
A Tumor-Homing Peptide Platform Enhances Drug Solubility, Improves Blood–Brain Barrier Permeability and Targets Glioblastoma

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
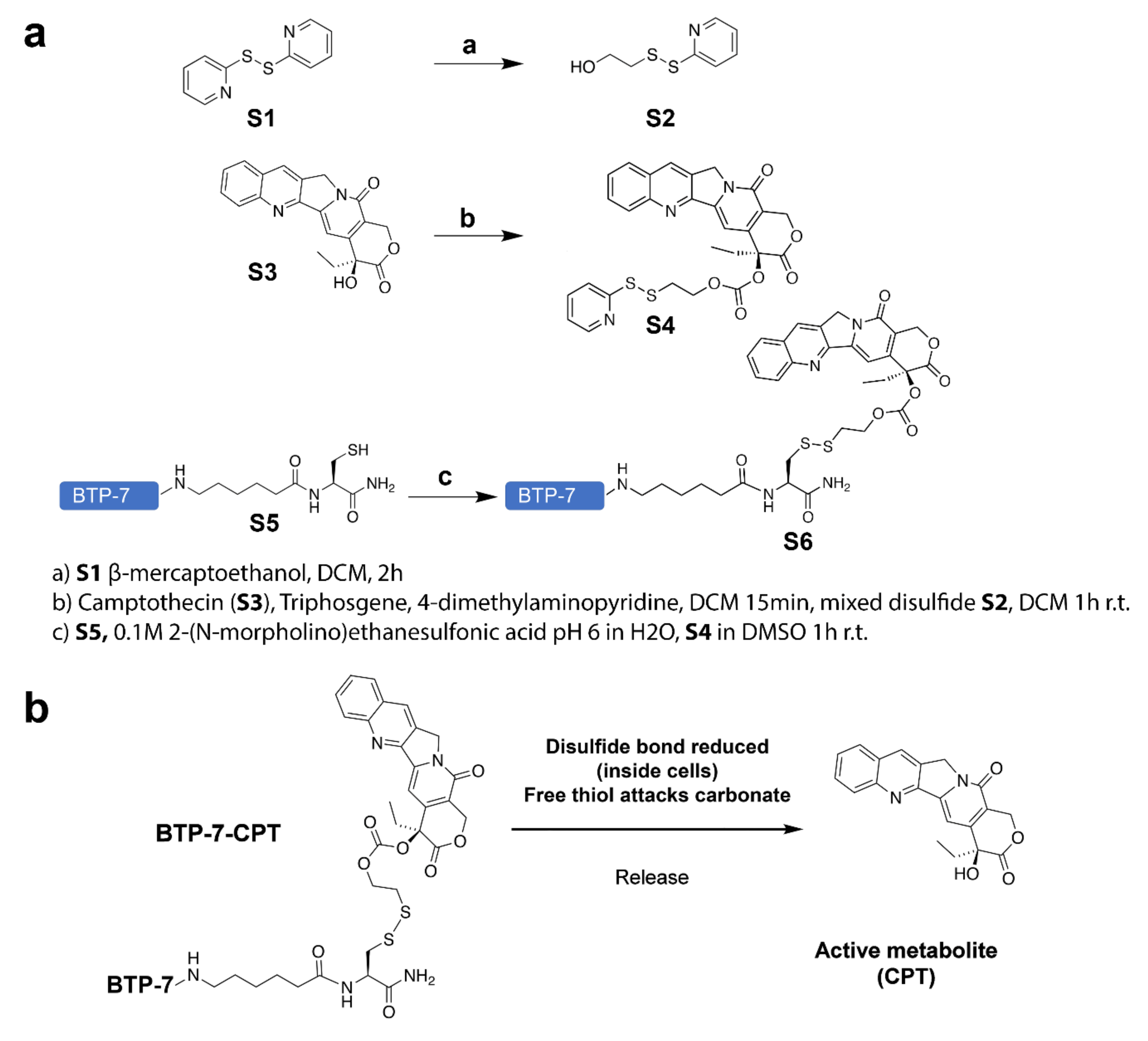
2.2. Synthesis of BTP-7-CPT
2.3. Analysis of Compound Solubility
2.4. Western Blot Analysis
2.5. Cell Viability Assay
2.6. Permeability Analysis In Vitro Using Human BBB Organoids
2.7. Ex Vivo Serum Stability Assay
2.8. Intracranial GBM Implantation and Efficacy Studies
2.9. Ex Vivo Immunofluorescence Staining of GBM Tissue Sections
2.10. Statistical Analysis
2.11. Use of Human Specimens
3. Results
3.1. Conjugation of CPT with BTP-7 Enhances Drug Solubility
3.1.1. Synthesis of BTP-7-CPT
3.1.2. BTP-7 Conjugation Improves CPT Solubility
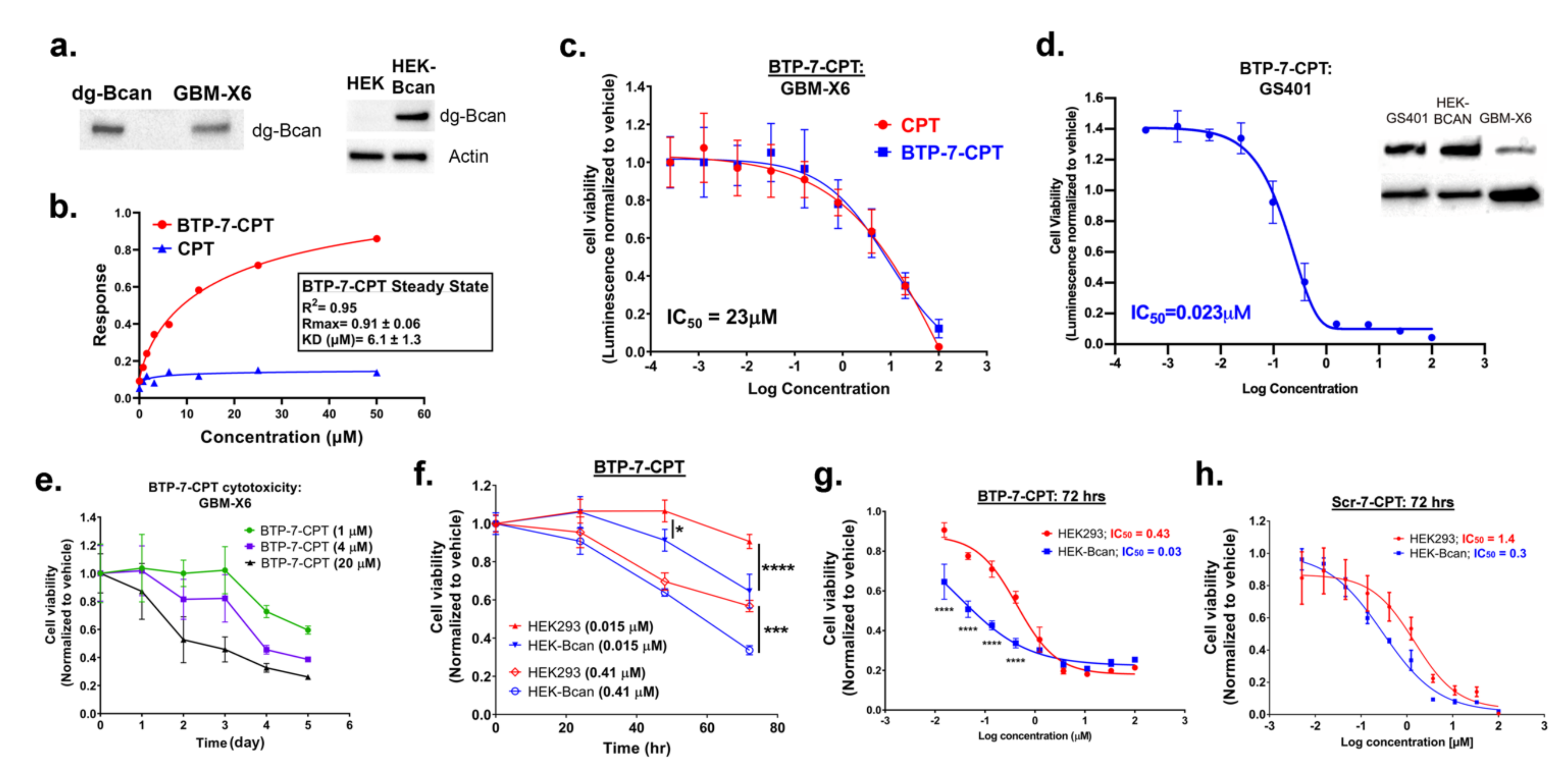
3.2. BTP-7-CPT Binds dg-Bcan Protein
3.3. Cytotoxicity of BTP-7-CPT
3.4. Analysis of BBB Permeability
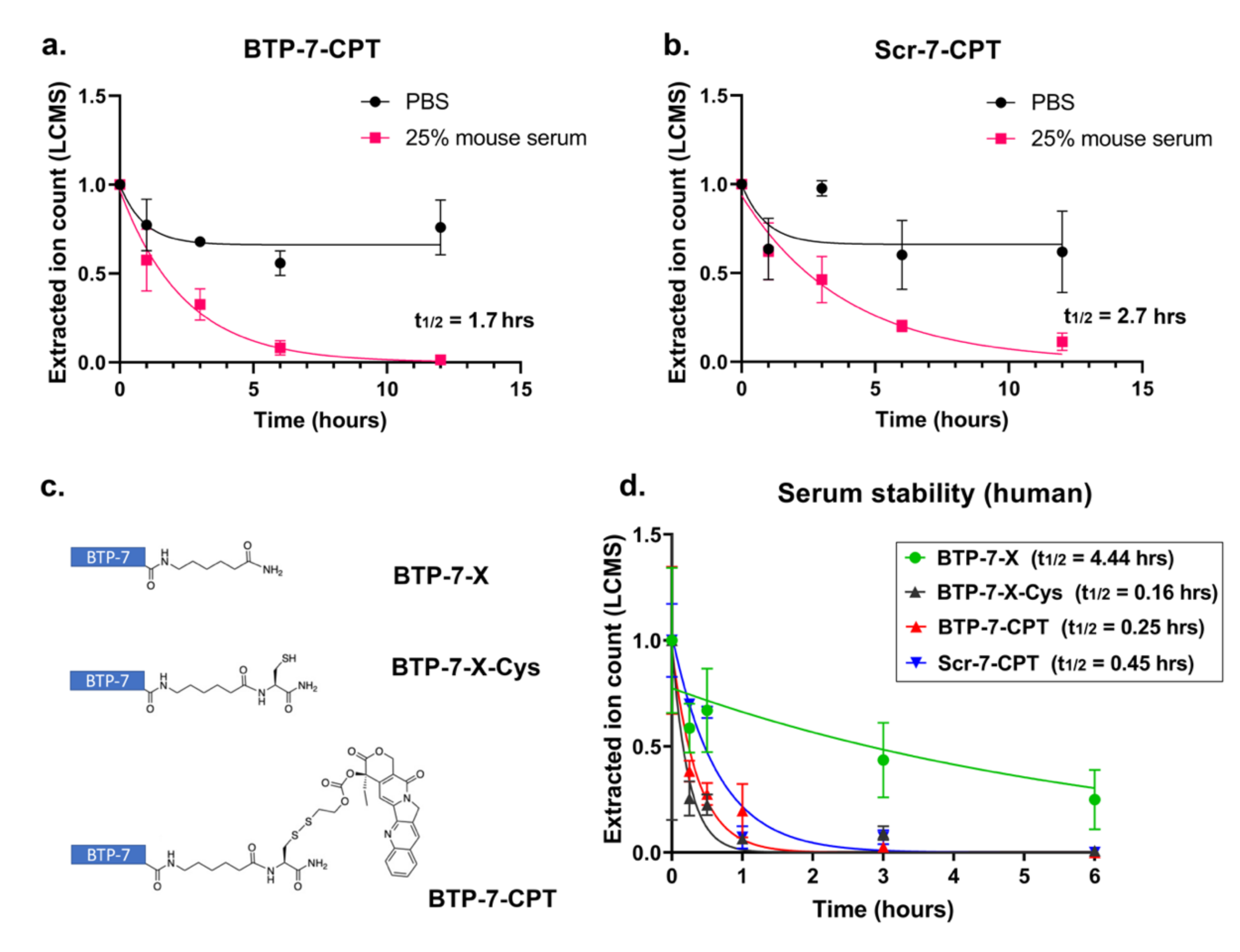
3.5. Stability of Peptide–Drug Conjugate in Serum
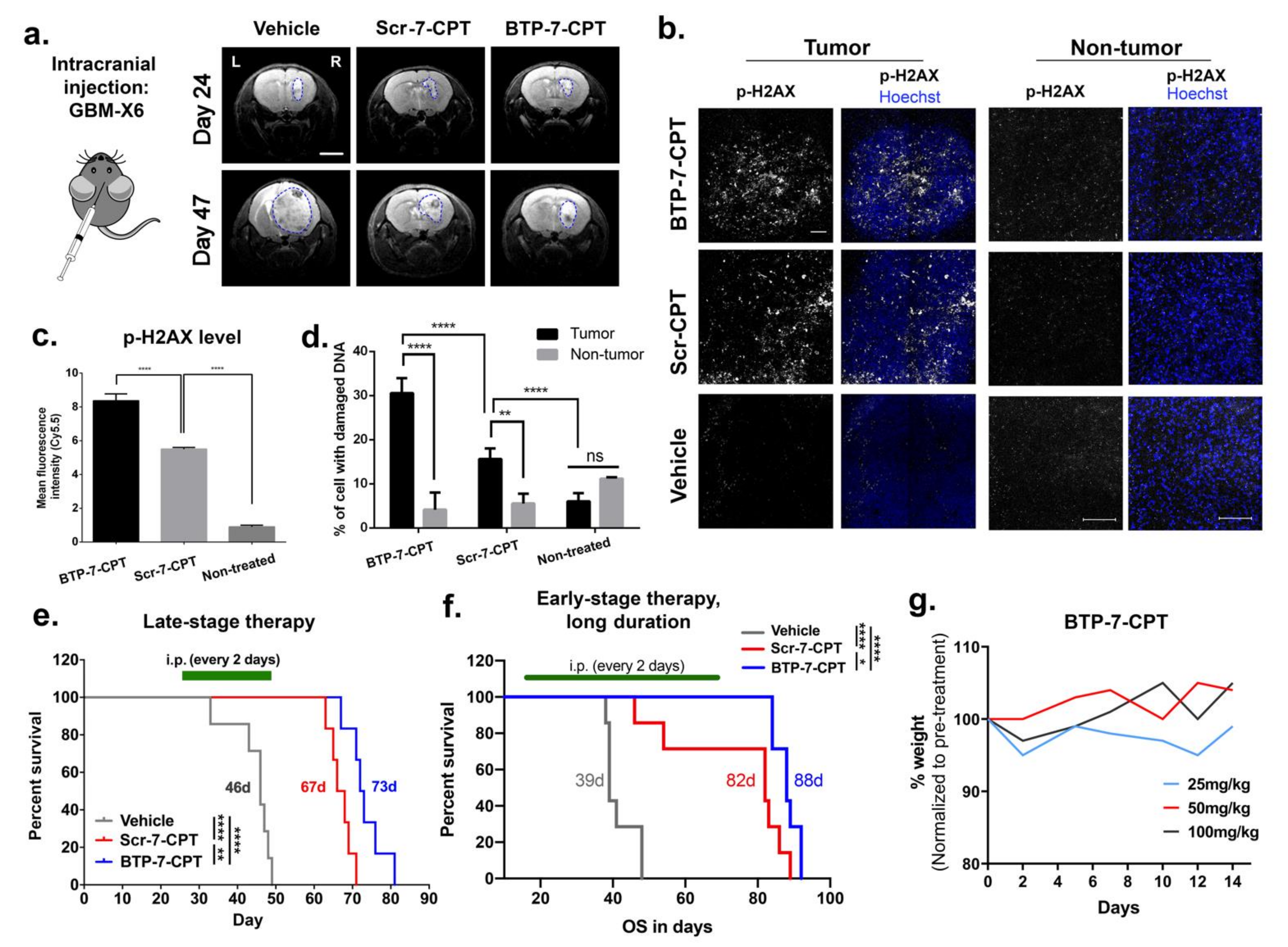
3.6. Delivery of CPT to Intracranial GBM Tumor Using BTP-7
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011–2015. Neuro-Oncology 2018, 20, iv1–iv86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, K.K. A Critical Overview of Targeted Therapies for Glioblastoma. Front. Oncol. 2018, 8, 419. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N. Molecular Pathology of Malignant Gliomas. Annu. Rev. Pathol. 2006, 1, 97–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löscher, W.; Potschka, H. Drug Resistance in Brain Diseases and the Role of Drug Efflux Transporters. Nat. Rev. Neurosci. 2005, 6, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Gala, U.H.; Miller, D.A.; Williams, R.O. Harnessing the Therapeutic Potential of Anticancer Drugs through Amorphous Solid Dispersions. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1873, 188319. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma Targeted Therapy: Updated Approaches from Recent Biological Insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [Green Version]
- Raavé, R.; van Kuppevelt, T.H.; Daamen, W.F. Chemotherapeutic Drug Delivery by Tumoral Extracellular Matrix Targeting. J. Control. Release 2018, 274, 1–8. [Google Scholar] [CrossRef]
- Jaworski, D.M.; Kelly, G.M.; Hockfield, S. BEHAB, a New Member of the Proteoglycan Tandem Repeat Family of Hyaluronan-Binding Proteins That Is Restricted to the Brain. J. Cell Biol. 1994, 125, 495–509. [Google Scholar] [CrossRef]
- Viapiano, M.S.; Bi, W.L.; Piepmeier, J.; Hockfield, S.; Matthews, R.T. Novel Tumor-Specific Isoforms of BEHAB/Brevican Identified in Human Malignant Gliomas. Cancer Res. 2005, 65, 6726–6733. [Google Scholar] [CrossRef] [Green Version]
- Jaworski, D.M.; Kelly, G.M.; Piepmeier, J.M.; Hockfield, S. BEHAB (Brain Enriched Hyaluronan Binding) Is Expressed in Surgical Samples of Glioma and in Intracranial Grafts of Invasive Glioma Cell Lines. Cancer Res. 1996, 56, 2293–2298. [Google Scholar] [PubMed]
- Lu, R.; Wu, C.; Guo, L.; Liu, Y.; Mo, W.; Wang, H.; Ding, J.; Wong, E.T.; Yu, M. The Role of Brevican in Glioma: Promoting Tumor Cell Motility in Vitro and in Vivo. BMC Cancer 2012, 12, 607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spreckelsen, N.; Fadzen, C.M.; Hartrampf, N.; Ghotmi, Y.; Wolfe, J.M.; Dubey, S.; Yang, B.Y.; Kijewski, M.F.; Wang, S.; Farquhar, C.; et al. Targeting Glioblastoma Using a Novel Peptide Specific to a Deglycosylated Isoform of Brevican. Adv. Ther. 2021, 4, 2000244. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.T.; Nakanishi, Y.; Kroll, D.J.; Griset, A.P.; Carnahan, M.A.; Wathier, M.; Oberlies, N.H.; Manikumar, G.; Wani, M.C.; Grinstaff, M.W. Dendrimer-Encapsulated Camptothecins: Increased Solubility, Cellular Uptake, and Cellular Retention Affords Enhanced Anticancer Activity in Vitro. Cancer Res. 2006, 66, 11913–11921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberlies, N.H.; Kroll, D.J. Camptothecin and Taxol: Historic Achievements in Natural Products Research. J. Nat. Prod. 2004, 67, 129–135. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor Stem Cells Derived from Glioblastomas Cultured in BFGF and EGF More Closely Mirror the Phenotype and Genotype of Primary Tumors than Do Serum-Cultured Cell Lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Henne, W.A.; Doorneweerd, D.D.; Hilgenbrink, A.R.; Kularatne, S.A.; Low, P.S. Synthesis and Activity of a Folate Peptide Camptothecin Prodrug. Bioorg. Med. Chem. Lett. 2006, 16, 5350–5355. [Google Scholar] [CrossRef]
- Giannini, C.; Sarkaria, J.N.; Saito, A.; Uhm, J.H.; Galanis, E.; Carlson, B.L.; Schroeder, M.A.; James, C.D. Patient Tumor EGFR and PDGFRA Gene Amplifications Retained in an Invasive Intracranial Xenograft Model of Glioblastoma Multiforme. Neuro-Oncology 2005, 7, 164–176. [Google Scholar] [CrossRef]
- Cho, C.-F.; Wolfe, J.M.; Fadzen, C.M.; Calligaris, D.; Hornburg, K.; Chiocca, E.A.; Agar, N.Y.R.; Pentelute, B.L.; Lawler, S.E. Blood-Brain-Barrier Spheroids as an in Vitro Screening Platform for Brain-Penetrating Agents. Nat. Commun. 2017, 8, 15623. [Google Scholar] [CrossRef]
- Bergmann, S.; Lawler, S.E.; Qu, Y.; Fadzen, C.M.; Wolfe, J.M.; Regan, M.S.; Pentelute, B.L.; Agar, N.Y.R.; Cho, C.-F. Blood-Brain-Barrier Organoids for Investigating the Permeability of CNS Therapeutics. Nat. Protoc. 2018, 13, 2827–2843. [Google Scholar] [CrossRef] [Green Version]
- Saito, G.; Swanson, J.A.; Lee, K.-D. Drug Delivery Strategy Utilizing Conjugation via Reversible Disulfide Linkages: Role and Site of Cellular Reducing Activities. Adv. Drug Deliv. Rev. 2003, 55, 199–215. [Google Scholar] [CrossRef]
- Bailly, C. Irinotecan: 25 Years of Cancer Treatment. Pharmacol. Res. 2019, 148, 104398. [Google Scholar] [CrossRef] [PubMed]
- Friedman, G.K.; Haas, M.C.; Kelly, V.M.; Markert, J.M.; Gillespie, G.Y.; Cassady, K.A. Hypoxia Moderates γ(1)34.5-Deleted Herpes Simplex Virus Oncolytic Activity in Human Glioma Xenoline Primary Cultures. Transl. Oncol. 2012, 5, 200–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Liu, C.; Wang, P.; Ghazwani, M.; Xu, J.; Huang, Y.; Ma, X.; Zhang, P.; Li, S. The Self-Assembling Camptothecin-Tocopherol Prodrug: An Effective Approach for Formulating Camptothecin. Biomaterials 2015, 62, 176–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldwirt, L.; Beccaria, K.; Carpentier, A.; Farinotti, R.; Fernandez, C. Irinotecan and Temozolomide Brain Distribution: A Focus on ABCB1. Cancer Chemother. Pharm. 2014, 74, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Polivka, J.; Polivka, J.; Holubec, L.; Kubikova, T.; Priban, V.; Hes, O.; Pivovarcikova, K.; Treskova, I. Advances in Experimental Targeted Therapy and Immunotherapy for Patients with Glioblastoma Multiforme. Anticancer Res. 2017, 37, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Wu, W.; Bi, H.; Yang, D.; Zhang, C. Glioblastoma Precision Therapy: From the Bench to the Clinic. Cancer Lett. 2020, 475, 79–91. [Google Scholar] [CrossRef]
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational Analysis Reveals the Origin and Therapy-Driven Evolution of Recurrent Glioma. Science 2014, 343, 189–193. [Google Scholar] [CrossRef] [Green Version]
- Frischknecht, R.; Seidenbecher, C.I. Brevican: A Key Proteoglycan in the Perisynaptic Extracellular Matrix of the Brain. Int. J. Biochem. Cell Biol. 2012, 44, 1051–1054. [Google Scholar] [CrossRef]
- Hu, B.; Kong, L.L.; Matthews, R.T.; Viapiano, M.S. The Proteoglycan Brevican Binds to Fibronectin after Proteolytic Cleavage and Promotes Glioma Cell Motility. J. Biol. Chem. 2008, 283, 24848–24859. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, C.A.; Bi, W.L.; Viapiano, M.S.; Matthews, R.T. Brevican Knockdown Reduces Late-Stage Glioma Tumor Aggressiveness. J. Neurooncol. 2014, 120, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsidulko, A.Y.; Kazanskaya, G.M.; Volkov, A.M.; Suhovskih, A.V.; Kiselev, R.S.; Kobozev, V.V.; Gaytan, A.S.; Krivoshapkin, A.L.; Aidagulova, S.V.; Grigorieva, E.V. Chondroitin Sulfate Content and Decorin Expression in Glioblastoma Are Associated with Proliferative Activity of Glioma Cells and Disease Prognosis. Cell Tissue Res 2020, 379, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Virga, J.; Bognár, L.; Hortobágyi, T.; Zahuczky, G.; Csősz, É.; Kalló, G.; Tóth, J.; Hutóczki, G.; Reményi-Puskár, J.; Steiner, L.; et al. Prognostic Role of the Expression of Invasion-Related Molecules in Glioblastoma. J. Neurol. Surg. A Cent. Eur. Neurosurg. 2016, 78, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Viapiano, M.S.; Matthews, R.T. From Barriers to Bridges: Chondroitin Sulfate Proteoglycans in Neuropathology. Trends Mol. Med. 2006, 12, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Viapiano, M.S.; Hockfield, S.; Matthews, R.T. BEHAB/Brevican Requires ADAMTS-Mediated Proteolytic Cleavage to Promote Glioma Invasion. J. Neurooncol. 2008, 88, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Glass, R.; Synowitz, M.; Kronenberg, G.; Walzlein, J.-H.; Markovic, D.S.; Wang, L.-P.; Gast, D.; Kiwit, J.; Kempermann, G.; Kettenmann, H. Glioblastoma-Induced Attraction of Endogenous Neural Precursor Cells Is Associated with Improved Survival. J. Neurosci. 2005, 25, 2637–2646. [Google Scholar] [CrossRef] [Green Version]
- Grigorieva, E.V. Radiation Effects on Brain Extracellular Matrix. Front. Oncol. 2020, 10, 576701. [Google Scholar] [CrossRef]
- Gupta, K.; Burns, T.C. Radiation-Induced Alterations in the Recurrent Glioblastoma Microenvironment: Therapeutic Implications. Front. Oncol. 2018, 8, 503. [Google Scholar] [CrossRef] [Green Version]
- Virga, J.; Szivos, L.; Hortobágyi, T.; Chalsaraei, M.K.; Zahuczky, G.; Steiner, L.; Tóth, J.; Reményi-Puskár, J.; Bognár, L.; Klekner, A. Extracellular Matrix Differences in Glioblastoma Patients with Different Prognoses. Oncol. Lett. 2019, 17, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Ladner, R.C.; Sato, A.K.; Gorzelany, J.; Souza, M. Phage Display-Derived Peptides as Therapeutic Alternatives to Antibodies. Drug Discov. Today 2004, 9, 525–529. [Google Scholar] [CrossRef]
- Zhang, X.-X.; Eden, H.S.; Chen, X. Peptides in Cancer Nanomedicine: Drug Carriers, Targeting Ligands and Protease Substrates. J. Control. Release 2012, 159, 2–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruger, R.P. The Coming Peptide Tidal Wave. Cell 2017, 171, 497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venditto, V.J.; Simanek, E.E. Cancer Therapies Utilizing the Camptothecins: A Review of the in Vivo Literature. Mol. Pharm. 2010, 7, 307–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalim, M.; Chen, J.; Wang, S.; Lin, C.; Ullah, S.; Liang, K.; Ding, Q.; Chen, S.; Zhan, J. Intracellular Trafficking of New Anticancer Therapeutics: Antibody–Drug Conjugates. Drug Des. Dev. Ther. 2017, 11, 2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B. Development of Potent Monoclonal Antibody Auristatin Conjugates for Cancer Therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable Linkers in Antibody-Drug Conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, C.-F.; Farquhar, C.E.; Fadzen, C.M.; Scott, B.; Zhuang, P.; von Spreckelsen, N.; Loas, A.; Hartrampf, N.; Pentelute, B.L.; Lawler, S.E. A Tumor-Homing Peptide Platform Enhances Drug Solubility, Improves Blood–Brain Barrier Permeability and Targets Glioblastoma. Cancers 2022, 14, 2207. https://doi.org/10.3390/cancers14092207
Cho C-F, Farquhar CE, Fadzen CM, Scott B, Zhuang P, von Spreckelsen N, Loas A, Hartrampf N, Pentelute BL, Lawler SE. A Tumor-Homing Peptide Platform Enhances Drug Solubility, Improves Blood–Brain Barrier Permeability and Targets Glioblastoma. Cancers. 2022; 14(9):2207. https://doi.org/10.3390/cancers14092207
Chicago/Turabian StyleCho, Choi-Fong, Charlotte E. Farquhar, Colin M. Fadzen, Benjamin Scott, Pei Zhuang, Niklas von Spreckelsen, Andrei Loas, Nina Hartrampf, Bradley L. Pentelute, and Sean E. Lawler. 2022. "A Tumor-Homing Peptide Platform Enhances Drug Solubility, Improves Blood–Brain Barrier Permeability and Targets Glioblastoma" Cancers 14, no. 9: 2207. https://doi.org/10.3390/cancers14092207
APA StyleCho, C.-F., Farquhar, C. E., Fadzen, C. M., Scott, B., Zhuang, P., von Spreckelsen, N., Loas, A., Hartrampf, N., Pentelute, B. L., & Lawler, S. E. (2022). A Tumor-Homing Peptide Platform Enhances Drug Solubility, Improves Blood–Brain Barrier Permeability and Targets Glioblastoma. Cancers, 14(9), 2207. https://doi.org/10.3390/cancers14092207