
Impact of the Tumor Microenvironment for Esophageal Tumor Development—An Opportunity for Prevention?


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
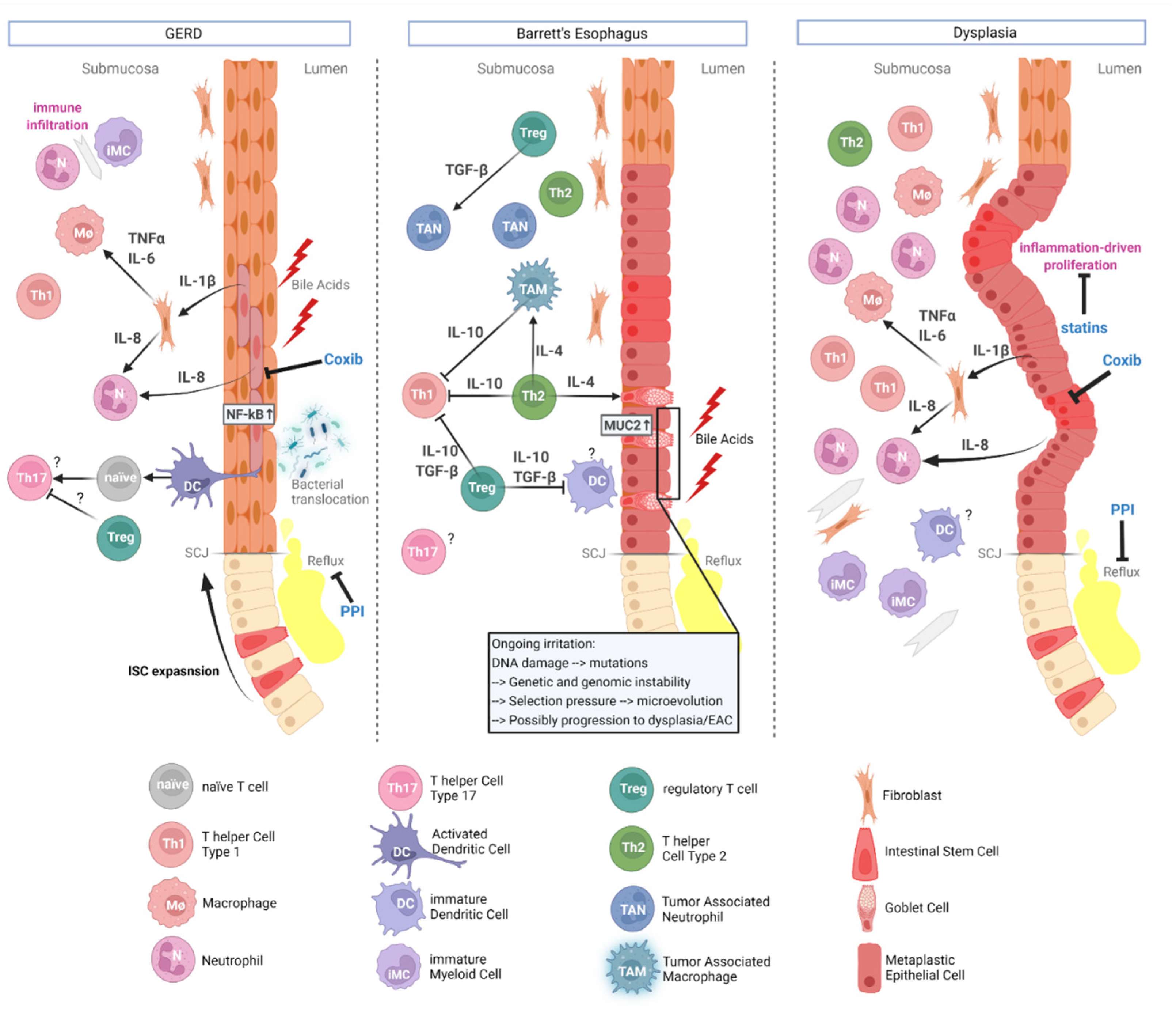
2. Pathophysiology and Carcinogenesis of EAC
3. The Tumor Microenvironment Fuels Esophageal Carcinogenesis
3.1. The Mesenchymal Contribution
3.2. The Ignition of Immune Reactions
3.3. Dendritic Cells
3.4. T Cells
3.5. MDSCs
3.6. TAMs
3.7. Neutrophils
3.8. NK Cells
3.9. Secretory Molecules
3.10. Microbiome
3.11. Search Strategy
4. The TME as an Opportunity for Cancer Prevention
TME as a Target for Chemoprevention
5. Research Perspectives
6. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Quante, M.; Varga, J.; Wang, T.C.; Greten, F.R. The Gastrointestinal Tumor Microenvironment. Gastroenterology 2013, 145, 63–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hold, G.L.; El-Omar, M.E. Genetic aspects of inflammation and cancer. Biochem. J. 2008, 410, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Koulaouzidis, A.; Marlicz, W.; Lok, V.; Chu, C.; Ngai, C.H.; Zhang, L.; Chen, P.; Wang, S.; Yuan, J.; et al. Global Burden, Risk Factors, and Trends of Esophageal Cancer: An Analysis of Cancer Registries from 48 Countries. Cancers 2021, 13, 141. [Google Scholar] [CrossRef]
- Hvid-Jensen, F.; Pedersen, L.; Drewes, A.M.; Sørensen, H.T.; Funch-Jensen, P. Incidence of adenocarcinoma among patients with Barrett’s esophagus. N. Engl. J. Med. 2011, 365, 1375–1383. [Google Scholar] [CrossRef] [Green Version]
- Coleman, H.G.; Bhat, S.; Johnston, B.T.; McManus, D.; Gavin, A.T.; Murray, L.J. Tobacco smoking increases the risk of high-grade dysplasia and cancer among patients with Barrett’s esophagus. Gastroenterology 2012, 142, 233–240. [Google Scholar] [CrossRef] [Green Version]
- O’Donovan, M.; Fitzgerald, R.C. Screening for Barrett’s esophagus: Are new high-volume methods feasible? Dig. Dis. Sci. 2018, 63, 2105–2114. [Google Scholar] [CrossRef] [Green Version]
- Qumseya, B.J.; Bukannan, A.; Gendy, S.; Ahemd, Y.; Sultan, S.; Bain, P.; Gross, S.A.; Iyer, P.; Wani, S. Systematic review and meta-analysis of prevalence and risk factors for Barrett’s esophagus. Gastrointest. Endosc. 2019, 90, 707–717.e701. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Ebbing, E.A.; van der Zalm, A.P.; Steins, A.; Creemers, A.; Hermsen, S.; Rentenaar, R.; Klein, M.; Waasdorp, C.; Hooijer, G.K.J.; Meijer, S.L.; et al. Stromal-derived interleukin 6 drives epithelial-to-mesenchymal transition and therapy resistance in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2019, 116, 2237–2242. [Google Scholar] [CrossRef] [Green Version]
- Quante, M.; Bhagat, G.; Abrams, J.A.; Marache, F.; Good, P.; Lee, M.D.; Lee, Y.; Friedman, R.; Asfaha, S.; Dubeykovskaya, Z.; et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer Cell 2012, 21, 36–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayin, S.I.; Baumeister, T.; Wang, T.C.; Quante, M. Origins of metaplasia in the esophagus: Is this a GE junction stem cell disease? Dig. Dis. Sci. 2018, 63, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, M.; O’Sullivan, K.; O’Hanlon, C.; O’Sullivan, J.; Lysaght, J.; Reynolds, J. The esophagitis to adenocarcinoma sequence; the role of inflammation. Cancer Lett. 2014, 345, 182–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, H.; Ha, K.; Hornick, J.L.; Madha, S.; Cejas, P.; Jajoo, K.; Singh, P.; Polak, P.; Lee, H.; Shivdasani, R.A. Hybrid Stomach-Intestinal Chromatin States Underlie Human Barrett’s Metaplasia. Gastroenterology 2021, 161, 924–939.e911. [Google Scholar] [CrossRef]
- Nowicki-Osuch, K.; Zhuang, L.; Jammula, S.; Bleaney, C.W.; Mahbubani, K.T.; Devonshire, G.; Katz-Summercorn, A.; Eling, N.; Wilbrey-Clark, A.; Madissoon, E.; et al. Molecular phenotyping reveals the identity of Barrett’s esophagus and its malignant transition. Science 2021, 373, 760–767. [Google Scholar] [CrossRef]
- Barrett, N.R. Chronic peptic ulcer of the oesophagus and ‘oesophagitis’. Br. J. Surg. 1950, 38, 175–182. [Google Scholar] [CrossRef]
- Quante, M.; Wang, T.C.; Bass, A.J. Adenocarcinoma of the oesophagus: Is it gastric cancer? Gut 2022. [Google Scholar] [CrossRef]
- Schmidt, M.; Hackett, R.J.; Baker, A.-M.; McDonald, S.A.; Quante, M.; Graham, T.A. Evolutionary dynamics in Barrett oesophagus: Implications for surveillance, risk stratification and therapy. Nat. Rev. Gastroenterol. Hepatol. 2021, 19, 95–111. [Google Scholar] [CrossRef]
- Proaño-Vasco, A.; Baumeister, T.; Metwaly, A.; Reitmeier, S.; Kleigrewe, K.; Meng, C.; Gigl, M.; Engleitner, T.; Öllinger, R.; Rad, R. High-Fructose Diet Alters Intestinal Microbial Profile and Correlates with Early Tumorigenesis in a Mouse Model of Barrett’s Esophagus. Microorganisms 2021, 9, 2432. [Google Scholar] [CrossRef]
- Cho, K.R.; Vogelstein, B. Genetic alterations in the adenoma–carcinoma sequence. Cancer 1992, 70, 1727–1731. [Google Scholar] [CrossRef]
- Pectasides, E.; Stachler, M.D.; Derks, S.; Liu, Y.; Maron, S.; Islam, M.; Alpert, L.; Kwak, H.; Kindler, H.; Polite, B.; et al. Genomic Heterogeneity as a Barrier to Precision Medicine in Gastroesophageal Adenocarcinoma. Cancer Discov. 2018, 8, 37–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stachler, M.D.; Camarda, N.D.; Deitrick, C.; Kim, A.; Agoston, A.T.; Odze, R.D.; Hornick, J.L.; Nag, A.; Thorner, A.R.; Ducar, M. Detection of mutations in Barrett’s esophagus before progression to high-grade dysplasia or adenocarcinoma. Gastroenterology 2018, 155, 156–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Galipeau, P.C.; Paulson, T.G.; Sanchez, C.A.; Arnaudo, J.; Liu, K.; Sather, C.L.; Kostadinov, R.L.; Odze, R.D.; Kuhner, M.K. Temporal and spatial evolution of somatic chromosomal alterations: A case-cohort study of Barrett’s esophagus. Cancer Prev. Res. 2014, 7, 114–127. [Google Scholar] [CrossRef] [Green Version]
- Maley, C.C.; Galipeau, P.C.; Li, X.; Sanchez, C.A.; Paulson, T.G.; Reid, B.J. Selectively advantageous mutations and hitchhikers in neoplasms: p16 lesions are selected in Barrett’s esophagus. Cancer Res. 2004, 64, 3414–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busslinger, G.A.; de Barbanson, B.; Oka, R.; Weusten, B.L.; de Maat, M.; van Hillegersberg, R.; Brosens, L.A.; van Boxtel, R.; van Oudenaarden, A.; Clevers, H. Molecular characterization of Barrett’s esophagus at single-cell resolution. Proc. Natl. Acad. Sci. USA 2021, 118, e2113061118. [Google Scholar] [CrossRef]
- Dai, J.Y.; Wang, X.; Buas, M.F.; Zhang, C.; Ma, J.; Wei, B.; Li, Y.; Zhao, B.; Hyun, T.S.; Chen, X. Whole-genome sequencing of esophageal adenocarcinoma in Chinese patients reveals distinct mutational signatures and genomic alterations. Commun. Biol. 2018, 1, 174. [Google Scholar] [CrossRef]
- Kastelein, F.; Biermann, K.; Steyerberg, E.W.; Verheij, J.; Kalisvaart, M.; Looijenga, L.H.J.; Stoop, H.A.; Walter, L.; Kuipers, E.J.; Spaander, M.C.W.; et al. Aberrant p53 protein expression is associated with an increased risk of neoplastic progression in patients with Barrett’s oesophagus. Gut 2013, 62, 1676–1683. [Google Scholar] [CrossRef]
- Song, S.; Ajani, J.A.; Honjo, S.; Maru, D.M.; Chen, Q.; Scott, A.W.; Heallen, T.R.; Xiao, L.; Hofstetter, W.L.; Weston, B. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 2014, 74, 4170–4182. [Google Scholar] [CrossRef] [Green Version]
- Ajani, J.A.; Xu, Y.; Huo, L.; Wang, R.; Li, Y.; Wang, Y.; Pizzi, M.P.; Scott, A.; Harada, K.; Ma, L. YAP1 mediates gastric adenocarcinoma peritoneal metastases that are attenuated by YAP1 inhibition. Gut 2021, 70, 55–66. [Google Scholar] [CrossRef]
- Verbeek, R.E.; Leenders, M.; Ten Kate, F.J.; van Hillegersberg, R.; Vleggaar, F.P.; van Baal, J.W.; van Oijen, M.G.; Siersema, P.D. Surveillance of Barrett’s esophagus and mortality from esophageal adenocarcinoma: A population-based cohort study. Am. J. Gastroenterol. 2014, 109, 1215–1222. [Google Scholar] [CrossRef] [PubMed]
- Kerkar, S.P.; Restifo, N.P. Cellular Constituents of Immune Escape within the Tumor Microenvironment. Cancer Res. 2012, 72, 3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anand, A.; Fang, H.-Y.; Mohammad-Shahi, D.; Ingermann, J.; Baumeister, T.; Strangmann, J.; Schmid, R.M.; Wang, T.C.; Quante, M. Elimination of NF-kappaB signaling in Vimentin+ stromal cells attenuates tumorigenesis in a mouse model of Barrett’s Esophagus. Carcinogenesis 2021, 42, 405–413. [Google Scholar] [CrossRef]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.; Takashi, S.; Baik, G.H.; Shibata, W.; Diprete, B.; Betz, K.S.; et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charbord, P. Bone marrow mesenchymal stem cells: Historical overview and concepts. Hum. Gene Ther. 2010, 21, 1045–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drake, L.E.; Macleod, K.F. Tumour suppressor gene function in carcinoma-associated fibroblasts: From tumour cells via EMT and back again? J. Pathol. 2014, 232, 283–288. [Google Scholar] [CrossRef]
- Hanley, C.J.; Noble, F.; Ward, M.; Bullock, M.; Drifka, C.; Mellone, M.; Manousopoulou, A.; Johnston, H.E.; Hayden, A.; Thirdborough, S. A subset of myofibroblastic cancer-associated fibroblasts regulate collagen fiber elongation, which is prognostic in multiple cancers. Oncotarget 2016, 7, 6159. [Google Scholar] [CrossRef] [Green Version]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and Cancer associated fibroblasts in the reactive stroma and its relation to Cancer biology. J. Exp. Clin. Cancer Res. 2019, 38, 115. [Google Scholar] [CrossRef] [Green Version]
- Jedryka, M.; Chrobak, A.; Chelmonska-Soyta, A.; Gawron, D.; Halbersztadt, A.; Wojnar, A.; Kornafel, J. Matrix metalloproteinase (MMP)-2 and MMP-9 expression in tumor infiltrating CD3 lymphocytes from women with endometrial cancer. Int. J. Gynecol. Cancer 2012, 22, 1303–1309. [Google Scholar] [CrossRef]
- Garalla, H.M.; Lertkowit, N.; Tiszlavicz, L.; Reisz, Z.; Holmberg, C.; Beynon, R.; Simpson, D.; Varga, A.; Kumar, J.D.; Dodd, S. Matrix metalloproteinase (MMP)-7 in Barrett’s esophagus and esophageal adenocarcinoma: Expression, metabolism, and functional significance. Physiol. Rep. 2018, 6, e13683. [Google Scholar] [CrossRef]
- Duda, D.G.; Duyverman, A.M.; Kohno, M.; Snuderl, M.; Steller, E.J.; Fukumura, D.; Jain, R.K. Malignant cells facilitate lung metastasis by bringing their own soil. Proc. Natl. Acad. Sci. USA 2010, 107, 21677–21682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.T.; Sun, W.; Zhang, J.T.; Fan, Y.Z. Cancer-associated fibroblast regulation of tumor neo-angiogenesis as a therapeutic target in cancer. Oncol. Lett. 2019, 17, 3055–3065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- McQuaid, K.R.; Laine, L.; Fennerty, M.B.; Souza, R.; Spechler, S.J. Systematic review: The role of bile acids in the pathogenesis of gastro-oesophageal reflux disease and related neoplasia. Aliment. Pharmacol. Ther. 2011, 34, 146–165. [Google Scholar] [CrossRef]
- Rieder, F.; Cheng, L.; Harnett, K.M.; Chak, A.; Cooper, G.S.; Isenberg, G.; Ray, M.; Katz, J.A.; Catanzaro, A.; O’Shea, R. Gastroesophageal reflux disease–associated esophagitis induces endogenous cytokine production leading to motor abnormalities. Gastroenterology 2007, 132, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Münch, N.S.; Fang, H.Y.; Ingermann, J.; Maurer, H.C.; Anand, A.; Kellner, V.; Sahm, V.; Wiethaler, M.; Baumeister, T.; Wein, F.; et al. High-Fat Diet Accelerates Carcinogenesis in a Mouse Model of Barrett’s Esophagus via Interleukin 8 and Alterations to the Gut Microbiome. Gastroenterology 2019, 157, 492–506.e2. [Google Scholar] [CrossRef] [Green Version]
- Medema, J.P.; Vermeulen, L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature 2011, 474, 318–326. [Google Scholar] [CrossRef]
- Zeki, S.S.; Graham, T.A.; Wright, N.A. Stem cells and their implications for colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Kunze, B.; Wein, F.; Fang, H.-Y.; Anand, A.; Baumeister, T.; Strangmann, J.; Gerland, S.; Ingermann, J.; Münch, N.S.; Wiethaler, M. Notch signaling mediates differentiation in Barrett’s esophagus and promotes progression to adenocarcinoma. Gastroenterology 2020, 159, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Pan, B.; Liu, L.; Hu, X.; Sun, X.; Zhu, Y.; Zhang, T.; Wei, D.; Guo, Y.; Shan, J. Promotion of esophageal adenocarcinoma metastasis via Wnt/ß-catenin signal pathway by sorting nexins 3. J. Gastroenterol. Hepatol. 2020, 35, 2131–2139. [Google Scholar] [CrossRef] [PubMed]
- Che, D.; Zhang, S.; Jing, Z.; Shang, L.; Jin, S.; Liu, F.; Shen, J.; Li, Y.; Hu, J.; Meng, Q. Macrophages induce EMT to promote invasion of lung cancer cells through the IL-6-mediated COX-2/PGE2/β-catenin signalling pathway. Mol. Immunol. 2017, 90, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.-C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Galon, J.; Marincola, F.M.; Thurin, M.; Trinchieri, G.; Fox, B.A.; Gajewski, T.F.; Ascierto, P.A. The immune score as a new possible approach for the classification of cancer. J. Transl. Med. 2012, 10, 1. [Google Scholar] [CrossRef]
- Broussard, E.K.; Disis, M.L. TNM staging in colorectal cancer: T is for T cell and M is for memory. Am. Soc. Clin. Oncol. 2011, 29, 601–603. [Google Scholar] [CrossRef]
- Conroy, M.J.; Kennedy, S.A.; Doyle, S.L.; Hayes, B.; Kavanagh, M.; van der Stok, E.P.; O’Sullivan, K.; Cathcart, M.-C.; Reynolds, J.V.; Lysaght, J. A study of the immune infiltrate and patient outcomes in esophageal cancer. Carcinogenesis 2021, 42, 395–404. [Google Scholar] [CrossRef]
- Segal, N.H.; Parsons, D.W.; Peggs, K.S.; Velculescu, V.; Kinzler, K.W.; Vogelstein, B.; Allison, J.P. Epitope Landscape in Breast and Colorectal Cancer. Cancer Res. 2008, 68, 889. [Google Scholar] [CrossRef] [Green Version]
- Lagisetty, K.H.; McEwen, D.P.; Nancarrow, D.J.; Schiebel, J.G.; Ferrer-Torres, D.; Ray, D.; Frankel, T.L.; Lin, J.; Chang, A.C.; Kresty, L.A. Immune determinants of Barrett’s progression to esophageal adenocarcinoma. JCI Insight 2021, 6, e143888. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Tran, D.; Killingsworth, M.C.; Buckland, M.; Lord, R.V. Dendritic cells in Barrett’s esophagus and esophageal adenocarcinoma. J. Gastrointest. Surg. 2009, 13, 44–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, A.; Maradey-Romero, C.; Fass, R. The relationship between eosinophilic esophagitis and esophageal cancer. Dis. Esophagus 2017, 30, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravi, K.; Codipilly, D.C.; Sunjaya, D.; Fang, H.; Arora, A.S.; Katzka, D.A. Esophageal lichen planus is associated with a significant increase in risk of squamous cell carcinoma. Clin. Gastroenterol. Hepatol. 2019, 17, 1902–1903.e1901. [Google Scholar] [CrossRef]
- Romani, N.; Ratzinger, G.; Pfaller, K.; Salvenmoser, W.; Stössel, H.; Koch, F.; Stoitzner, P. Migration of dendritic cells into lymphatics—The langerhans cell example: Routes, regulation, and relevance. In International Review of Cytology; Academic Press: Cambridge, MA, USA, 2001; Volume 207, pp. 237–270. [Google Scholar]
- Veglia, F.; Tyurin, V.A.; Mohammadyani, D.; Blasi, M.; Duperret, E.K.; Donthireddy, L.; Hashimoto, A.; Kapralov, A.; Amoscato, A.; Angelini, R.; et al. Lipid bodies containing oxidatively truncated lipids block antigen cross-presentation by dendritic cells in cancer. Nat. Commun. 2017, 8, 2122. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. The tumor-draining lymph node as an immune-privileged site. Immunol. Rev. 2006, 213, 146–158. [Google Scholar] [CrossRef]
- Almand, B.; Resser, J.R.; Lindman, B.; Nadaf, S.; Clark, J.I.; Kwon, E.D.; Carbone, D.P.; Gabrilovich, D.I. Clinical significance of defective dendritic cell differentiation in cancer. Clin. Cancer Res. 2000, 6, 1755–1766. [Google Scholar]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Kollmann, D.; Ignatova, D.; Jedamzik, J.; Chang, Y.-T.; Jomrich, G.; Baierl, A.; Kazakov, D.; Michal, M.; French, L.E.; Hoetzenecker, W. PD-L1 expression is an independent predictor of favorable outcome in patients with localized esophageal adenocarcinoma. Oncoimmunology 2018, 7, e1435226. [Google Scholar] [CrossRef]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.-j.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+ CD25− naive T cells to CD4+ CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.H.; Lin, Z.Y.; Huang, C.J.; Shih, M.C.; Chuang, W.L. Management of bilateral adrenal metastases from hepatocellular carcinoma: A case report. Kaohsiung J. Med. Sci. 2005, 21, 371–376. [Google Scholar] [CrossRef] [Green Version]
- McShane, R.; Arya, S.; Stewart, A.J.; Caie, P.D.; Bates, M. Prognostic features of the tumour microenvironment in oesophageal adenocarcinoma. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1876, 188598. [Google Scholar] [CrossRef] [PubMed]
- Erdman, S.E.; Rao, V.P.; Poutahidis, T.; Ihrig, M.M.; Ge, Z.; Feng, Y.; Tomczak, M.; Rogers, A.B.; Horwitz, B.H.; Fox, J.G. CD4 CD25 Regulatory lymphocytes require Il10 to interrupt colon carcinogenesis in mice. Cancer Res. 2003, 63, 6042–6050. [Google Scholar] [PubMed]
- Erdman, S.E.; Poutahidis, T.; Tomczak, M.; Rogers, A.B.; Cormier, K.; Plank, B.; Horwitz, B.H.; Fox, J.G. CD4+ CD25+ Regulatory T Lymphocytes Inhibit Microbially Induced Colon Cancer in Rag2-Deficient Mice. Am. J. Pathol. 2003, 162, 691–702. [Google Scholar] [CrossRef] [Green Version]
- Erdman, S.E.; Sohn, J.J.; Rao, V.P.; Nambiar, P.R.; Ge, Z.; Fox, J.G.; Schauer, D.B. CD4+CD25+ regulatory lymphocytes induce regression of intestinal tumors in ApcMin/+ mice. Cancer Res. 2005, 65, 3998–4004. [Google Scholar] [CrossRef] [Green Version]
- Gounaris, E.; Blatner, N.R.; Dennis, K.; Magnusson, F.; Gurish, M.F.; Strom, T.B.; Beckhove, P.; Gounari, F.; Khazaie, K. T-Regulatory Cells Shift from a Protective Anti-Inflammatory to a Cancer-Promoting Proinflammatory Phenotype in Polyposis. Cancer Res. 2009, 69, 5490–5497. [Google Scholar] [CrossRef] [Green Version]
- Maniati, E.; Soper, R.; Hagemann, T. Up for Mischief? IL-17/Th17 in the tumour microenvironment. Oncogene 2010, 29, 5653–5662. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Hu, Q.; Mao, C.; Jiao, Z.; Wang, S.; Yu, L.; Xu, Y.; Dai, D.; Yin, L.; Xu, H. Increased IL-17-producing CD4+ T cells in patients with esophageal cancer. Cell. Immunol. 2012, 272, 166–174. [Google Scholar] [CrossRef]
- Bannister, J.R.; Khan, A.L.; Eccleston, D.W.; Deol-Poonia, R.K.; Hughes, S.F. Interleukin-17 expression in the Barrett’s metaplasia-dysplasia-adenocarcinoma sequence. Int. Sch. Res. Not. 2012, 2012, 578149. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.; Hellstrom, I.; Yip, Y.Y.; Sjögren, H.O.; Hellstrom, K.E. Tumor regression and cure depends on sustained Th1 responses. J. Immunother. 2018, 41, 369. [Google Scholar] [CrossRef] [PubMed]
- Ostrand-Rosenberg, S. Immune surveillance: A balance between protumor and antitumor immunity. Curr. Opin. Genet. Dev. 2008, 18, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, R.; Onwuegbusi, B.; Bajaj-Elliott, M.; Saeed, I.; Burnham, W.; Farthing, M. Diversity in the oesophageal phenotypic response to gastro-oesophageal reflux: Immunological determinants. Gut 2002, 50, 451–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohata, Y.; Fujiwara, Y.; Machida, H.; Okazaki, H.; Yamagami, H.; Tanigawa, T.; Watanabe, K.; Watanabe, T.; Tominaga, K.; Wei, M. Role of Th-2 cytokines in the development of Barrett’s esophagus in rats. J. Gastroenterol. 2011, 46, 883–893. [Google Scholar] [CrossRef] [PubMed]
- van Sandick, J.W.; Boermeester, M.A.; Gisbertz, S.S.; Ten Berge, I.J.; Out, T.A.; van der Pouw Kraan, T.C.; van Lanschot, J.J.B. Lymphocyte subsets and T h 1/T h 2 immune responses in patients with adenocarcinoma of the oesophagus or oesophagogastric junction: Relation to pTNM stage and clinical outcome. Cancer Immunol. Immunother. 2003, 52, 617–624. [Google Scholar] [CrossRef]
- O’riordan, J.; Abdel-Latif, M.; Ravi, N.; McNamara, D.; Byrne, P.; McDonald, G.; Keeling, P.; Kelleher, D.; Reynolds, J. Proinflammatory cytokine and nuclear factor kappa-B expression along the inflammation–metaplasia–dysplasia–adenocarcinoma sequence in the esophagus. Off. J. Am. Coll. Gastroenterol.|ACG 2005, 100, 1257–1264. [Google Scholar] [CrossRef]
- Watanabe, S.; Deguchi, K.; Zheng, R.; Tamai, H.; Wang, L.X.; Cohen, P.A.; Shu, S. Tumor-induced CD11b+Gr-1+ myeloid cells suppress T cell sensitization in tumor-draining lymph nodes. J. Immunol. 2008, 181, 3291–3300. [Google Scholar] [CrossRef] [Green Version]
- Marx, J. Cancer immunology. Cancer’s bulwark against immune attack: MDS cells. Science 2008, 319, 154–156. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Arimura, S.; Matsunaga, A.; Kitamura, T.; Aoki, K.; Aoki, M.; Taketo, M.M. Reduced level of smoothened suppresses intestinal tumorigenesis by down-regulation of Wnt signaling. Gastroenterology 2009, 137, 629–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Biyajima, K.; Aoki, M.; Oshima, M.; Taketo, M.M. Matrix metalloproteinase 7 is required for tumor formation, but dispensable for invasion and fibrosis in SMAD4-deficient intestinal adenocarcinomas. Lab. Investig. 2009, 89, 98–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, T.; Kometani, K.; Hashida, H.; Matsunaga, A.; Miyoshi, H.; Hosogi, H.; Aoki, M.; Oshima, M.; Hattori, M.; Takabayashi, A.; et al. SMAD4-deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nat. Genet. 2007, 39, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Taketo, M.M. Role of bone marrow-derived cells in colon cancer: Lessons from mouse model studies. J Gastroenterol 2009, 44, 93–102. [Google Scholar] [CrossRef]
- Weber, R.; Groth, C.; Lasser, S.; Arkhypov, I.; Petrova, V.; Altevogt, P.; Utikal, J.; Umansky, V. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell. Immunol. 2021, 359, 104254. [Google Scholar] [CrossRef]
- Karakasheva, T.A.; Lin, E.W.; Tang, Q.; Qiao, E.; Waldron, T.J.; Soni, M.; Klein-Szanto, A.J.; Sahu, V.; Basu, D.; Ohashi, S.; et al. IL-6 Mediates Cross-Talk between Tumor Cells and Activated Fibroblasts in the Tumor Microenvironment. Cancer Res. 2018, 78, 4957–4970. [Google Scholar] [CrossRef] [Green Version]
- Asfaha, S.; Dubeykovskiy, A.N.; Tomita, H.; Yang, X.; Stokes, S.; Shibata, W.; Friedman, R.A.; Ariyama, H.; Dubeykovskaya, Z.A.; Muthupalani, S.; et al. Mice That Express Human Interleukin-8 Have Increased Mobilization of Immature Myeloid Cells, Which Exacerbates Inflammation and Accelerates Colon Carcinogenesis. Gastroenterology 2013, 144, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Bae, J.-S. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediat. Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [Green Version]
- Eisinger, S.; Sarhan, D.; Boura, V.F.; Ibarlucea-Benitez, I.; Tyystjärvi, S.; Oliynyk, G.; Arsenian-Henriksson, M.; Lane, D.; Wikström, S.L.; Kiessling, R. Targeting a scavenger receptor on tumor-associated macrophages activates tumor cell killing by natural killer cells. Proc. Natl. Acad. Sci. USA 2020, 117, 32005–32016. [Google Scholar] [CrossRef]
- Cao, W.; Peters, J.H.; Nieman, D.; Sharma, M.; Watson, T.; Yu, J. Macrophage subtype predicts lymph node metastasis in oesophageal adenocarcinoma and promotes cancer cell invasion in vitro. Br. J. Cancer 2015, 113, 738–746. [Google Scholar] [CrossRef] [Green Version]
- Yakupova, E.I.; Maleev, G.V.; Krivtsov, A.V.; Plotnikov, E.Y. Macrophage polarization in hypoxia and ischemia/reperfusion: Insights into the role of energetic metabolism. Exp. Biol. Med. 2022, 15353702221080130. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, M.; Liu, S.; Guo, J.; Lu, Y.; Cheng, J.; Liu, J. Macrophage-derived extracellular vesicles: Diverse mediators of pathology and therapeutics in multiple diseases. Cell Death Dis. 2020, 11, 924. [Google Scholar] [CrossRef] [PubMed]
- Piccard, H.; Muschel, R.; Opdenakker, G. On the dual roles and polarized phenotypes of neutrophils in tumor development and progression. Crit. Rev. Oncol./Hematol. 2012, 82, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, A.; Brooks, M.W.; Houshyar, S.; Reinhardt, F.; Ardolino, M.; Fessler, E.; Chen, M.B.; Krall, J.A.; DeCock, J.; Zervantonakis, I.K.; et al. Neutrophils Suppress Intraluminal NK Cell–Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discov. 2016, 6, 630–649. [Google Scholar] [CrossRef] [Green Version]
- Mandó, P.; Rivero, S.G.; Rizzo, M.M.; Pinkasz, M.; Levy, E.M. Targeting ADCC: A different approach to HER2 breast cancer in the immunotherapy era. Breast 2021, 60, 15–25. [Google Scholar] [CrossRef]
- Dahlberg, C.I.; Sarhan, D.; Chrobok, M.; Duru, A.D.; Alici, E. Natural killer cell-based therapies targeting cancer: Possible strategies to gain and sustain anti-tumor activity. Front. Immunol. 2015, 6, 605. [Google Scholar] [CrossRef] [Green Version]
- Svensson, M.C.; Warfvinge, C.F.; Fristedt, R.; Hedner, C.; Borg, D.; Eberhard, J.; Micke, P.; Nodin, B.; Leandersson, K.; Jirström, K. The integrative clinical impact of tumor-infiltrating T lymphocytes and NK cells in relation to B lymphocyte and plasma cell density in esophageal and gastric adenocarcinoma. Oncotarget 2017, 8, 72108. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Z.-J.; Gao, J.-J.; Hua, S.-H.; Chen, D.-Y.; Wang, W.-H.; Wang, H.; Wang, X.-H.; Xu, H.-X. Correlation between circulating myeloid-derived suppressor cells and Th17 cells in esophageal cancer. World J. Gastroenterol. WJG 2012, 18, 5454. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, R.; Wu, J.; Pu, Y.; Yin, X.; Cheng, Y.; Wu, J.; Feng, C.; Luo, Y.; Zhang, J. Interleukin-17A promotes esophageal adenocarcinoma cell invasiveness through ROS-dependent, NF-κB-mediated MMP-2/9 activation. Oncol. Rep. 2017, 37, 1779–1785. [Google Scholar] [CrossRef] [Green Version]
- Sekikawa, A.; Fukui, H.; Suzuki, K.; Karibe, T.; Fujii, S.; Ichikawa, K.; Tomita, S.; Imura, J.; Shiratori, K.; Chiba, T. Involvement of the IL-22/REG Iα axis in ulcerative colitis. Lab. Investig. 2010, 90, 496–505. [Google Scholar] [CrossRef] [Green Version]
- Kortylewski, M.; Xin, H.; Kujawski, M.; Lee, H.; Liu, Y.; Harris, T.; Drake, C.; Pardoll, D.; Yu, H. Regulation of the IL-23 and IL-12 balance by Stat3 signaling in the tumor microenvironment. Cancer Cell 2009, 15, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Laurence, A.; Kanno, Y.; Pacher-Zavisin, M.; Zhu, B.M.; Tato, C.; Yoshimura, A.; Hennighausen, L.; O’Shea, J.J. Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 8137–8142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Bhat, A.A.; Lu, H.; Soutto, M.; Capobianco, A.; Rai, P.; Zaika, A.; El-Rifai, W. Exposure of Barrett’s and esophageal adenocarcinoma cells to bile acids activates EGFR–STAT3 signaling axis via induction of APE1. Oncogene 2018, 37, 6011–6024. [Google Scholar] [CrossRef]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef]
- Snider, E.J.; Freedberg, D.E.; Abrams, J.A. Potential Role of the Microbiome in Barrett’s Esophagus and Esophageal Adenocarcinoma. Dig. Dis. Sci. 2016, 61, 2217–2225. [Google Scholar] [CrossRef] [Green Version]
- Arora, M.; Poe, S.L.; Oriss, T.B.; Krishnamoorthy, N.; Yarlagadda, M.; Wenzel, S.E.; Billiar, T.R.; Ray, A.; Ray, P. TLR4/MyD88-induced CD11b+Gr-1 int F4/80+ non-migratory myeloid cells suppress Th2 effector function in the lung. Mucosal Immunol. 2010, 3, 578–593. [Google Scholar] [CrossRef] [Green Version]
- Snider, E.J.; Compres, G.; Freedberg, D.E.; Khiabanian, H.; Nobel, Y.R.; Stump, S.; Uhlemann, A.C.; Lightdale, C.J.; Abrams, J.A. Alterations to the Esophageal Microbiome Associated with Progression from Barrett’s Esophagus to Esophageal Adenocarcinoma. Cancer Epidemiol. Prev. Biomark. 2019, 28, 1687–1693. [Google Scholar] [CrossRef] [Green Version]
- Guccione, C.; Yadlapati, R.; Shah, S.; Knight, R.; Curtius, K. Challenges in Determining the Role of Microbiome Evolution in Barrett’s Esophagus and Progression to Esophageal Adenocarcinoma. Microorganisms 2021, 9, 2003. [Google Scholar] [CrossRef]
- Lv, J.; Guo, L.; Liu, J.-J.; Zhao, H.-P.; Zhang, J.; Wang, J.-H. Alteration of the esophageal microbiota in Barrett’s esophagus and esophageal adenocarcinoma. World J. Gastroenterol. 2019, 25, 2149. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.K.; Krishnan, K.; Samala, N.; Singh, J.; Cluley, J.; Perla, S.; Howden, C.W. The incidence of oesophageal adenocarcinoma in non-dysplastic Barrett’s oesophagus: A meta-analysis. Gut 2012, 61, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, N.J.; Falk, G.W.; Iyer, P.G.; Gerson, L.B. ACG clinical guideline: Diagnosis and management of Barrett’s esophagus. Off. J. Am. Coll. Gastroenterol.|ACG 2016, 111, 30–50. [Google Scholar] [CrossRef] [PubMed]
- Dam, A.N.; Klapman, J. A narrative review of Barrett’s esophagus in 2020, molecular and clinical update. Ann. Transl. Med. 2020, 8, 1107. [Google Scholar] [CrossRef]
- Iqbal, U.; Siddique, O.; Ovalle, A.; Anwar, H.; Moss, S.F. Safety and efficacy of a minimally invasive cell sampling device (‘Cytosponge’) in the diagnosis of esophageal pathology: A systematic review. Eur. J. Gastroenterol. Hepatol. 2018, 30, 1261–1269. [Google Scholar] [CrossRef]
- Glamour, B.K.; Alaber, O.; Cioffi, G.; Chandar, A.K.; Barnholtz-Sloan, J.; Brock, W.; Falk, G.W.; Canto, M.I.; Wang, J.S.; Iyer, P.G. Age of diagnosis in familial Barrett’s associated neoplasia. Fam. Cancer 2022, 21, 115–120. [Google Scholar] [CrossRef]
- Ross-Innes, C.S.; Becq, J.; Warren, A.; Cheetham, R.K.; Northen, H.; O’Donovan, M.; Malhotra, S.; Di Pietro, M.; Ivakhno, S.; He, M. Whole-genome sequencing provides new insights into the clonal architecture of Barrett’s esophagus and esophageal adenocarcinoma. Nat. Genet. 2015, 47, 1038–1046. [Google Scholar] [CrossRef]
- Bird–Lieberman, E.L.; Dunn, J.M.; Coleman, H.G.; Lao–Sirieix, P.; Oukrif, D.; Moore, C.E.; Varghese, S.; Johnston, B.T.; Arthur, K.; McManus, D.T. Population-based study reveals new risk-stratification biomarker panel for Barrett’s esophagus. Gastroenterology 2012, 143, 927–935.e923. [Google Scholar] [CrossRef]
- Verbeek, R.E.; Spittuler, L.F.; Peute, A.; van Oijen, M.G.; Fiebo, J.; Vermeijden, J.R.; Oberndorff, A.; van Baal, J.W.; Siersema, P.D. Familial clustering of Barrett’s esophagus and esophageal adenocarcinoma in a European cohort. Clin. Gastroenterol. Hepatol. 2014, 12, 1656–1663.e1651. [Google Scholar] [CrossRef]
- Fang, H.-Y.; Münch, N.S.; Schottelius, M.; Ingermann, J.; Liu, H.; Schauer, M.; Stangl, S.; Multhoff, G.; Steiger, K.; Gerngroß, C.; et al. CXCR4 is a potential target for diagnostic PET/CT imaging in Barrett’s dysplasia and esophageal adenocarcinoma. Clin. Cancer Res. 2018, 24, 1048–1061. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, D.J.; Januszewicz, W.; Ali, S.; Fitzgerald, R.C.; di Pietro, M.; Bohndiek, S.E. Spectral Endoscopy Enhances Contrast for Neoplasia in Surveillance of Barrett’s Esophagus. Cancer Res. 2021, 81, 3415. [Google Scholar] [CrossRef] [PubMed]
- de Jongh, S.J.; Voskuil, F.J.; Schmidt, I.; Karrenbeld, A.; Kats-Ugurlu, G.; Meersma, G.J.; Westerhof, J.; Witjes, M.J.H.; van Dam, G.M.; Robinson, D.J.; et al. C-Met targeted fluorescence molecular endoscopy in Barrett’s esophagus patients and identification of outcome parameters for phase-I studies. Theranostics 2020, 10, 5357–5367. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.-Y.; Stangl, S.; Marcazzan, S.; Carvalho, M.J.B.; Baumeister, T.; Anand, A.; Strangmann, J.; Huspenina, J.S.; Wang, T.C.; Schmid, R.M.; et al. Targeted Hsp70 fluorescence molecular endoscopy detects dysplasia in Barrett’s esophagus. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 2049–2063. [Google Scholar] [CrossRef] [PubMed]
- Marcazzan, S.; Braz Carvalho, M.J.; Konrad, M.; Strangmann, J.; Tenditnaya, A.; Baumeister, T.; Schmid, R.M.; Wester, H.J.; Ntziachristos, V.; Gorpas, D.; et al. CXCR4 peptide-based fluorescence endoscopy in a mouse model of Barrett’s esophagus. EJNMMI Res. 2022, 12, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Pan, Z. Influence of microbiota on immunity and immunotherapy for gastric and esophageal cancers. Gastroenterol. Rep. 2020, 8, 206–214. [Google Scholar] [CrossRef]
- Ajayi, T.A.; Cantrell, S.; Spann, A.; Garman, K.S. Barrett’s esophagus and esophageal cancer: Links to microbes and the microbiome. PLoS Pathog. 2018, 14, e1007384. [Google Scholar] [CrossRef]
- Boursi, B.; Mamtani, R.; Haynes, K.; Yang, Y.-X. Recurrent antibiotic exposure may promote cancer formation–another step in understanding the role of the human microbiota? Eur. J. Cancer 2015, 51, 2655–2664. [Google Scholar] [CrossRef] [Green Version]
- Baumeister, T.; Ingermann, J.; Marcazzan, S.; Fang, H.Y.; Oellinger, R.; Rad, R.; Engleitner, T.; Kleigrewe, K.; Anand, A.; Strangmann, J.; et al. Anti-inflammatory chemoprevention attenuates the phenotype in a mouse model of esophageal adenocarcinoma. Carcinogenesis 2021, 42, 1068–1078. [Google Scholar] [CrossRef]
- Jankowski, J.A.Z.; de Caestecker, J.; Love, S.B.; Reilly, G.; Watson, P.; Sanders, S.; Ang, Y.; Morris, D.; Bhandari, P.; Brooks, C.; et al. Esomeprazole and aspirin in Barrett’s oesophagus (AspECT): A randomised factorial trial. Lancet 2018, 392, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Singh, A.G.; Singh, P.P.; Murad, M.H.; Iyer, P.G. Statins are associated with reduced risk of esophageal cancer, particularly in patients with Barrett’s esophagus: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2013, 11, 620–629. [Google Scholar] [CrossRef] [Green Version]
- Kastelein, F.; Spaander, M.C.; Biermann, K.; Steyerberg, E.W.; Kuipers, E.J.; Bruno, M.J.; Group, P.-S. Nonsteroidal anti-inflammatory drugs and statins have chemopreventative effects in patients with Barrett’s esophagus. Gastroenterology 2011, 141, 2000–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartzberg, L.; Kim, E.S.; Liu, D.; Schrag, D. Precision oncology: Who, how, what, when, and when not? Am. Soc. Clin. Oncol. Educ. Book 2017, 37, 160–169. [Google Scholar] [CrossRef] [PubMed]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective Derivation of a Living Organoid Biobank of Colorectal Cancer Patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, J.; Xie, Y.; Qu, L.; Wang, L.; Zhou, S.; Wang, Y.; Fan, Z.; Yang, S.; Jiao, S. A nomogram-based immunoprofile predicts overall survival for previously untreated patients with esophageal squamous cell carcinoma after esophagectomy. J. Immunother. Cancer 2018, 6, 100. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borgmann, M.; Quante, M. Impact of the Tumor Microenvironment for Esophageal Tumor Development—An Opportunity for Prevention? Cancers 2022, 14, 2246. https://doi.org/10.3390/cancers14092246
Borgmann M, Quante M. Impact of the Tumor Microenvironment for Esophageal Tumor Development—An Opportunity for Prevention? Cancers. 2022; 14(9):2246. https://doi.org/10.3390/cancers14092246
Chicago/Turabian StyleBorgmann, Martin, and Michael Quante. 2022. "Impact of the Tumor Microenvironment for Esophageal Tumor Development—An Opportunity for Prevention?" Cancers 14, no. 9: 2246. https://doi.org/10.3390/cancers14092246
APA StyleBorgmann, M., & Quante, M. (2022). Impact of the Tumor Microenvironment for Esophageal Tumor Development—An Opportunity for Prevention? Cancers, 14(9), 2246. https://doi.org/10.3390/cancers14092246