
Extrachromosomal Circular DNA from TCGA Tumors Is Generated from Common Genomic Loci, Is Characterized by Self-Homology and DNA Motifs near Circle Breakpoints


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Generating Random Circular DNA for Control Comparisons
2.3. Pan-Cancer Genome-Wide Analysis of Circle Location
2.4. Identifying Circles with Genes and Circles without Genes
2.5. Gene Expression Analysis
2.6. Homology and Microhomology Analysis
2.7. Motif Enrichment
3. Results
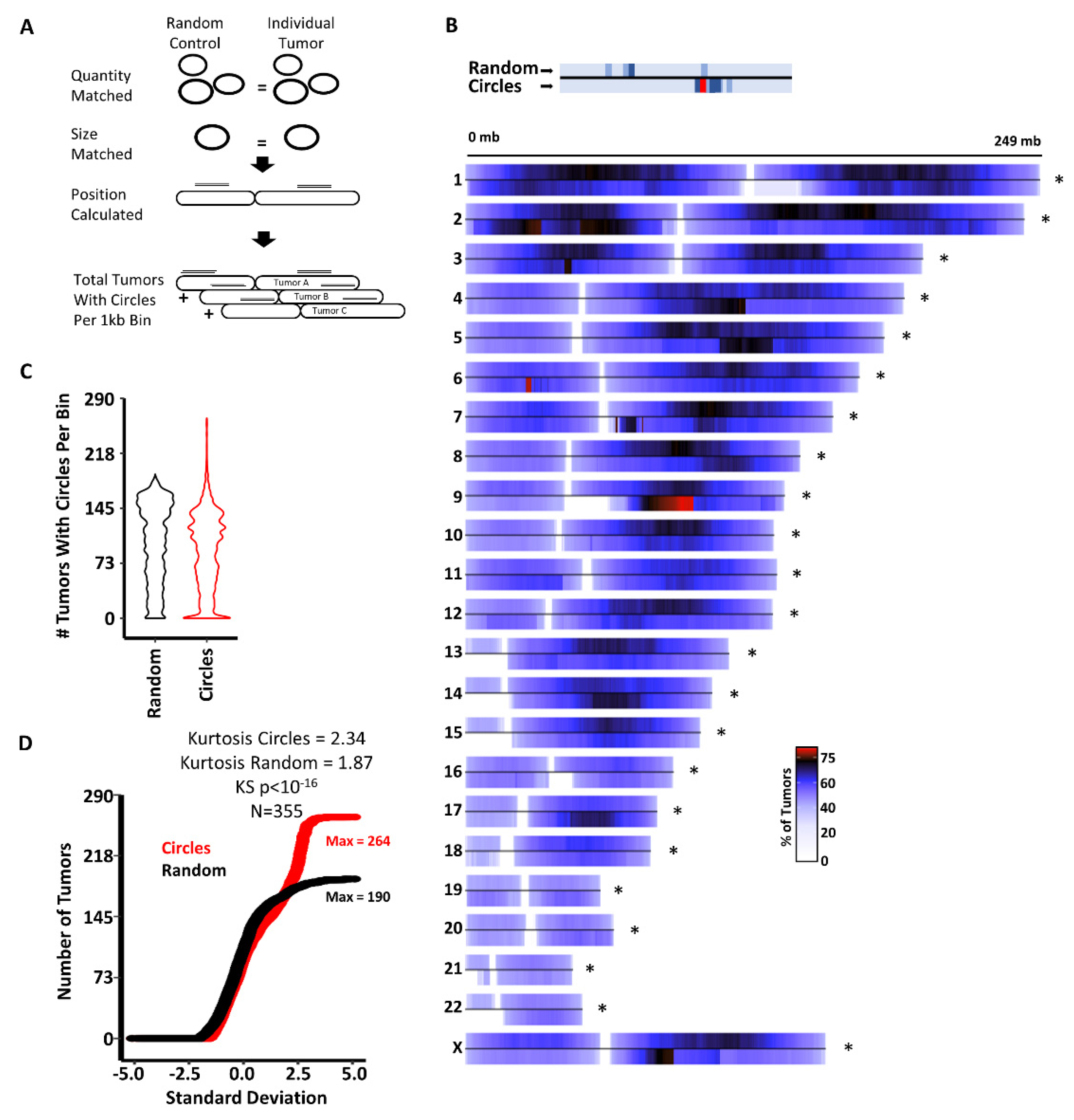
3.1. Circles Are Commonly Generated at Specific Genomic Loci Independent of Cancer Type
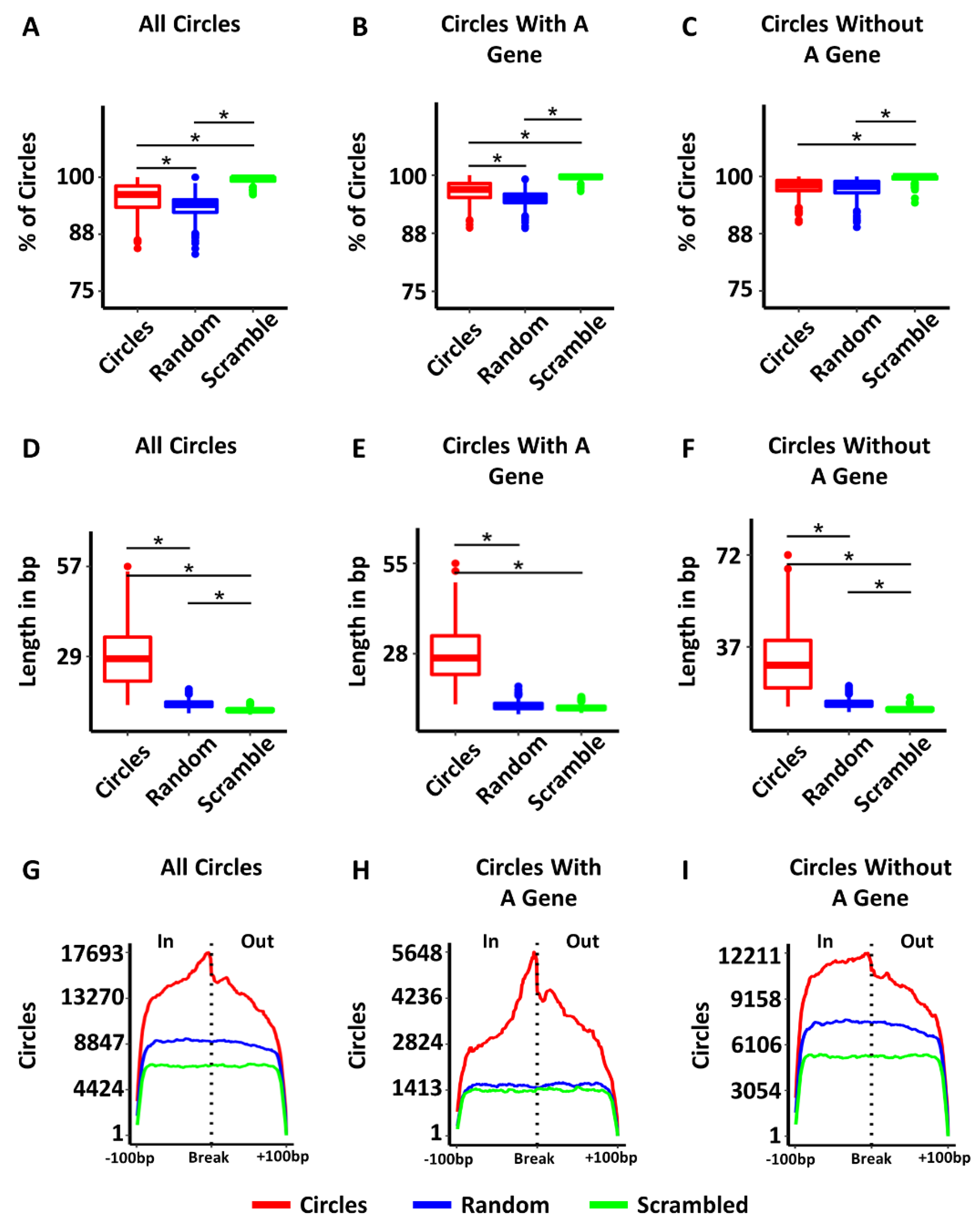
3.2. Characteristics of Circular DNAs from TCGA Samples
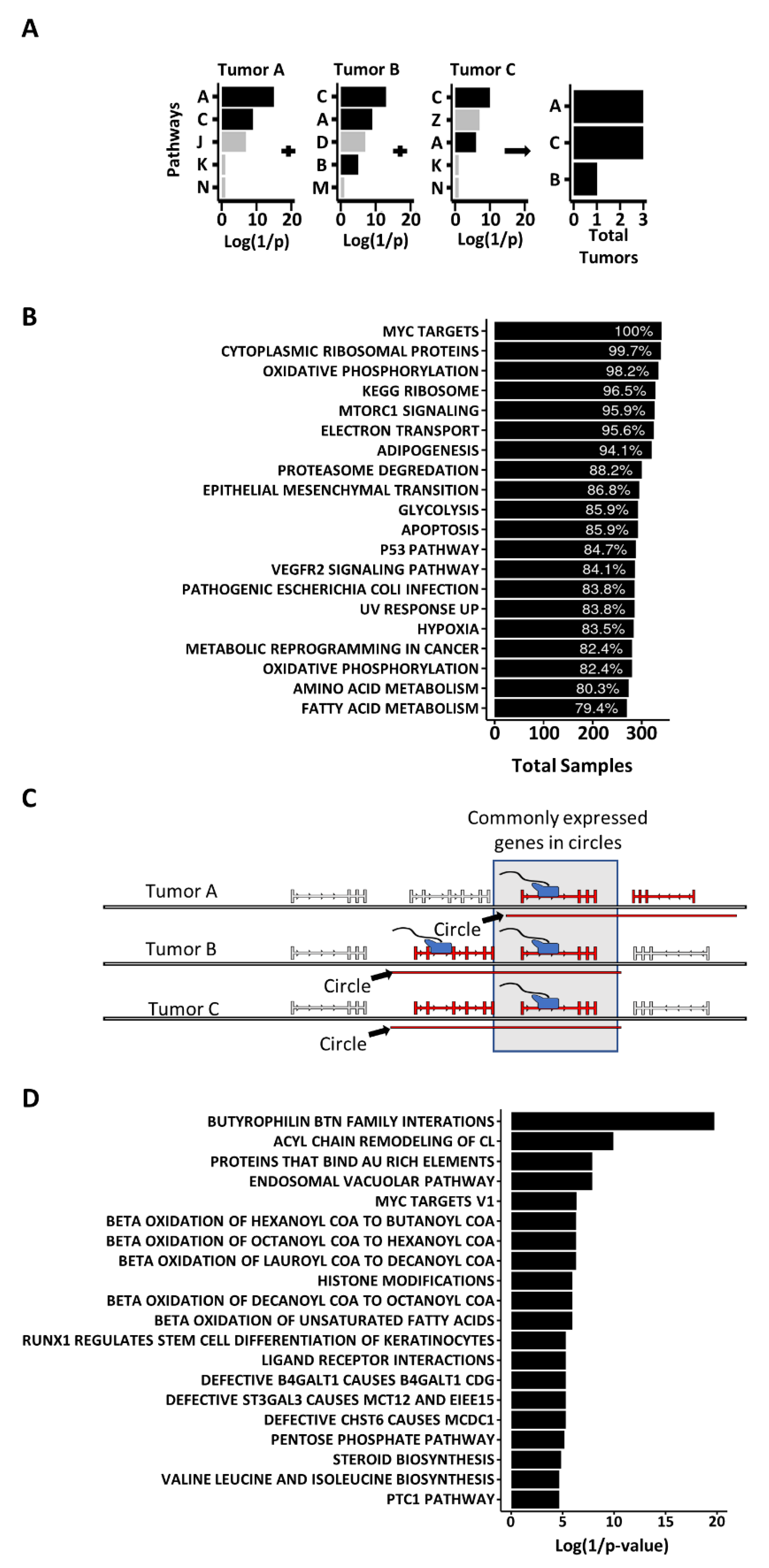
3.3. Genes on Circles Are More Likely to Be Expressed and Are More Highly Expressed, than Other Genes in the Genome
3.4. Some Circles Are Self-Homologous
3.5. Most Circles Are Self-Microhomologous
3.6. Circle Ends Are Enriched in Transcription Factor DNA Binding Motifs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hotta, Y.; Bassel, A. Molecular size and circularity of DNA in cells of mammals and higher plants. Proc. Natl. Acad. Sci. USA 1965, 53, 356–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radloff, R.; Bauer, W.; Vinograd, J. A dye-buoyant-density method for the detection and isolation of closed circular duplex DNA: The closed circular DNA in HeLa cells. Proc. Natl. Acad. Sci. USA 1967, 57, 1514–1521. [Google Scholar] [CrossRef] [Green Version]
- Zuo, S.; Yi, Y.; Wang, C.; Li, X.; Zhou, M.; Peng, Q.; Zhou, J.; Yang, Y.; He, Q. Extrachromosomal Circular DNA (eccDNA): From Chaos to Function. Front. Cell Dev. Biol. 2022, 9, 792555. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.J. Circles with a Point: New Insights into Oncogenic Extrachromosomal DNA. Cancer Cell 2020, 37, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Zhang, H.; Zhou, Y.; Shi, J. Extrachromosomal circular DNA: A new potential role in cancer progression. J. Transl. Med. 2021, 19, 257. [Google Scholar] [CrossRef]
- Yan, Y.; Guo, G.; Huang, J.; Gao, M.; Zhu, Q.; Zeng, S.; Gong, Z.; Xu, Z. Current understanding of extrachromosomal circular DNA in cancer pathogenesis and therapeutic resistance. J. Hematol. Oncol. 2020, 13, 124. [Google Scholar] [CrossRef]
- Møller, H.D.; Parsons, L.; Jørgensen, T.S.; Botstein, D.; Regenberg, B. Extrachromosomal circular DNA is common in yeast. Proc. Natl. Acad. Sci. USA 2015, 112, E3114–E3122. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Nguyen, N.-P.; Turner, K.; Wu, S.; Gujar, A.D.; Luebeck, J.; Liu, J.; Deshpande, V.; Rajkumar, U.; Namburi, S.; et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat. Genet. 2020, 52, 891–897. [Google Scholar] [CrossRef]
- Mukherjee, K.; Storici, F. A Mechanism of Gene Amplification Driven by Small DNA Fragments. PLoS Genet. 2012, 8, e1003119. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, M.; Djekidel, M.N.; Chen, H.; Liu, D.; Alt, F.W.; Zhang, Y. eccDNAs are apoptotic products with high innate immunostimulatory activity. Nature 2021, 599, 308–314. [Google Scholar] [CrossRef]
- Dillon, L.W.; Kumar, P.; Shibata, Y.; Wang, Y.-H.; Willcox, S.; Griffith, J.D.; Pommier, Y.; Takeda, S.; Dutta, A. Production of Extrachromosomal MicroDNAs Is Linked to Mismatch Repair Pathways and Transcriptional Activity. Cell Rep. 2015, 11, 1749–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, S.M.; De Rose, M.; Gaudray, P.; Moore, C.M.; VanDevanter, D.; Von Hoff, D.D.; Wahl, G.M. Double minute chromosomes can be produced from precursors derived from a chromosomal deletion. Mol. Cell. Biol. 1988, 8, 1525–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahl, G.M.; Vincent, B.R.D.S.; De Rose, M. Effect of chromosomal position on amplification of transfected genes in animal cells. Nature 1984, 307, 516–520. [Google Scholar] [CrossRef] [PubMed]
- Koche, R.P.; Rodriguez-Fos, E.; Helmsauer, K.; Burkert, M.; MacArthur, I.C.; Maag, J.; Chamorro, R.; Munoz-Perez, N.; Puiggròs, M.; Garcia, H.D.; et al. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nat. Genet. 2019, 52, 29–34. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; McGill, J.R.; Forseth, B.J.; Davidson, K.K.; Bradley, T.P.; Van Devanter, D.R.; Wahl, G.M. Elimination of extrachromosomally amplified MYC genes from human tumor cells reduces their tumorigenicity. Proc. Natl. Acad. Sci. USA 1992, 89, 8165–8169. [Google Scholar] [CrossRef] [Green Version]
- Turner, K.M.; Deshpande, V.; Beyter, D.; Koga, T.; Rusert, J.; Lee, C.; Li, B.; Arden, K.; Ren, B.; Nathanson, D.A.; et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017, 543, 122–125. [Google Scholar] [CrossRef]
- Morton, A.; Dogan-Artun, N.; Faber, Z.; MacLeod, G.; Bartels, C.F.; Piazza, M.; Allan, K.C.; Mack, S.C.; Wang, X.; Gimple, R.C.; et al. Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell 2019, 179, 1330–1341.e13. [Google Scholar] [CrossRef]
- Wu, S.; Turner, K.M.; Nguyen, N.-P.; Raviram, R.; Erb, M.; Santini, J.; Luebeck, J.; Rajkumar, U.; Diao, Y.; Li, B.; et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 2019, 575, 699–703. [Google Scholar] [CrossRef]
- Weiser, A.; Hung, K.; Chang, H. Oncogene Convergence in Extrachromosomal DNA Hubs. Cancer Discov. 2022, 12, 1–4. [Google Scholar] [CrossRef]
- Nathanson, D.A.; Gini, B.; Mottahedeh, J.; Visnyei, K.; Koga, T.; Gomez, G.; Eskin, A.; Hwang, K.; Wang, J.; Masui, K.; et al. Targeted Therapy Resistance Mediated by Dynamic Regulation of Extrachromosomal Mutant EGFR DNA. Science 2014, 343, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Møller, H.D.; Mohiyuddin, M.; Prada-Luengo, I.; Sailani, M.R.; Halling, J.F.; Plomgaard, P.; Maretty, L.; Hansen, A.J.; Snyder, M.P.; Pilegaard, H.; et al. Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat. Commun. 2018, 9, 1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Kiran, S.; Saha, S.; Su, Z.; Paulsen, T.; Chatrath, A.; Shibata, Y.; Shibata, E.; Dutta, A. ATAC-Seq Identifies Thousands of Extrachromosomal Circular DNA in Cancer and Cell Lines. Sci. Adv. 2020, 6, eaba2489. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Bafna, V.; Chang, H.Y.; Mischel, P.S. Extrachromosomal DNA: An Emerging Hallmark in Human Cancer. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 367–386. [Google Scholar] [CrossRef]
- Bailey, C.; Shoura, M.; Mischel, P.; Swanton, C. Extrachromosomal DNA—Relieving heredity constraints, accelerating tumour evolution. Ann. Oncol. 2020, 31, 884–893. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Jiao, X.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef] [Green Version]
- Seol, J.-H.; Shim, E.Y.; Lee, S.E. Microhomology-mediated end joining: Good, bad and ugly. Mutat. Res. Mol. Mech. Mutagen. 2017, 809, 81–87. [Google Scholar] [CrossRef]
- Wang, H.; Xu, X. Microhomology-mediated end joining: New players join the team. Cell Biosci. 2017, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Sfeir, A.; Symington, L.S. Microhomology-Mediated End Joining: A Back-up Survival Mechanism or Dedicated Pathway? Trends Biochem. Sci. 2015, 40, 701–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarreal, D.D.; Lee, K.; Deem, A.; Shim, E.Y.; Malkova, A.; Lee, S.E. Microhomology Directs Diverse DNA Break Repair Pathways and Chromosomal Translocations. PLoS Genet. 2012, 8, e1003026. [Google Scholar] [CrossRef] [Green Version]
- A Castro-Mondragon, J.; Riudavets-Puig, R.; Rauluseviciute, I.; Lemma, R.B.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Pérez, N.M.; et al. JASPAR 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2021, 50, D165–D173. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheffield, N.C.; Bock, C. LOLA: Enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics 2015, 32, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Han, Y.; Meng, J.; Zhong, B.; Chen, J.; Zhang, H.; Qin, J.; Pang, J.; Liu, L. Small extrachromosomal circular DNA (eccDNA): Major functions in evolution and cancer. Mol. Cancer 2021, 20, 113. [Google Scholar] [CrossRef]
- Ouedraogo, M.; Bettembourg, C.; Bretaudeau, A.; Sallou, O.; Diot, C.; Demeure, O.; Lecerf, F. The Duplicated Genes Database: Identification and Functional Annotation of Co-Localised Duplicated Genes across Genomes. PLoS ONE 2012, 7, e50653. [Google Scholar] [CrossRef] [Green Version]
- Patterson-Fortin, J.; D’Andrea, A.D. Exploiting the Microhomology-Mediated End-Joining Pathway in Cancer Therapy. Cancer Res. 2020, 80, 4593–4600. [Google Scholar] [CrossRef]
- Uhlén, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, 2507. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, D.R.; Chinnaiyan, A.M. Integrative analysis of the cancer transcriptome. Nat. Genet. 2005, 37, S31–S37. [Google Scholar] [CrossRef] [Green Version]
- Cieślik, M.; Chinnaiyan, A.M. Cancer transcriptome profiling at the juncture of clinical translation. Nat. Rev. Genet. 2017, 19, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Marnef, A.; Cohen, S.; Legube, G. Transcription-Coupled DNA Double-Strand Break Repair: Active Genes Need Special Care. J. Mol. Biol. 2017, 429, 1277–1288. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Ui, A.; Chiba, N.; Yasui, A. Relationship among DNA double-strand break (DSB), DSB repair, and transcription prevents genome instability and cancer. Cancer Sci. 2020, 111, 1443–1451. [Google Scholar] [CrossRef]
- Schwer, B.; Wei, P.-C.; Chang, A.N.; Kao, J.; Du, Z.; Meyers, R.M.; Alt, F.W. Transcription-associated processes cause DNA double-strand breaks and translocations in neural stem/progenitor cells. Proc. Natl. Acad. Sci. USA 2016, 113, 2258–2263. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tatman, P.D.; Black, J.C. Extrachromosomal Circular DNA from TCGA Tumors Is Generated from Common Genomic Loci, Is Characterized by Self-Homology and DNA Motifs near Circle Breakpoints. Cancers 2022, 14, 2310. https://doi.org/10.3390/cancers14092310
Tatman PD, Black JC. Extrachromosomal Circular DNA from TCGA Tumors Is Generated from Common Genomic Loci, Is Characterized by Self-Homology and DNA Motifs near Circle Breakpoints. Cancers. 2022; 14(9):2310. https://doi.org/10.3390/cancers14092310
Chicago/Turabian StyleTatman, Philip D., and Joshua C. Black. 2022. "Extrachromosomal Circular DNA from TCGA Tumors Is Generated from Common Genomic Loci, Is Characterized by Self-Homology and DNA Motifs near Circle Breakpoints" Cancers 14, no. 9: 2310. https://doi.org/10.3390/cancers14092310
APA StyleTatman, P. D., & Black, J. C. (2022). Extrachromosomal Circular DNA from TCGA Tumors Is Generated from Common Genomic Loci, Is Characterized by Self-Homology and DNA Motifs near Circle Breakpoints. Cancers, 14(9), 2310. https://doi.org/10.3390/cancers14092310