LDH-A—Modulation and the Variability of LDH Isoenzyme Profiles in Murine Gliomas: A Link with Metabolic and Growth Responses


 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. Generation of LDH-A Knock-Down and Control Cell Lines
2.3. Western Blotting
2.4. LDH Enzyme Activity
2.5. Measurement of Changes in Cellular Bioenergetics in Real Time
2.6. mRNA Gene Expression Profile Analysis
2.7. Proliferation Assay In Vitro
2.8. Animal Models
2.9. LDH Zymography
2.10. Immunohistochemical Staining and Image Analyses
2.11. Statistical Analysis
3. Results
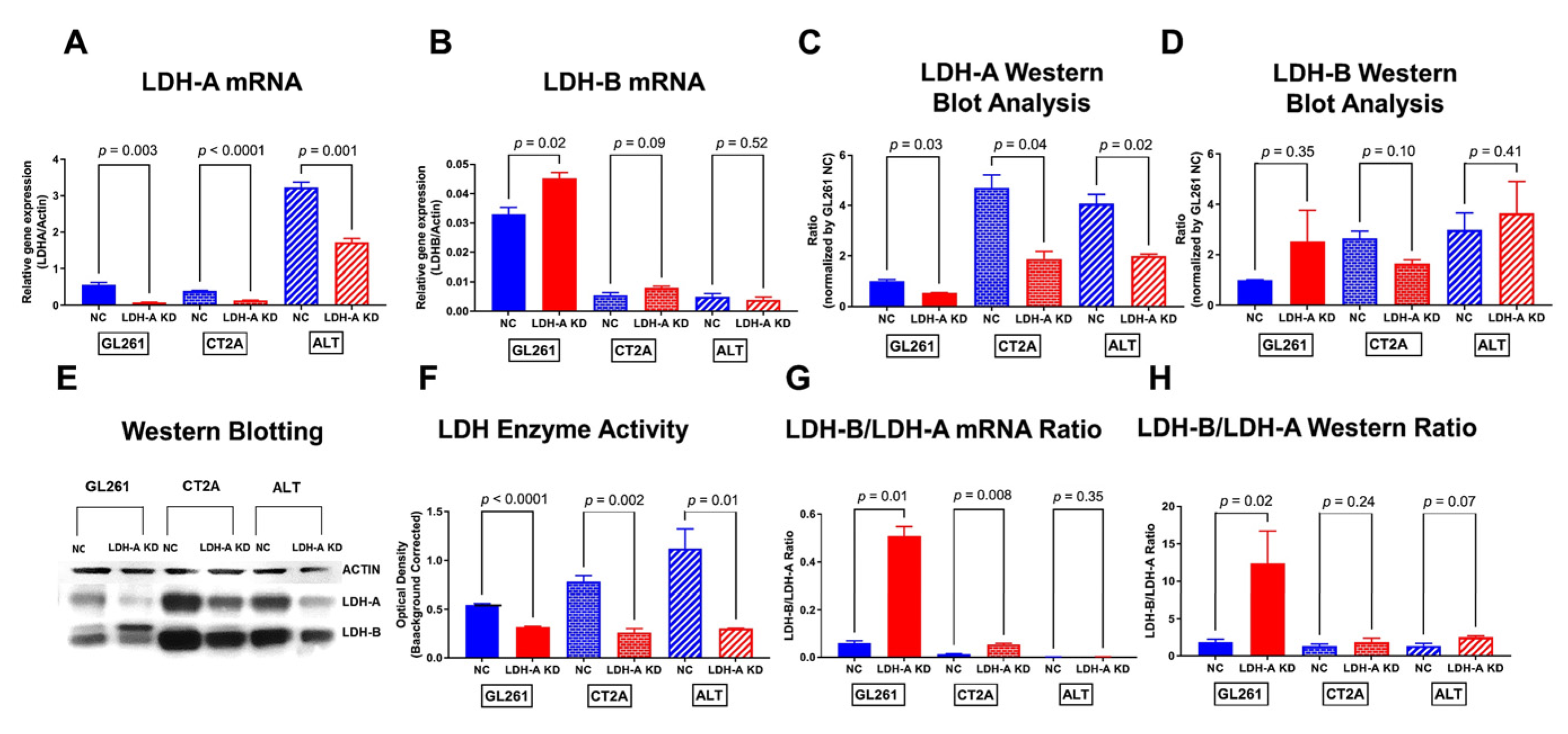
3.1. Effects of LDH-A Knock-Down on Murine Glioma Cells
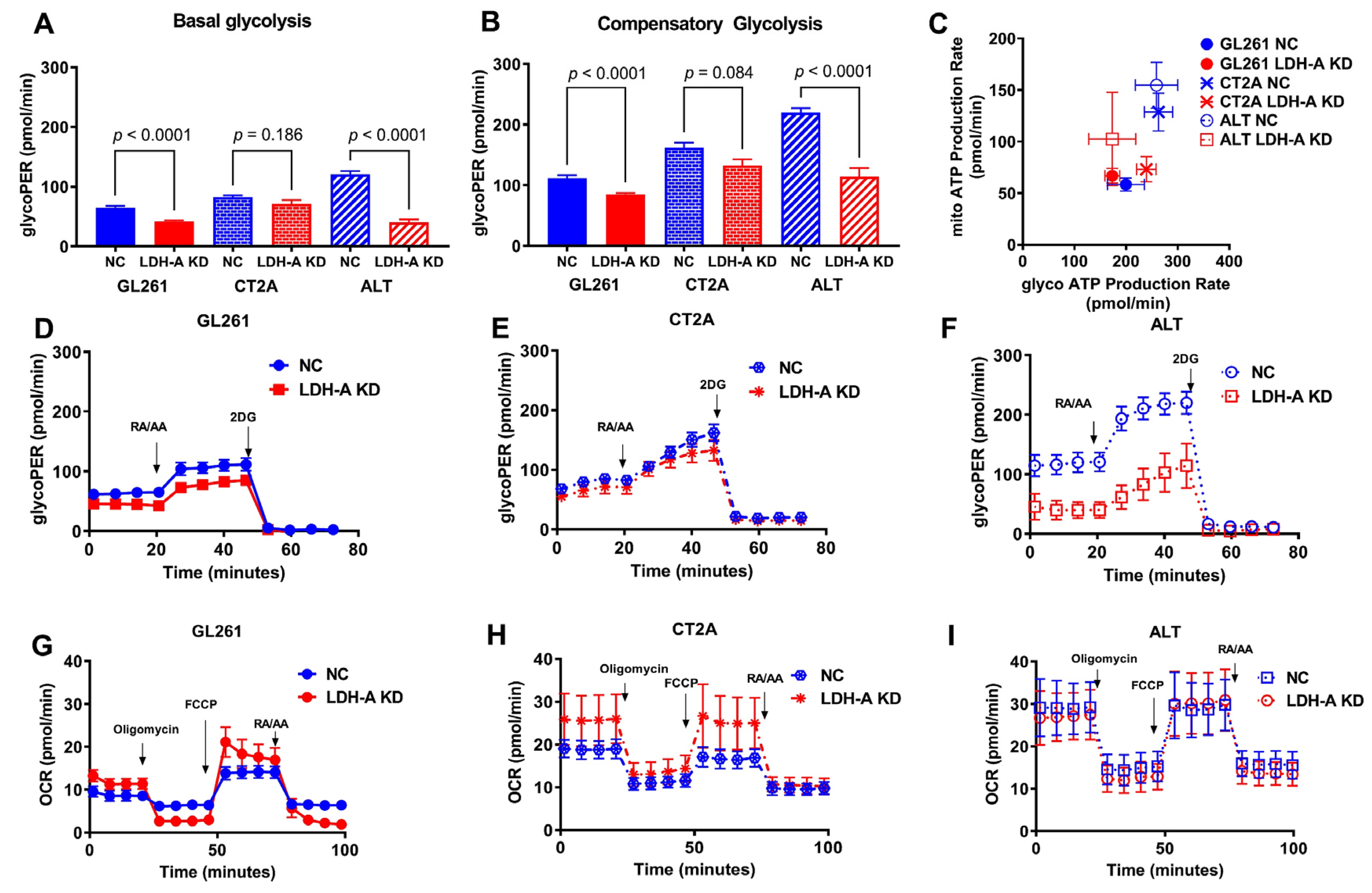
3.2. Changes in Metabolic Properties of Glioma Cells Following LDH-A Knock-Down
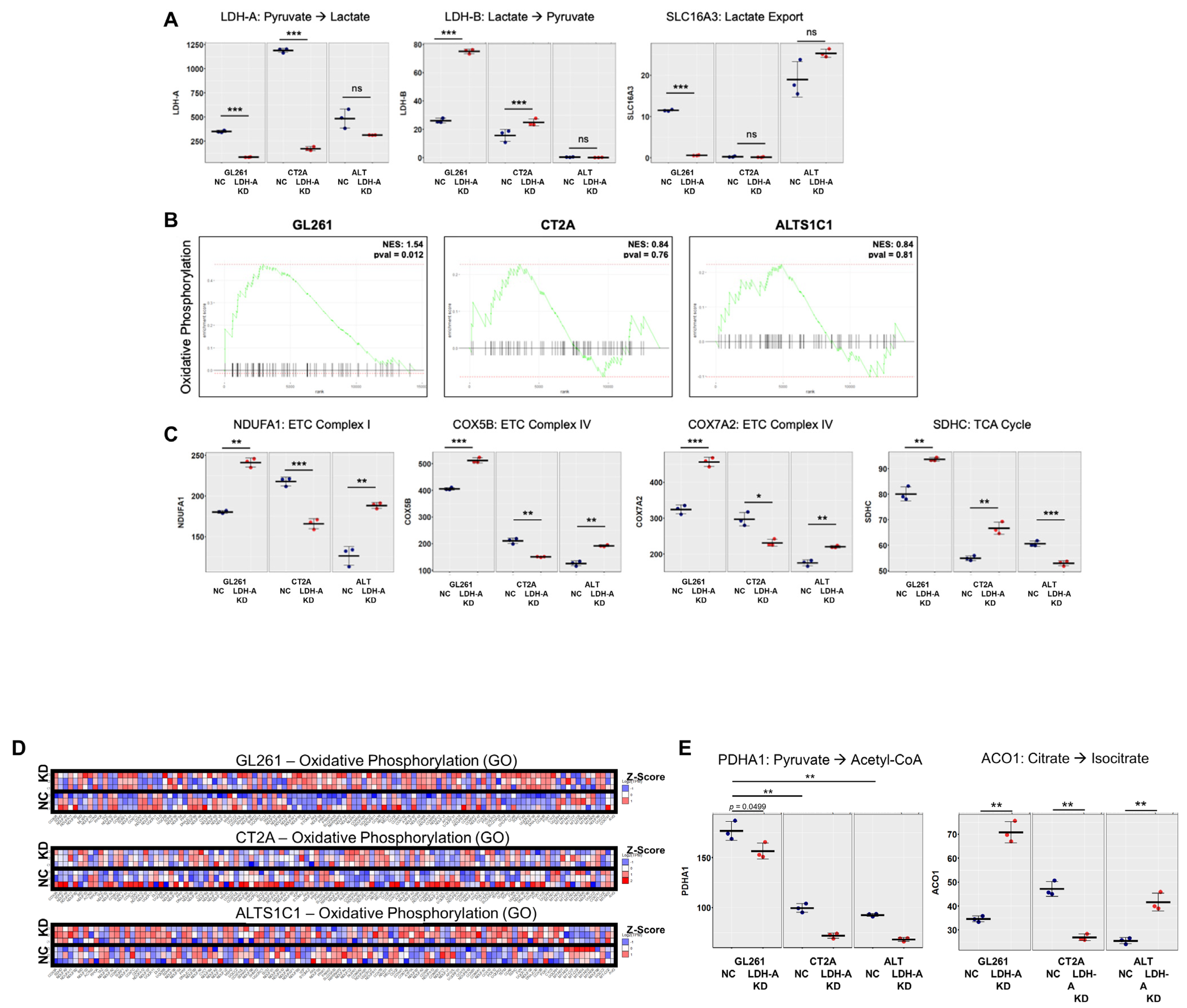
3.3. Changes in mRNA Expression Levels of Glioma Cells Following LDH-A Knock-Down
3.4. Effects of LDH-A Knock-Down In Vitro and on Tumor Growth Profiles in Immune-Competent and Incompetent Animals
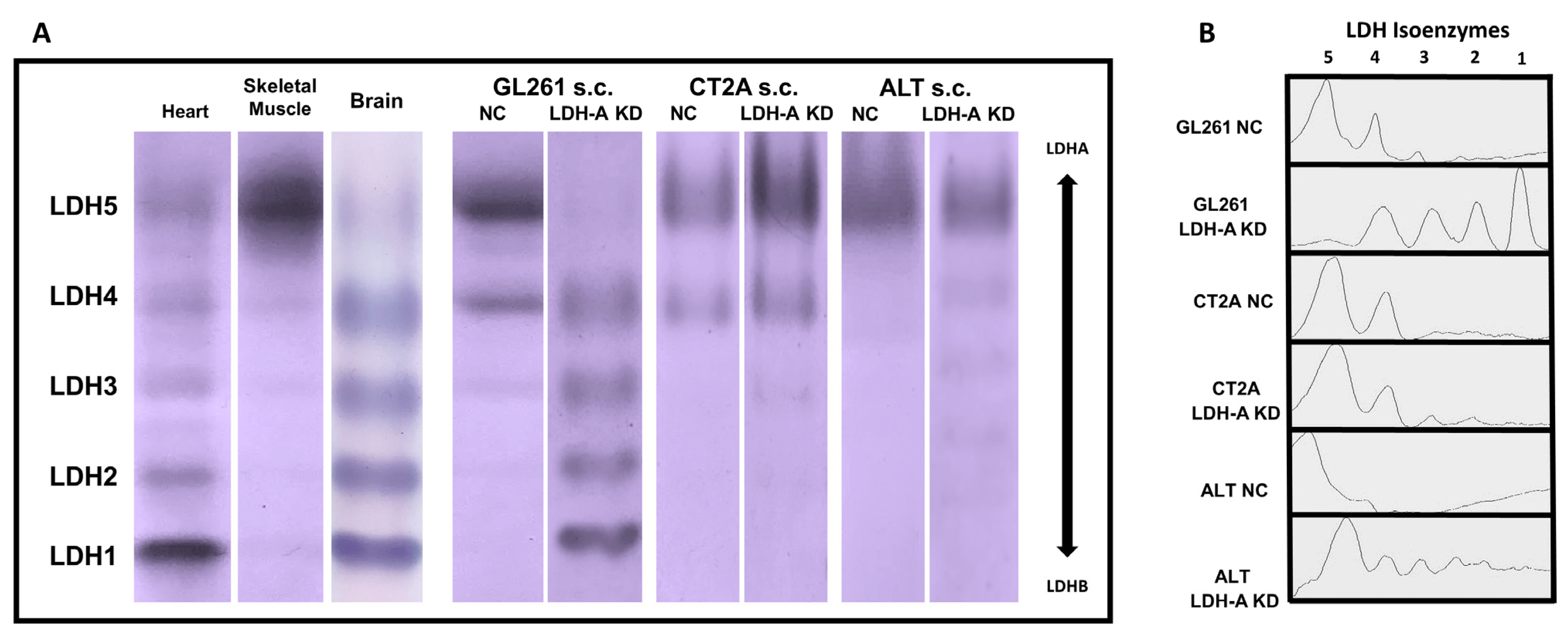
3.5. LDH Isoenzyme Pattern of Subcutaneously Located Murine Glioblastoma Tumors
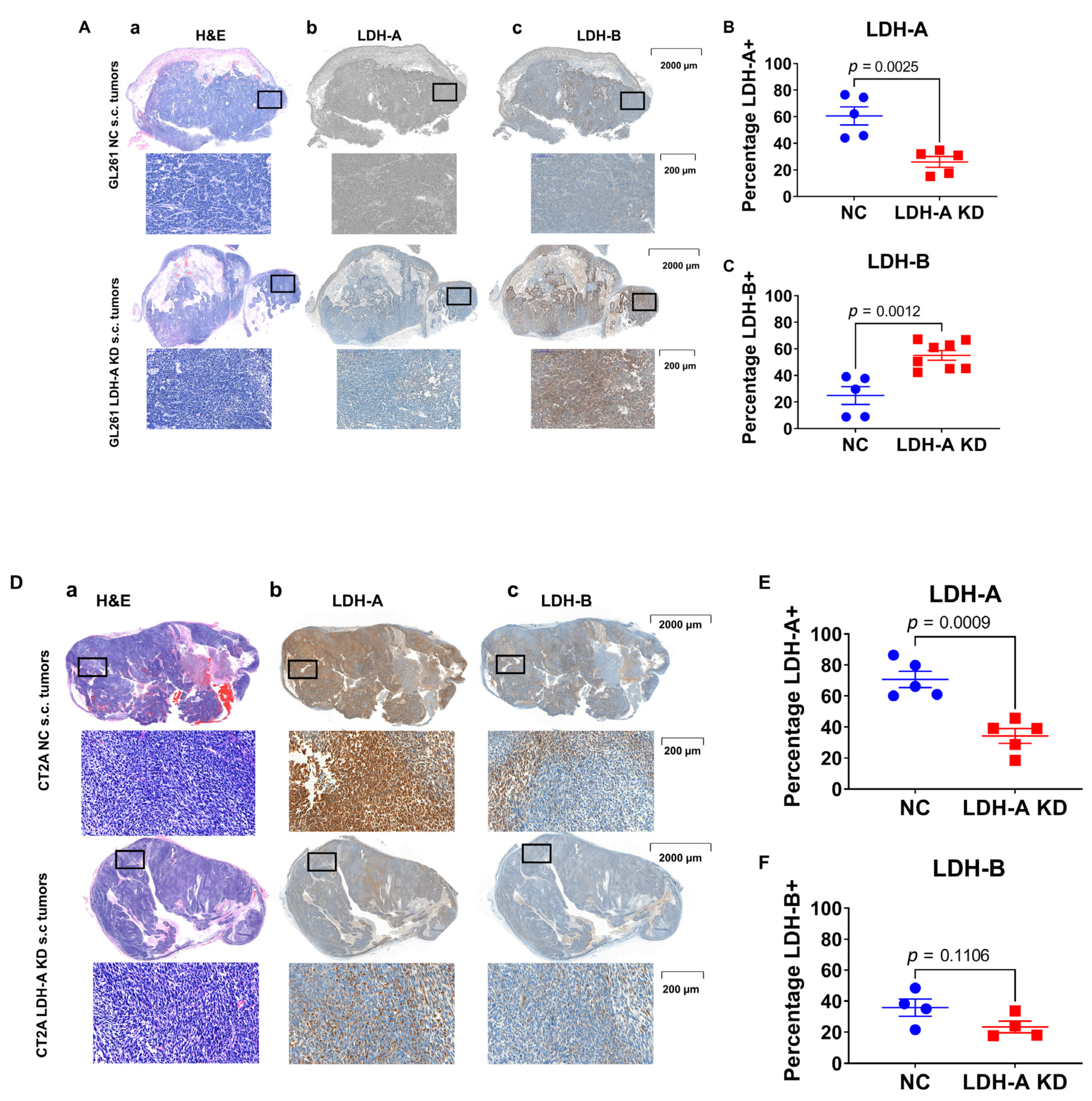
3.6. LDH-A and LDH-B Expression Patterns in Tumors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brooks, G.A. The Science and Translation of Lactate Shuttle Theory. Cell Metab. 2018, 27, 757–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.J.; Mahieu, N.G.; Huang, X.J.; Singh, M.; Crawford, P.A.; Johnson, S.L.; Gross, R.W.; Schaefer, J.; Patti, G.J. Lactate metabolism is associated with mammalian mitochondria. Nat. Chem. Biol. 2016, 12, 937–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinowitz, J.D.; Enerback, S. Lactate: The ugly duckling of energy metabolism. Nat. Metab. 2020, 2, 566–571. [Google Scholar] [CrossRef] [PubMed]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef] [Green Version]
- Chirasani, S.R.; Leukel, P.; Gronwald, W.; Gottfried, E.; Stadler, K.; Bogdahn, U.; Hau, P.; Kreutz, M.; Grauer, O.M. Diclofenac Inhibits Lactate Formation and Efficiently Counteracts Local Immune Suppression in a Murine Glioma Model. Neuro-Oncology 2012, 14, 23. [Google Scholar] [CrossRef]
- Griguer, C.E.; Oliva, C.R.; Gillespie, G.Y. Glucose metabolism heterogeneity in human and mouse malignant glioma cell lines. J. Neurooncol. 2005, 74, 123–133. [Google Scholar] [CrossRef]
- Daniele, S.; Giacomelli, C.; Zappelli, E.; Granchi, C.; Trincavelli, M.L.; Minutolo, F.; Martini, C. Lactate dehydrogenase-A inhibition induces human glioblastoma multiforme stem cell differentiation and death. Sci. Rep. 2015, 5, 15556. [Google Scholar] [CrossRef] [Green Version]
- Koukourakis, M.I.; Kakouratos, C.; Kalamida, D.; Bampali, Z.; Mavropoulou, S.; Sivridis, E.; Giatromanolaki, A. Hypoxia-inducible proteins HIF1alpha and lactate dehydrogenase LDH5, key markers of anaerobic metabolism, relate with stem cell markers and poor post-radiotherapy outcome in bladder cancer. Int. J. Radiat. Biol. 2016, 92, 353–363. [Google Scholar] [CrossRef]
- Serganova, I.; Rizwan, A.; Khanin, R.; Ni, X.; Karabeber, H.; Zakian, K.; Thakur, S.; Koutcher, J.A.; Blasberg, R. Linking LDH-A, metastases and lactate identified by MRS imaging. J. Nucl. Med. 2013, 54, 22–23. [Google Scholar]
- Cui, J.J.; Shi, M.; Xie, D.C.; Wei, D.Y.; Jia, Z.L.; Zheng, S.J.; Gao, Y.; Huang, S.Y.; Xie, K.P. FOXM1 Promotes the Warburg Effect and Pancreatic Cancer Progression via Transactivation of LDHA Expression. Clin. Cancer Res. 2014, 20, 2595–2606. [Google Scholar] [CrossRef] [Green Version]
- Rizwan, A.; Serganova, I.; Khanin, R.; Karabeber, H.; Ni, X.; Thakur, S.; Zakian, K.L.; Blasberg, R.; Koutcher, J.A. Relationships between LDH-A, lactate, and metastases in 4T1 breast tumors. Clin. Cancer Res. 2013, 19, 5158–5169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serganova, I.; Cohen, I.J.; Vemuri, K.; Shindo, M.; Maeda, M.; Mane, M.; Moroz, E.; Khanin, R.; Satagopan, J.; Koutcher, J.A.; et al. LDH-A regulates the tumor microenvironment via HIF-signaling and modulates the immune response. PLoS ONE 2018, 13, e0203965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, D.M.; Goodfriend, T.L.; Kaplan, N.O. Lactic dehydrogenases: Functions of the two types rates of synthesis of the two major forms can be correlated with metabolic differentiation. Science 1964, 143, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Hussien, R.; Brooks, G.A. Mitochondrial and plasma membrane lactate transporter and lactate dehydrogenase isoform expression in breast cancer cell lines. Physiol. Genom. 2011, 43, 255–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanska, K.; Orzechowski, A. Unappreciated Role of LDHA and LDHB to Control Apoptosis and Autophagy in Tumor Cells. Int. J. Mol. Sci. 2019, 20, 2085. [Google Scholar] [CrossRef] [Green Version]
- Brooks, G.A. Cell-cell and intracellular lactate shuttles. J. Physiol. 2009, 587, 5591–5600. [Google Scholar] [CrossRef]
- Rong, Y.; Wu, W.; Ni, X.; Kuang, T.; Jin, D.; Wang, D.; Lou, W. Lactate dehydrogenase A is overexpressed in pancreatic cancer and promotes the growth of pancreatic cancer cells. Tumour Biol. 2013, 34, 1523–1530. [Google Scholar] [CrossRef]
- Li, L.; Kang, L.; Zhao, W.; Feng, Y.Y.; Liu, W.P.; Wang, T.; Mai, H.X.; Huang, J.; Chen, S.Y.; Liang, Y.C.; et al. miR-30a-5p suppresses breast tumor growth and metastasis through inhibition of LDHA-mediated Warburg effect. Cancer Lett. 2017, 400, 89–98. [Google Scholar] [CrossRef]
- Xian, Z.Y.; Liu, J.M.; Chen, Q.K.; Chen, H.Z.; Ye, C.J.; Xue, J.; Yang, H.Q.; Li, J.L.; Liu, X.F.; Kuang, S.J. Inhibition of LDHA suppresses tumor progression in prostate cancer. Tumor Biol. 2015, 36, 8093–8100. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.C.; Zhou, Z.J.; Wang, J.Z.; Yu, H.; Lu, H.C.; Yuan, B.R.; Han, J.; Zhou, R.; Zhang, X.L.; Yang, X.; et al. Prognostic Value of Lactate Dehydrogenase Expression in Different Cancers: A Meta-Analysis. Am. J. Med. Sci. 2019, 358, 412–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, R.H.; Zhou, X.; Yu, Z.H.; Liu, J.J.; Huang, G. Low Expression of LDHB Correlates With Unfavorable Survival in Hepatocellular Carcinoma Strobe-Compliant Article. Medicine 2015, 94, e1583. [Google Scholar] [CrossRef] [PubMed]
- Dennison, J.B.; Molina, J.R.; Mitra, S.; Gonzalez-Angulo, A.M.; Balko, J.M.; Kuba, M.G.; Sanders, M.E.; Pinto, J.A.; Gomez, H.L.; Arteaga, C.L.; et al. Lactate Dehydrogenase B: A Metabolic Marker of Response to Neoadjuvant Chemotherapy in Breast Cancer. Clin. Cancer Res. 2013, 19, 3703–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chesnelong, C.; Chaumeil, M.M.; Blough, M.D.; Al-Najjar, M.; Stechishin, O.D.; Chan, J.A.; Pieper, R.O.; Ronen, S.M.; Weiss, S.; Luchman, H.A.; et al. Lactate dehydrogenase A silencing in IDH mutant gliomas. Neuro-Oncol. 2014, 16, 686–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef] [Green Version]
- De Haas, T.; Hasselt, N.; Troost, D.; Caron, H.; Popovic, M.; Zadravec-Zaletel, L.; Grajkowska, W.; Perek, M.; Osterheld, M.C.; Ellison, D.; et al. Molecular risk stratification of medulloblastoma patients based on immunohistochemical analysis of MYC, LDHB, and CCNB1 expression. Clin. Cancer Res. 2008, 14, 4154–4160. [Google Scholar] [CrossRef] [Green Version]
- Masatomo Maeda, M.K.; Mane, M.M.; Cohen, I.J.; Shindo, M.; Vemuri, K.; Serganova, I.; Blasberg, R. Genetic and drug inhibition of LDH-A: Effects on murine gliomas. Cancers 2022, in press.
- Szatmari, T.; Lumniczky, K.; Desaknai, S.; Trajcevski, S.; Hidvegi, E.J.; Hamada, H.; Safrany, G. Detailed characterization of the mouse glioma 261 tumor model for experimental glioblastoma therapy. Cancer Sci. 2006, 97, 546–553. [Google Scholar] [CrossRef]
- Jacobs, V.L.; Valdes, P.A.; Hickey, W.F.; De Leo, J.A. Current review of in vivo GBM rodent models: Emphasis on the CNS-1 tumour model. Asn Neuro 2011, 3, e00063. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.C.; Hong, J.H.; Hsueh, C.; Chiang, C.S. Tumor-secreted SDF-1 promotes glioma invasiveness and TAM tropism toward hypoxia in a murine astrocytoma model. Lab. Investig. 2012, 92, 151–162. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Elabbadi, M.; Roy, M.L. Ganglioside Distribution in Murine Neural Tumors. Mol. Chem. Neuropathol. 1992, 17, 147–167. [Google Scholar] [CrossRef]
- Mane, M.M.; Cohen, I.J.; Ackerstaff, E.; Shalaby, K.; Ijoma, J.N.; Ko, M.; Maeda, M.; Albeg, A.S.; Vemuri, K.; Satagopan, J.; et al. Lactate Dehydrogenase A Depletion Alters MyC-CaP Tumor Metabolism, Microenvironment, and CAR T Cell Therapy. Mol. Ther.-Oncolytics 2020, 18, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Moroz, E.; Carlin, S.; Dyomina, K.; Burke, S.; Thaler, H.T.; Blasberg, R.; Serganova, I. Real-Time Imaging of HIF-1 alpha Stabilization and Degradation. PLoS ONE 2009, 4, e5077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.J.; Qiu, N.H.; Zhang, L.; Zhong, G.S.; Zeng, F.C. A more universal and stable method for lactate dehydrogenase isoenzyme test. Anal. Methods 2019, 11, 4173–4183. [Google Scholar] [CrossRef]
- Forkasiewicz, A.; Dorociak, M.; Stach, K.; Szelachowski, P.; Tabola, R.; Augoff, K. The usefulness of lactate dehydrogenase measurements in current oncological practice. Cell. Mol. Biol. Lett. 2020, 25, 35. [Google Scholar] [CrossRef] [PubMed]
- Deoca, F.M.; Macy, M.L.; Shannon, J.E. Isoenzyme Characterization of Animal Cell Cultures. Proc. Soc. Exp. Biol. Med. 1969, 132, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Barnett, H. The Staining of Lactic Dehydrogenase Isoenzymes after Electrophoretic Separation on Cellulose Acetate. J. Clin. Pathol. 1964, 17, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Arganda-Carreras, I.; Kaynig, V.; Rueden, C.; Eliceiri, K.W.; Schindelin, J.; Cardona, A.; Sebastian Seung, H. Trainable Weka Segmentation: A machine learning tool for microscopy pixel classification. Bioinformatics 2017, 33, 2424–2426. [Google Scholar] [CrossRef]
- Zappasodi, R.; Serganova, I.; Cohen, I.J.; Maeda, M.; Shindo, M.; Senbabaoglu, Y.; Watson, M.J.; Leftin, A.; Maniyar, R.; Verma, S.; et al. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature 2021, 591, 652–658. [Google Scholar] [CrossRef]
- Sure, V.N.; Sakamuri, S.S.V.P.; Sperling, J.A.; Evans, W.R.; Merdzo, I.; Mostany, R.; Murfee, W.L.; Busija, D.W.; Katakam, P.V.G. A novel high-throughput assay for respiration in isolated brain microvessels reveals impaired mitochondrial function in the aged mice. Geroscience 2018, 40, 365–375. [Google Scholar] [CrossRef]
- Fritz, V.; Fajas, L. Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene 2010, 29, 4369–4377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Acosta, M.L.; Fletcher, E.L.; Azizoglu, S.; Foster, L.E.; Farber, D.B.; Kalloniatis, M. Early markers of retinal degeneration in rd/rd mice. Mol. Vis. 2005, 11, 717–728. [Google Scholar]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markert, C.L.; Shaklee, J.B.; Whitt, G.S. Evolution of a gene. Multiple genes for LDH isozymes provide a model of the evolution of gene structure, function and regulation. Science 1975, 189, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Read, J.A.; Winter, V.J.; Eszes, C.M.; Sessions, R.B.; Brady, R.L. Structural basis for altered activity of M- and H-isozyme forms of human lactate dehydrogenase. Proteins 2001, 43, 175–185. [Google Scholar] [CrossRef]
- Farhana, A.; Lappin, S.L. Biochemistry, Lactate Dehydrogenase; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Le, A.; Cooper, C.R.; Gouw, A.M.; Dinavahi, R.; Maitra, A.; Deck, L.M.; Royer, R.E.; Vander Jagt, D.L.; Semenza, G.L.; Dang, C.V. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. USA 2010, 107, 2037–2042. [Google Scholar] [CrossRef] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.Y.; Ma, L.; Qian, J.; Xu, J.; Xu, T.P.; Pang, L.J.; Zhou, H.; Shu, Y.Q.; Zhou, J.W. The molecular mechanism and clinical significance of LDHA in HER2-mediated progression of gastric cancer. Am. J. Transl. Res. 2018, 10, 2055. [Google Scholar]
- Fantin, V.R.; St-Pierre, J.; Leder, P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance (volume 9, pages 425, 2006). Cancer Cell 2006, 10, 172. [Google Scholar] [CrossRef] [Green Version]
- Koukourakis, M.I.; Giatromanolaki, A.; Simopoulos, C.; Polychronidis, A.; Sivridis, E. Lactate dehydrogenase 5 (LDH5) relates to up-regulated hypoxia inducible factor pathway and metastasis in colorectal cancer. Clin. Exp. Metastasis 2005, 22, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, M.I.; Giatromanolaki, A.; Sivridis, E.; Gatter, K.C.; Harris, A.L. Lactate dehydrogenase 5 expression in operable colorectal cancer: Strong association with survival and activated vascular endothelial growth factor pathway—A report of the tumour angiogenesis research group. J. Clin. Oncol. 2006, 24, 4301–4308. [Google Scholar] [CrossRef] [PubMed]
- McCleland, M.L.; Adler, A.S.; Shang, Y.L.; Hunsaker, T.; Truong, T.; Peterson, D.; Torres, E.; Li, L.; Haley, B.; Stephan, J.P.; et al. An Integrated Genomic Screen Identifies LDHB as an Essential Gene for Triple-Negative Breast Cancer. Cancer Res. 2012, 72, 5812–5823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCleland, M.L.; Adler, A.S.; Deming, L.; Cosino, E.; Lee, L.; Blackwood, E.M.; Solon, M.; Tao, J.; Li, L.; Shames, D.; et al. Lactate Dehydrogenase B Is Required for the Growth of KRAS-Dependent Lung Adenocarcinomas. Clin. Cancer Res. 2013, 19, 773–784. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Xie, H.; Scicluna, P.; Lee, L.; Bjornhagen, V.; Hoog, A.; Larsson, C.; Lui, W.O. MiR-375 Regulation of LDHB Plays Distinct Roles in Polyomavirus-Positive and -Negative Merkel Cell Carcinoma. Cancers 2018, 10, 443. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Han, J.; Jang, Y.; Kim, S.J.; Lee, M.J.; Ryu, M.J.; Kweon, G.R.; Heo, J.Y. High-capacity glycolytic and mitochondrial oxidative metabolisms mediate the growth ability of glioblastoma. Int. J. Oncol. 2015, 47, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shindo, M.; Maeda, M.; Myat, K.; Mane, M.M.; Cohen, I.J.; Vemuri, K.; Albeg, A.S.; Serganova, I.; Blasberg, R. LDH-A—Modulation and the Variability of LDH Isoenzyme Profiles in Murine Gliomas: A Link with Metabolic and Growth Responses. Cancers 2022, 14, 2303. https://doi.org/10.3390/cancers14092303
Shindo M, Maeda M, Myat K, Mane MM, Cohen IJ, Vemuri K, Albeg AS, Serganova I, Blasberg R. LDH-A—Modulation and the Variability of LDH Isoenzyme Profiles in Murine Gliomas: A Link with Metabolic and Growth Responses. Cancers. 2022; 14(9):2303. https://doi.org/10.3390/cancers14092303
Chicago/Turabian StyleShindo, Masahiro, Masatomo Maeda, Ko Myat, Mayuresh M. Mane, Ivan J. Cohen, Kiranmayi Vemuri, Avi S. Albeg, Inna Serganova, and Ronald Blasberg. 2022. "LDH-A—Modulation and the Variability of LDH Isoenzyme Profiles in Murine Gliomas: A Link with Metabolic and Growth Responses" Cancers 14, no. 9: 2303. https://doi.org/10.3390/cancers14092303
APA StyleShindo, M., Maeda, M., Myat, K., Mane, M. M., Cohen, I. J., Vemuri, K., Albeg, A. S., Serganova, I., & Blasberg, R. (2022). LDH-A—Modulation and the Variability of LDH Isoenzyme Profiles in Murine Gliomas: A Link with Metabolic and Growth Responses. Cancers, 14(9), 2303. https://doi.org/10.3390/cancers14092303