.; Tooley, K.L.; Sun, W.Y.; Moldenhauer, L.M.; Cockshell, M.P.; Gliddon, B.L.; Moretti, P.A.; Tigyi, G.; Pitson, S.M.; et al. Sphingosine 1-phosphate is a ligand for peroxisome proliferator-activated receptor-γ that regulates neoangiogenesis. FASEB J. 2015, 29, 3638–3653. [Google Scholar] [PubMed]
- Pyne, N.J.; Pyne, S. Recent advances in the role of sphingosine 1-phosphate in cancer. FEBS Lett. 2020, 594, 3583–3601. [Google Scholar] [CrossRef]
- Park, M.A.; Mitchell, C.; Zhang, G.; Yacoub, A.; Allegood, J.; Häussinger, D.; Reinehr, R.; Larner, A.; Spiegel, S.; Fisher, P.B.; et al. Vorinostat and Sorafenib Increase CD95 Activation in Gastrointestinal Tumor Cells through a Ca2+-De novo Ceramide-PP2A-Reactive Oxygen Species–Dependent Signaling Pathway. Cancer Res. 2010, 70, 6313–6324. [Google Scholar] [CrossRef] [Green Version]
- Nganga, R.; Oleinik, N.; Ogretmen, B. Mechanisms of ceramide-dependent cancer cell death. Adv. Cancer Res. 2018, 140, 1–25. [Google Scholar] [PubMed]
- Mizrachi, A.; Ben-Aharon, I.; Li, H.; Bar-Joseph, H.; Bodden, C.; Hikri, E.; Popovtzer, A.; Shalgi, R.; Haimovitz-Friedman, A. Chemotherapy-induced acute vascular injury involves intracellular generation of ROS via activation of the acid sphingomyelinase pathway. Cell. Signal. 2021, 82, 109969. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Dent, P. Prior exposure of pancreatic tumors to [sorafenib + vorinostat] enhances the efficacy of an anti-PD-1 antibody. Cancer Biol. Ther. 2019, 20, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saddoughi, S.A.; Garrett-Mayer, E.; Chaudhary, U.; O’Brien, P.E.; Afrin, L.B.; Day, T.A.; Gillespie, M.B.; Sharma, A.K.; Wilhoit, C.S.; Bostick, R.; et al. Results of a phase II trial of gemcitabine plus doxorubicin in patients with recurrent head and neck cancers: Serum C18-ceramide as a novel biomarker for monitoring response. Clin. Cancer Res. 2011, 17, 6097–6105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senkal, C.E.; Ponnusamy, S.; Rossi, M.J.; Bialewski, J.; Sinha, D.; Jiang, J.C.; Jazwinski, S.M.; Hannun, Y.A.; Ogretmen, B. Role of human longevity assurance gene 1 and C18-ceramide in chemotherapy-induced cell death in human head and neck squamous cell carcinomas. Mol. Cancer Ther. 2007, 6, 712–722. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Yin, X.; Allan, R.; Lu, D.D.; Maurer, C.W.; Haimovitz-Friedman, A.; Fuks, Z.; Shaham, S.; Kolesnick, R. Ceramide biogenesis is required for radiation-induced apoptosis in the germ line of C. elegans. Science 2008, 322, 110–115. [Google Scholar] [CrossRef] [Green Version]
- Ch’ang, H.-J.; Maj, J.G.; Paris, F.; Xing, H.R.; Zhang, J.; Truman, J.-P.; Cardon-Cardo, C.; Haimovitz-Friedman, A.; Kolesnick, R.; Fuks, Z. ATM regulates target switching to escalating doses of radiation in the intestines. Nat. Med. 2005, 11, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Rotolo, J.; Stancevic, B.; Zhang, J.; Hua, G.; Fuller, J.; Yin, X.; Haimovitz-Friedman, A.; Kim, K.; Qian, M.; Cardó-Vila, M.; et al. Anti-ceramide antibody prevents the radiation gastrointestinal syndrome in mice. J. Clin. Investig. 2012, 122, 1786–1790. [Google Scholar] [CrossRef]
- Bodo, S.; Campagne, C.; Thin, T.H.; Higginson, D.S.; Vargas, H.A.; Hua, G.; Fuller, J.D.; Ackerstaff, E.; Russell, J.; Zhang, Z.; et al. Single-dose radiotherapy disables tumor cell homologous recombination via ischemia/reperfusion injury. J. Clin. Investig. 2019, 129, 786–801. [Google Scholar] [CrossRef] [Green Version]
- Yura, Y.; Masui, A.; Hamada, M. Inhibitors of Ceramide-and Sphingosine-Metabolizing Enzymes as Sensitizers in Radiotherapy and Chemotherapy for Head and Neck Squamous Cell Carcinoma. Cancers 2020, 12, 2062. [Google Scholar] [CrossRef] [PubMed]
- Priceman, S.J.; Shen, S.; Wang, L.; Deng, J.; Yue, C.; Kujawski, M.; Yu, H. S1PR1 is crucial for accumulation of regulatory T cells in tumors via STAT3. Cell Rep. 2014, 6, 992–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbloom, B.E.; Weinreb, N.J.; Zimran, A.; Kacena, K.A.; Charrow, J.; Ward, E. Gaucher disease and cancer incidence: A study from the Gaucher Registry. Blood 2005, 105, 4569–4572. [Google Scholar] [CrossRef]
- Nair, S.; Branagan, A.R.; Liu, J.; Boddupalli, C.S.; Mistry, P.K.; Dhodapkar, M.V. Clonal immunoglobulin against lysolipids in the origin of myeloma. N. Engl. J. Med. 2016, 374, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Pandey, M.K.; Burrow, T.A.; Rani, R.; Martin, L.J.; Witte, D.; Setchell, K.D.; Mckay, M.A.; Magnusen, A.F.; Zhang, W.; Liou, B.; et al. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 2017, 543, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Sofi, M.H.; Heinrichs, J.; Dany, M.; Nguyen, H.; Dai, M.; Bastian, D.; Schutt, S.; Wu, Y.; Daenthanasanmak, A.; Gencer, S.; et al. Ceramide synthesis regulates T cell activity and GVHD development. JCI Insight 2017, 2, e91701. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.; Kuril, S.; Bastian, D.; Kim, J.; Zhang, M.; Vaena, S.G.; Dany, M.; Dai, M.; Heinrichs, J.L.; Daenthanasanmak, A.; et al. Complement C3a and C5a receptors promote GVHD by suppressing mitophagy in recipient dendritic cells. JCI Insight 2018, 3, e121697. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; He, Y.-W. The complement receptors C3aR and C5aR are a new class of immune checkpoint receptor in cancer immunotherapy. Front. Immunol. 2019, 10, 1574. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Sun, S.-N.; Liu, Q.; Yu, Y.-Y.; Guo, J.; Wang, K.; Xing, B.-C.; Zheng, Q.-F.; Campa, M.J.; Patz, E.F.; et al. Autocrine complement inhibits IL10-dependent T-cell–mediated antitumor immunity to promote tumor progression. Cancer Discov. 2016, 6, 1022–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajona, D.; Ortiz-Espinosa, S.; Moreno, H.; Lozano, T.; Pajares, M.J.; Agorreta, J.; Bértolo, C.; Lasarte, J.J.; Vicent, S.; Hoehlig, K.; et al. A combined PD-1/C5a blockade synergistically protects against lung cancer growth and metastasis. Cancer Discov. 2017, 7, 694–703. [Google Scholar] [CrossRef] [Green Version]
- Roumenina, L.T.; Daugan, M.V.; Petitprez, F.; Sautès-Fridman, C.; Fridman, W.H. Context-dependent roles of complement in cancer. Nat. Rev. Cancer 2019, 19, 698–715. [Google Scholar] [CrossRef] [Green Version]
- Ratajczak, M.Z.; Kim, C.; Wu, W.; Shin, D.M.; Bryndza, E.; Kucia, M.; Ratajczak, J. The role of innate immunity in trafficking of hematopoietic stem cells—an emerging link between activation of complement cascade and chemotactic gradients of bioactive sphingolipids. Curr. Top. Innate Immun. II 2012, 946, 37–54. [Google Scholar]
- Lei, Y.-C.; Lu, C.-L.; Chen, L.; Ge, K.; Yang, L.-L.; Li, W.; Wu, Y.-H. C5a/C5aR pathway is essential for up-regulating SphK1 expression through p38-MAPK activation in acute liver failure. World J. Gastroenterol. 2016, 22, 10148. [Google Scholar] [CrossRef] [PubMed]
- Bachmaier, K.; Guzman, E.; Kawamura, T.; Gao, X.; Malik, A.B. Sphingosine kinase 1 mediation of expression of the anaphylatoxin receptor C5L2 dampens the inflammatory response to endotoxin. PLoS ONE 2012, 7, e30742. [Google Scholar]
- Li, R.; Coulthard, L.G.; Wu, M.; Taylor, S.M.; Woodruff, T.M. C5L2: A controversial receptor of complement anaphylatoxin, C5a. FASEB J. 2013, 27, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wang, Q.; Shi, Y.; Peng, X.; Liu, H.; Peng, Y.; He, L. Resveratrol attenuates adriamycin-induced focal segmental glomerulosclerosis through C3aR/C5aR-sphingosine kinase 1 pathway. Pharmacology 2017, 100, 253–260. [Google Scholar] [CrossRef]
- Venant, H.; Rahmaniyan, M.; Jones, E.E.; Lu, P.; Lilly, M.B.; Garrett-Mayer, E.; Drake, R.R.; Kraveka, J.M.; Smith, C.D.; Voelkel-Johnson, C. The sphingosine kinase 2 inhibitor ABC294640 reduces the growth of prostate cancer cells and results in accumulation of dihydroceramides in vitro and in vivo. Mol. Cancer Ther. 2015, 14, 2744–2752. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.S.; Voelkel-Johnson, C.; Smith, C.D. Suppression of c-Myc and RRM2 expression in pancreatic cancer cells by the sphingosine kinase-2 inhibitor ABC294640. Oncotarget 2016, 7, 60181. [Google Scholar] [CrossRef]
- Kummetha Venkata, J.; An, N.; Stuart, R.; Costa, L.J.; Cai, H.; Coker, W.; Song, J.H.; Gibbs, K.; Matson, T.; Garrett-Mayer, E.; et al. Inhibition of sphingosine kinase 2 downregulates the expression of c-Myc and Mcl-1 and induces apoptosis in multiple myeloma. Blood J. Am. Soc. Hematol. 2014, 124, 1915–1925. [Google Scholar] [CrossRef] [Green Version]
- Britten, C.D.; Garrett-Mayer, E.; Chin, S.H.; Shirai, K.; Ogretmen, B.; Bentz, T.A.; Brisendine, A.; Anderton, K.; Cusack, S.L.; Maines, L.W.; et al. A phase I study of ABC294640, a first-in-class sphingosine kinase-2 inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2017, 23, 4642–4650. [Google Scholar] [CrossRef] [Green Version]
- Kucuk, O.; Smith, C.; Plasse, T.; Ogretmen, B.; Mehrotra, S.; Gourdin, T.S.; Bilen, M.A.; Carthon, B.C.; Nazha, B.; Goldman, J.; et al. Phase II Trial of Opaganib in Patients with Metastatic Castration-Resistant Prostate Cancer Progressing on Abiraterone or Enzalutamide (NCT04207255); American Society of Clinical Oncology: Alexandria, VA, USA, 2021. [Google Scholar]
- Cohen, J.A.; Barkhof, F.; Comi, G.; Hartung, H.-P.; Khatri, B.O.; Montalban, X.; Pelletier, J.; Capra, R.; Gallo, P.; Izquierdo, G.; et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 402–415. [Google Scholar] [CrossRef]
- Kappos, L.; Radue, E.-W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saddoughi, S.A.; Gencer, S.; Peterson, Y.K.; Ward, K.E.; Mukhopadhyay, A.; Oaks, J.; Bielawski, J.; Szulc, Z.M.; Thomas, R.J.; Selvam, S.P.; et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol. Med. 2013, 5, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Tian, F.; Ma, H.; Wang, H.; Yang, W.; Liu, Z.; Liao, A. FTY720 induces ferroptosis and autophagy via PP2A/AMPK pathway in multiple myeloma cells. Life Sci. 2020, 260, 118077. [Google Scholar] [CrossRef] [PubMed]
- Arriazu, E.; Pippa, R.; Odero, M.D. Protein phosphatase 2A as a therapeutic target in acute myeloid leukemia. Front. Oncol. 2016, 6, 78. [Google Scholar] [CrossRef] [Green Version]
- Pippa, R.; Dominguez, A.; Christensen, D.; Moreno-Miralles, I.; Blanco-Prieto, M.; Vitek, M.; Odero, M. Effect of FTY720 on the SET–PP2A complex in acute myeloid leukemia; SET binding drugs have antagonistic activity. Leukemia 2014, 28, 1915–1918. [Google Scholar] [CrossRef]
- Neviani, P.; Harb, J.G.; Oaks, J.J.; Santhanam, R.; Walker, C.J.; Ellis, J.J.; Ferenchak, G.; Dorrance, A.M.; Paisie, C.A.; Eiring, A.M.; et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Investig. 2013, 123, 4144–4157. [Google Scholar] [CrossRef]
- Neviani, P.; Santhanam, R.; Oaks, J.J.; Eiring, A.M.; Notari, M.; Blaser, B.W.; Liu, S.; Trotta, R.; Muthusamy, N.; Gambacorti-Passerini, C.; et al. FTY720, a new alternative for treating blast crisis chronic myelogenous leukemia and Philadelphia chromosome–positive acute lymphocytic leukemia. J. Clin. Investig. 2007, 117, 2408–2421. [Google Scholar] [CrossRef] [Green Version]
- Oaks, J.J.; Santhanam, R.; Walker, C.J.; Roof, S.; Harb, J.G.; Ferenchak, G.; Eisfeld, A.-K.; Van Brocklyn, J.R.; Briesewitz, R.; Saddoughi, S.A.; et al. Antagonistic activities of the immunomodulator and PP2A-activating drug FTY720 (Fingolimod, Gilenya) in Jak2-driven hematologic malignancies. Blood J. Am. Soc. Hematol. 2013, 122, 1923–1934. [Google Scholar] [CrossRef] [Green Version]
- Young, M.M.; Bui, V.; Chen, C.; Wang, H.-G. FTY720 induces non-canonical phosphatidylserine externalization and cell death in acute myeloid leukemia. Cell Death Dis. 2019, 10, 847. [Google Scholar] [CrossRef] [Green Version]
- Hirata, N.; Yamada, S.; Yanagida, S.; Ono, A.; Kanda, Y. FTY720 Inhibits Expansion of Breast Cancer Stem Cells via PP2A Activation. Int. J. Mol. Sci. 2021, 22, 7259. [Google Scholar] [CrossRef]
- Nganga, R.; Oleinik, N.; Kim, J.; Selvam, S.P.; De Palma, R.; Johnson, K.A.; Parikh, R.Y.; Gangaraju, V.; Peterson, Y.; Dany, M.; et al. Receptor-interacting Ser/Thr kinase 1 (RIPK1) and myosin IIA–dependent ceramidosomes form membrane pores that mediate blebbing and necroptosis. J. Biol. Chem. 2019, 294, 502–519. [Google Scholar] [CrossRef] [Green Version]
- De Palma, R.M.; Parnham, S.R.; Li, Y.; Oaks, J.J.; Peterson, Y.K.; Szulc, Z.M.; Roth, B.M.; Xing, Y.; Ogretmen, B. The NMR-based characterization of the FTY720-SET complex reveals an alternative mechanism for the attenuation of the inhibitory SET-PP2A interaction. FASEB J. 2019, 33, 7647–7666. [Google Scholar] [CrossRef] [PubMed]
- Morad, S.A.; Cabot, M.C. Ceramide-orchestrated signalling in cancer cells. Nat. Rev. Cancer 2013, 13, 51–65. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, M.; Ogretmen, B. The role of ceramide metabolism and signaling in the regulation of mitophagy and cancer therapy. Cancers 2021, 13, 2475. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Kitatani, K.; Toyoshima, M.; Ishibashi, M.; Usui, T.; Minato, J.; Egiz, M.; Shigeta, S.; Fox, T.; Deering, T.; et al. Ceramide nanoliposomes as a MLKL-dependent, necroptosis-inducing, chemotherapeutic reagent in ovarian cancer. Mol. Cancer Ther. 2018, 17, 50–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Companioni, O.; Mir, C.; Garcia-Mayea, Y.; LLeonart, M.E. Targeting Sphingolipids for Cancer Therapy. Front. Oncol. 2021, 11, 4295. [Google Scholar] [CrossRef] [PubMed]
- Ryland, L.K.; Doshi, U.A.; Shanmugavelandy, S.S.; Fox, T.E.; Aliaga, C.; Broeg, K.; Baab, K.T.; Young, M.; Khan, O.; Haakenson, J.K.; et al. C6-ceramide nanoliposomes target the Warburg effect in chronic lymphocytic leukemia. PLoS ONE 2013, 8, e84648. [Google Scholar]
- Li, G.; Liu, D.; Kimchi, E.T.; Kaifi, J.T.; Qi, X.; Manjunath, Y.; Liu, X.; Deering, T.; Avella, D.M.; Fox, T.; et al. Nanoliposome C6-ceramide increases the anti-tumor immune response and slows growth of liver tumors in mice. Gastroenterology 2018, 154, 1024–1036.e9. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ryland, L.; Yang, J.; Liao, A.; Aliaga, C.; Watts, R.; Tan, S.-F.; Kaiser, J.; Shanmugavelandy, S.S.; Rogers, A.; et al. Targeting of survivin by nanoliposomal ceramide induces complete remission in a rat model of NK-LGL leukemia. Blood J. Am. Soc. Hematol. 2010, 116, 4192–4201. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Fu, C.; Hu, Y.; Dong, C.; Song, Y.; Song, E. C6-ceramide nanoliposome suppresses tumor metastasis by eliciting PI3K and PKCζ tumor-suppressive activities and regulating integrin affinity modulation. Sci. Rep. 2015, 5, 9275. [Google Scholar] [CrossRef] [Green Version]
- Visentin, B.; Vekich, J.A.; Sibbald, B.J.; Cavalli, A.L.; Moreno, K.M.; Matteo, R.G.; Garland, W.A.; Lu, Y.; Yu, S.; Hall, H.S.; et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell 2006, 9, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, S.K.; Drabkin, H.A.; Reeves, J.A.; Hainsworth, J.D.; Hazel, S.E.; Paggiarino, D.A.; Wojciak, J.; Woodnutt, G.; Bhatt, R.S. A phase 2 study of the sphingosine-1-phosphate antibody sonepcizumab in patients with metastatic renal cell carcinoma. Cancer 2017, 123, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Kroll, A.; Cho, H.E.; Kang, M.H. Antineoplastic agents targeting sphingolipid pathways. Front. Oncol. 2020, 10, 833. [Google Scholar] [CrossRef] [PubMed]
- Dany, M.; Ogretmen, B. Ceramide induced mitophagy and tumor suppression. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 2834–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnute, M.E.; McReynolds, M.D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J.W.; Hall, T.; Chrencik, J.; Kraus, M.; Cronin, C.N.; et al. Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 2012, 444, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Ju, T.; Gao, D.; Fang, Z.-y. Targeting colorectal cancer cells by a novel sphingosine kinase 1 inhibitor PF-543. Biochem. Biophys. Res. Commun. 2016, 470, 728–734. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzymes | Metabolic Functions | Roles in Cancer | References |
|---|---|---|---|
| SPHK1 | S1P generation | Promotes tumor growth in melanoma, ovarian, and colitis-associated cancers | [18,19,20,21,22,23] |
| SPHK2 | S1P generation | Augments 5-FU chemotherapy resistance in human colorectal cancer and mediates FSH-induced cell proliferation in ovarian cancer | [22,23] |
| SGPL1 | Irreversibly breaks down S1P | Inhibits colon tumor formation and prevents S1P-induced migration and cell-colony formation in pediatric alveolar rhabdomyosarcoma | [24,25,26,27] |
| CERK | C1P generation | Promotes breast cancer growth and confers chemotherapy resistance to breast cancer cell lines | [31,32,33,34] |
| AC | Cleaves fatty acid moiety from ceramide | Overexpressed in several cancer types and mediates the switch between proliferative and invasive phenotype states in melanoma cells. It also confers resistance to cancer cell death | [36,37,38,39,40,41,42] |
| NC | Cleaves fatty acid moiety from ceramide | Inhibits cellular apoptosis in colon cancer cells and induces xenograft tumor growth | [44,45] |
| ACER3 | Cleaves fatty acid moiety from ceramide | Promotes tumor growth and inhibits apoptosis in HCC and AML cells | [57,58] |
| CerS1 | Synthesis of C18 ceramide | Inhibits HNSCC xenograft growth and induces cancer cell death | [69,70,71,72,73] |
| CerS2 | Synthesizes very-long-chain ceramides | Inhibits in vivo metastasis and invasiveness of ovarian cancer cells | [75] |
| Partially prevents programmed cell death induced by ionizing radiation in HeLa cells | [77] | ||
| CerS4 | Synthesis of C18–C20 ceramides | Inhibits A549 cancer cell migration and invasion | [79] |
| CerS6 | Generates C16 ceramide | Promotes cell proliferation in PDAC, HNSCC, and lung cancer cell lines | [71,83,84] |
| Has anti-proliferative and pro-apoptotic functions in polyploid giant cancer cells | [85] | ||
| Alk-SMase (ENPP7) | Ceramide generation | Reduces colon cancer progression in a mice model | [87] |
| ASMase (SMPD1) | Ceramide generation | ASMase’s induction in platelets induces B16F10 melanoma metastasis | [89,90,91] |
| NSMase2 (SMPD3) | Ceramide generation | Enhances the efficacy of anti-PD-1 antibody therapy in melanoma and breast cancer mouse models. It also inhibits tumor progression in oral squamous cell carcinoma | [92,93] |
| SMS2 (SGMS2) | Produces sphingomyelin and diacylglycerol | Promotes ovarian cancer cell growth and migration | [99] |
| Name | Sphingolipid Targets | Cancer Type | Stage | ClinicalTrials.gov Identifier |
|---|---|---|---|---|
| ABC294640 (Yeliva, opaganib) | SPHK2; DES | Prostate Cancer | Phase II | NCT04207255 |
| Multiple Myeloma | Phases I and II | NCT02757326 | ||
| Cholangiocarcinoma | Phase II | NCT03377179, NCT03414489 | ||
| Fingolimod (FTY720) (FDA approved for MS) | S1PR1 | Breast Carcinoma (treating paclitaxel-associated neuropathy) | Phase I | NCT03941743 |
| Glioblastoma & Anaplastic Astrocytoma (treating severe and persistent lymphopenia in patients undergoing radiation and chemotherapy) | Early Phase I | NCT02490930 | ||
| ASONEP™ (sonepcizumab/LT1009) | S1P | Solid Tumors | Phase I | NCT00661414 |
| Ceramide NanoLiposome | Ceramide inducer | Renal Cell Carcinoma | Phase II | NCT01762033 |
| Solid Tumors | Phase I | NCT02834611 | ||
| Acute Myeloid Leukemia | Phase I | NCT04716452 | ||
| Safingol | SPHK1 | Locally Advanced or Metastatic Solid Tumors | Phase I | NCT00084812 |
| Fluphenazine | ASMase | Multiple Myeloma and Plasma Cell Neoplasm | Phases I and II | NCT00335647 |
| Multiple Myeloma | Phase I | NCT00821301 | ||
| Desipramine | AC | Small Cell Lung Cancer and Neuroendocrine Tumors | Phase II | NCT01719861 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janneh, A.H.; Ogretmen, B. Targeting Sphingolipid Metabolism as a Therapeutic Strategy in Cancer Treatment. Cancers 2022, 14, 2183. https://doi.org/10.3390/cancers14092183
Janneh AH, Ogretmen B. Targeting Sphingolipid Metabolism as a Therapeutic Strategy in Cancer Treatment. Cancers. 2022; 14(9):2183. https://doi.org/10.3390/cancers14092183
Chicago/Turabian StyleJanneh, Alhaji H., and Besim Ogretmen. 2022. "Targeting Sphingolipid Metabolism as a Therapeutic Strategy in Cancer Treatment" Cancers 14, no. 9: 2183. https://doi.org/10.3390/cancers14092183