
Ready for Prime Time? Dendritic Cells in High-Grade Gliomas

{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Overview of DC Function in Antitumor Responses
3. What Is the Immunological Potential of DC in the CNS?
4. Dendritic Cell Infiltration of HGG: Who Are the Players and Where Are They?
5. HGG Antigen Drainage for the Initiation of Peripheral Immune Response
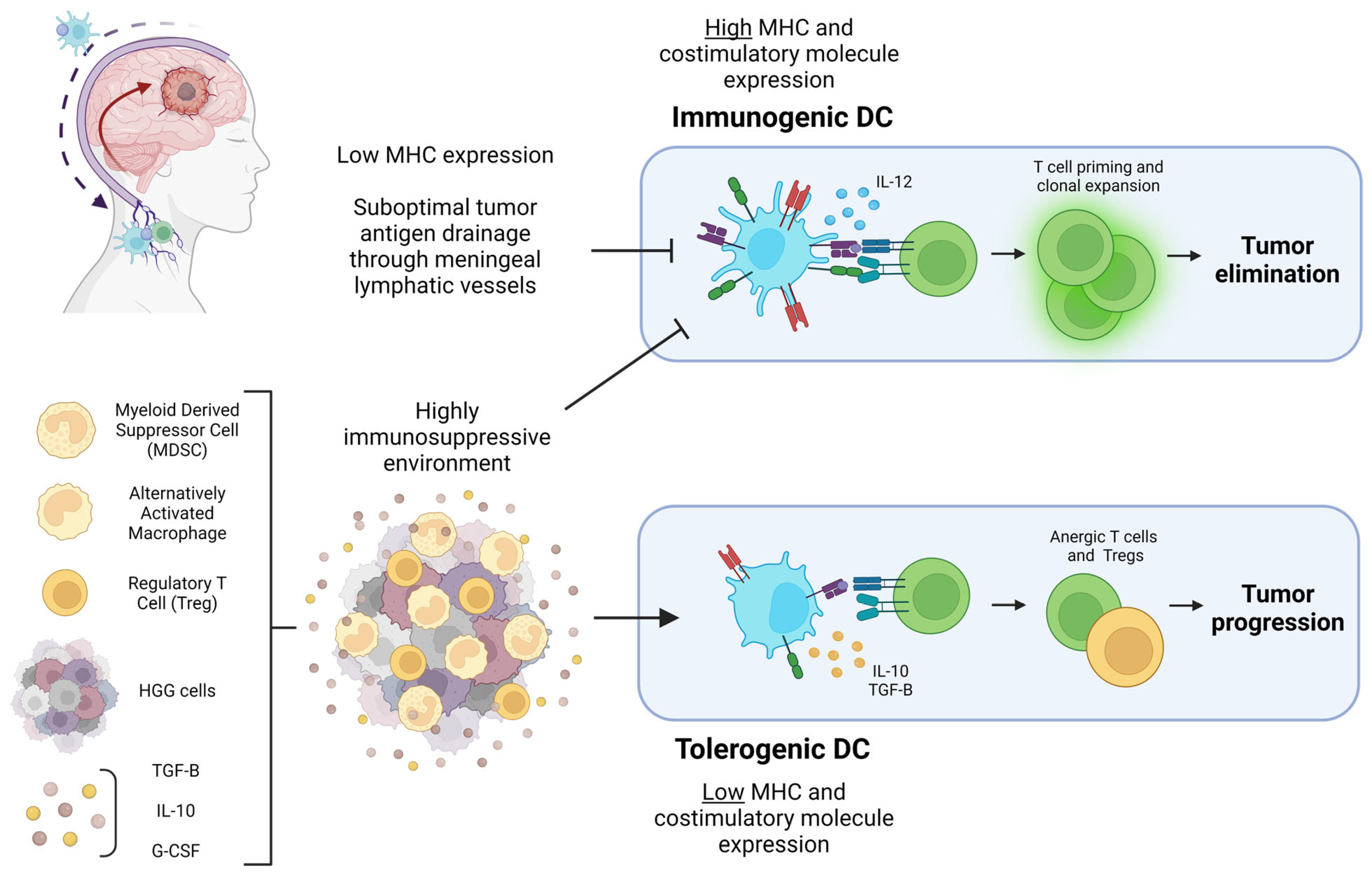
6. Dendritic Cell Activity within HGG: Immunogenic or Tolerogenic?
7. Dendritic Cell Subsets and Their Role in ICB-Mediated Antitumor Immune Responses: cDC1 or cDC2?
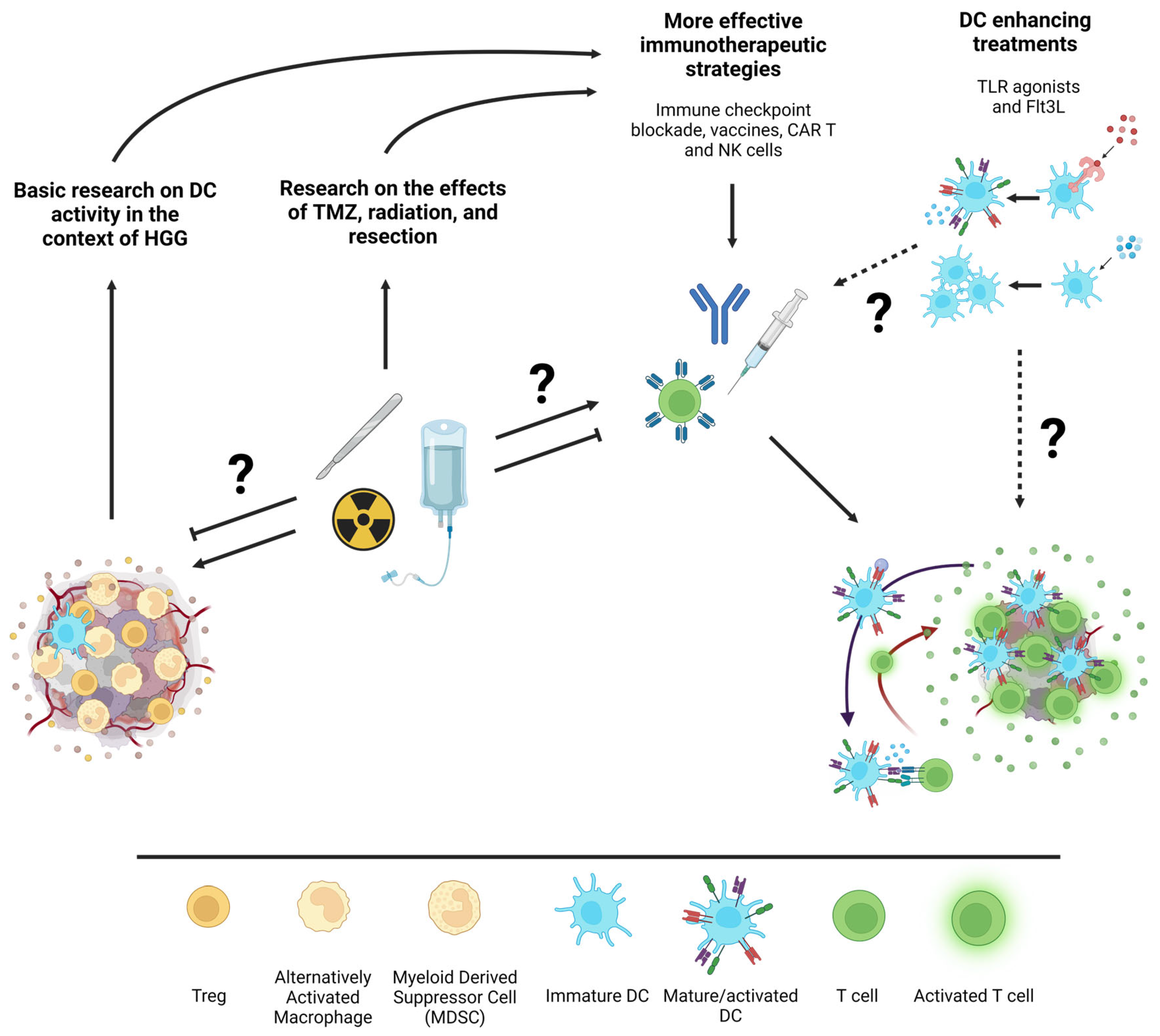
8. Future Direction: How Can Basic Research Best Support Effective Translation?
9. Conclusions/Final Comments
Author Contributions
Funding
Conflicts of Interest
References
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro. Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro. Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Dandy, W.E. Removal of Right Cerebral Hemisphere for Certain Tumors with Hemiplegia: Preliminary Report. J. Am. Med. Assoc. 1928, 90, 823–825. [Google Scholar] [CrossRef]
- Putavet, D.A.; de Keizer, P.L.J. Residual Disease in Glioma Recurrence: A Dangerous Liaison with Senescence. Cancers 2021, 13, 1560. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Cohn, Z.A. Identification of a Novel Cell Type in Peripheral Lymphoid Organs of Mice. I. Morphology, Quantitation, Tissue Distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef]
- Murphy, T.L.; Murphy, K.M. Dendritic Cells in Cancer Immunology. Cell. Mol. Immunol. 2022, 19, 3–13. [Google Scholar] [CrossRef]
- Bevan, M.J. Cross-Priming for a Secondary Cytotoxic Response to Minor H Antigens with H-2 Congenic Cells Which Do Not Cross-React in the Cytotoxic Assay. J. Exp. Med. 1976, 143, 1283–1288. [Google Scholar] [CrossRef]
- den Haan, J.M.; Lehar, S.M.; Bevan, M.J. CD8(+) but Not CD8(−) Dendritic Cells Cross-Prime Cytotoxic T Cells in vivo. J. Exp. Med. 2000, 192, 1685–1696. [Google Scholar] [CrossRef]
- Ferris, S.T.; Durai, V.; Wu, R.; Theisen, D.J.; Ward, J.P.; Bern, M.D.; Davidson, J.T.; Bagadia, P.; Liu, T.; Briseño, C.G.; et al. CDC1 Prime and Are Licensed by CD4+ T Cells to Induce Anti-Tumour Immunity. Nature 2020, 584, 624–629. [Google Scholar] [CrossRef]
- Heufler, C.; Koch, F.; Stanzl, U.; Topar, G.; Wysocka, M.; Trinchieri, G.; Enk, A.; Steinman, R.M.; Romani, N.; Schuler, G. Interleukin-12 Is Produced by Dendritic Cells and Mediates T Helper 1 Development as Well as Interferon-γ Production by T Helper 1 Cells. Eur. J. Immunol. 1996, 26, 659–668. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell 2019, 177, 556–571.e16. [Google Scholar] [CrossRef]
- Anderson, D.A.; Murphy, K.M.; Briseño, C.G. Development, Diversity, and Function of Dendritic Cells in Mouse and Human. Cold Spring Harb. Perspect. Biol. 2018, 10, a028613. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, P.F.; Alberti-Servera, L.; Eremin, A.; Grajales-Reyes, G.E.; Ivanek, R.; Tussiwand, R. Distinct Progenitor Lineages Contribute to the Heterogeneity of Plasmacytoid Dendritic Cells. Nat. Immunol. 2018, 19, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Lawrence, T.; Liang, Y. The Role of Plasmacytoid Dendritic Cells in Cancers. Front. Immunol. 2021, 12, 749190. [Google Scholar] [CrossRef]
- Aspord, C.; Leccia, M.-T.; Charles, J.; Plumas, J. Plasmacytoid Dendritic Cells Support Melanoma Progression by Promoting Th2 and Regulatory Immunity through OX40L and ICOSL. Cancer Immunol. Res. 2013, 1, 402–415. [Google Scholar] [CrossRef]
- Cools, N.; Ponsaerts, P.; Van Tendeloo, V.F.I.; Berneman, Z.N. Balancing between Immunity and Tolerance: An Interplay between Dendritic Cells, Regulatory T Cells, and Effector T Cells. J. Leukoc. Biol. 2007, 82, 1365–1374. [Google Scholar] [CrossRef]
- Schwartz, R.H.; Mueller, D.L.; Jenkins, M.K.; Quill, H. T-Cell Clonal Anergy. Cold Spring Harb. Symp. Quant. Biol. 1989, 54, 605–610. [Google Scholar] [CrossRef]
- Hawiger, D.; Inaba, K.; Dorsett, Y.; Guo, M.; Mahnke, K.; Rivera, M.; Ravetch, J.V.; Steinman, R.M.; Nussenzweig, M.C. Dendritic Cells Induce Peripheral T Cell Unresponsiveness under Steady State Conditions In Vivo. J. Exp. Med. 2001, 194, 769–779. [Google Scholar] [CrossRef]
- Wilson, N.S.; El-Sukkari, D.; Villadangos, J.A. Dendritic Cells Constitutively Present Self Antigens in Their Immature State In Vivo and Regulate Antigen Presentation by Controlling the Rates of MHC Class II Synthesis and Endocytosis. Blood 2004, 103, 2187–2195. [Google Scholar] [CrossRef]
- Akbari, O.; DeKruyff, R.H.; Umetsu, D.T. Pulmonary Dendritic Cells Producing IL-10 Mediate Tolerance Induced by Respiratory Exposure to Antigen. Nat. Immunol. 2001, 2, 725–731. [Google Scholar] [CrossRef]
- Gabrilovich, D.; Corak, J.; Ciernik, I.; Kavanaugh, D.; Carbone, D. Decreased Antigen Presentation by Dendritic Cells in Patients with Breast Cancer. Clin. Cancer Res. 1997, 3, 483–490. [Google Scholar] [PubMed]
- Pinzon-Charry, A.; Maxwell, T.; López, J.A. Dendritic Cell Dysfunction in Cancer: A Mechanism for Immunosuppression. Immunol. Cell. Biol. 2005, 83, 451–461. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Gabrilovich, D.I. Effect of Tumor-Derived Cytokines and Growth Factors on Differentiation and Immune Suppressive Features of Myeloid Cells in Cancer. Cancer Metastasis Rev. 2006, 25, 323–331. [Google Scholar] [CrossRef]
- Hishii, M.; Nitta, T.; Ishida, H.; Ebato, M.; Kurosu, A.; Yagita, H.; Sato, K.; Okumura, K. Human Glioma-Derived Interleukin-10 Inhibits Antitumor Immune Responses In Vitro. Neurosurgery 1995, 37, 1160–1166; discussion 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Todo, T.; Ino, Y.; Takahashi, M.; Miyazawa, K.; Miyazono, K. Autocrine TGF-Beta Signaling Maintains Tumorigenicity of Glioma-Initiating Cells through Sry-Related HMG-Box Factors. Cell Stem Cell 2009, 5, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Turkowski, K.; Brandenburg, S.; Mueller, A.; Kremenetskaia, I.; Bungert, A.D.; Blank, A.; Felsenstein, M.; Vajkoczy, P. VEGF as a Modulator of the Innate Immune Response in Glioblastoma. Glia 2018, 66, 161–174. [Google Scholar] [CrossRef]
- Mattila, S.; Tuominen, H.; Koivukangas, J.; Stenbäck, F. The Terminal Prostaglandin Synthases MPGES-1, MPGES-2, and CPGES Are All Overexpressed in Human Gliomas. Neuropathology 2009, 29, 156–165. [Google Scholar] [CrossRef]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.A.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- Nduom, E.K.; Weller, M.; Heimberger, A.B. Immunosuppressive Mechanisms in Glioblastoma. Neuro-Oncology 2015, 17, vii9–vii14. [Google Scholar] [CrossRef]
- Zong, J.; Keskinov, A.A.; Shurin, G.V.; Shurin, M.R. Tumor-Derived Factors Modulating Dendritic Cell Function. Cancer Immunol. Immunother. 2016, 65, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and Functional Features of Central Nervous System Lymphatic Vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.S.; Hunter, C.A. Protective and Pathological Immunity during Central Nervous System Infections. Immunity 2017, 46, 891–909. [Google Scholar] [CrossRef] [PubMed]
- Greter, M.; Heppner, F.L.; Lemos, M.P.; Odermatt, B.M.; Goebels, N.; Laufer, T.; Noelle, R.J.; Becher, B. Dendritic Cells Permit Immune Invasion of the CNS in an Animal Model of Multiple Sclerosis. Nat. Med. 2005, 11, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Anandasabapathy, N.; Victora, G.D.; Meredith, M.; Feder, R.; Dong, B.; Kluger, C.; Yao, K.; Dustin, M.L.; Nussenzweig, M.C.; Steinman, R.M.; et al. Flt3L Controls the Development of Radiosensitive Dendritic Cells in the Meninges and Choroid Plexus of the Steady-State Mouse Brain. J. Exp. Med. 2011, 208, 1695–1705. [Google Scholar] [CrossRef]
- Kivisäkk, P.; Imitola, J.; Rasmussen, S.; Elyaman, W.; Zhu, B.; Ransohoff, R.M.; Khoury, S.J. Localizing CNS Immune Surveillance: Meningeal APCs Activate T Cells during EAE. Ann. Neurol. 2009, 65, 457–469. [Google Scholar] [CrossRef]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395.e6. [Google Scholar] [CrossRef]
- Jordão, M.J.C.; Sankowski, R.; Brendecke, S.M.; Sagar; Locatelli, G.; Tai, Y.-H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; et al. Single-Cell Profiling Identifies Myeloid Cell Subsets with Distinct Fates during Neuroinflammation. Science 2019, 363, eaat7554. [Google Scholar] [CrossRef]
- Giles, D.A.; Duncker, P.C.; Wilkinson, N.M.; Washnock-Schmid, J.M.; Segal, B.M. CNS-Resident Classical DCs Play a Critical Role in CNS Autoimmune Disease. J. Clin. Investig. 2018, 128, 5322–5334. [Google Scholar] [CrossRef]
- Mundt, S.; Mrdjen, D.; Utz, S.G.; Greter, M.; Schreiner, B.; Becher, B. Conventional DCs Sample and Present Myelin Antigens in the Healthy CNS and Allow Parenchymal T Cell Entry to Initiate Neuroinflammation. Sci. Immunol. 2019, 4, eaau8380. [Google Scholar] [CrossRef]
- Liu, C.; Zhu, J.; Mi, Y.; Jin, T. Impact of Disease-Modifying Therapy on Dendritic Cells and Exploring Their Immunotherapeutic Potential in Multiple Sclerosis. J. Neuroinflamm. 2022, 19, 298. [Google Scholar] [CrossRef]
- Wolf, Y.; Shemer, A.; Levy-Efrati, L.; Gross, M.; Kim, J.-S.; Engel, A.; David, E.; Chappell-Maor, L.; Grozovski, J.; Rotkopf, R.; et al. Microglial MHC Class II Is Dispensable for Experimental Autoimmune Encephalomyelitis and Cuprizone-Induced Demyelination. Eur. J. Immunol. 2018, 48, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.; Ward, D.W.; Mattison, M.L. Immune Suppression, Gliomas, and Tuberculosis. Br. Med. J 1972, 1, 111. [Google Scholar] [CrossRef] [PubMed]
- Akasaki, Y.; Liu, G.; Chung, N.H.C.; Ehtesham, M.; Black, K.L.; Yu, J.S. Induction of a CD4+ T Regulatory Type 1 Response by Cyclooxygenase-2-Overexpressing Glioma. J. Immunol. 2004, 173, 4352–4359. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Curtin, J.F.; Zirger, J.M.; Xiong, W.; King, G.D.; Barcia, C.; Liu, C.; Puntel, M.; Goverdhana, S.; Lowenstein, P.R.; et al. Inflammatory and Anti-Glioma Effects of an Adenovirus Expressing Human Soluble Fms-like Tyrosine Kinase 3 Ligand (HsFlt3L): Treatment with HsFlt3L Inhibits Intracranial Glioma Progression. Mol. Ther. 2004, 10, 1071–1084. [Google Scholar] [CrossRef]
- Carenza, C.; Franzese, S.; Castagna, A.; Terzoli, S.; Simonelli, M.; Persico, P.; Bello, L.; Nibali, M.C.; Pessina, F.; Kunderfranco, P.; et al. Perioperative Corticosteroid Treatment Impairs Tumor-Infiltrating Dendritic Cells in Patients with Newly Diagnosed Adult-Type Diffuse Gliomas. Front. Immunol. 2023, 13, 1–13. [Google Scholar] [CrossRef]
- Pombo Antunes, A.R.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the Glioblastoma Immune Microenvironment as Basis for the Development of New Immunotherapeutic Strategies. eLife 2020, 9, e52176. [Google Scholar] [CrossRef]
- Brown, C.C.; Gudjonson, H.; Pritykin, Y.; Deep, D.; Lavallée, V.-P.; Mendoza, A.; Fromme, R.; Mazutis, L.; Ariyan, C.; Leslie, C.; et al. Transcriptional Basis of Mouse and Human Dendritic Cell Heterogeneity. Cell 2019, 179, 846–863.e24. [Google Scholar] [CrossRef]
- Yeo, A.T.; Rawal, S.; Delcuze, B.; Christofides, A.; Atayde, A.; Strauss, L.; Balaj, L.; Rogers, V.A.; Uhlmann, E.J.; Varma, H.; et al. Single-Cell RNA Sequencing Reveals Evolution of Immune Landscape during Glioblastoma Progression. Nat. Immunol. 2022, 23, 971–984. [Google Scholar] [CrossRef]
- Bowman-Kirigin, J.A.; Desai, R.; Saunders, B.T.; Wang, A.Z.; Schaettler, M.O.; Liu, C.J.; Livingstone, A.J.; Kobayashi, D.K.; Durai, V.; Kretzer, N.M.; et al. The Conventional Dendritic Cell 1 Subset Primes CD8+ T Cells and Traffics Tumor Antigen to Drive Antitumor Immunity in the Brain. Cancer Immunol. Res. 2023, 11, 20–37. [Google Scholar] [CrossRef]
- Friedrich, M.; Hahn, M.; Michel, J.; Sankowski, R.; Kilian, M.; Kehl, N.; Günter, M.; Bunse, T.; Pusch, S.; von Deimling, A.; et al. Dysfunctional Dendritic Cells Limit Antigen-Specific T Cell Response in Glioma. Neuro-Oncology 2022, 14, noac138. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.-R.; Choi, S.J.; Park, J.; Kwon, M.; Chowdhury, T.; Yu, H.J.; Kim, S.; Kang, H.; Kim, K.-M.; Park, S.-H.; et al. Spatial Immune Heterogeneity of Hypoxia-Induced Exhausted Features in High-Grade Glioma. Oncoimmunology 2022, 11, 2026019. [Google Scholar] [CrossRef] [PubMed]
- Garzon-Muvdi, T.; Theodros, D.; Luksik, A.S.; Maxwell, R.; Kim, E.; Jackson, C.M.; Belcaid, Z.; Ganguly, S.; Tyler, B.; Brem, H.; et al. Dendritic Cell Activation Enhances Anti-PD-1 Mediated Immunotherapy against Glioblastoma. Oncotarget 2018, 9, 20681–20697. [Google Scholar] [CrossRef]
- Song, E.; Mao, T.; Dong, H.; Boisserand, L.S.B.; Antila, S.; Bosenberg, M.; Alitalo, K.; Thomas, J.-L.; Iwasaki, A. VEGF-C-Driven Lymphatic Drainage Enables Immunosurveillance of Brain Tumours. Nature 2020, 577, 689–694. [Google Scholar] [CrossRef]
- Hu, X.; Deng, Q.; Ma, L.; Li, Q.; Chen, Y.; Liao, Y.; Zhou, F.; Zhang, C.; Shao, L.; Feng, J.; et al. Meningeal Lymphatic Vessels Regulate Brain Tumor Drainage and Immunity. Cell Res. 2020, 30, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Ma, L.; Xu, H.; Huo, Y.; Luo, J. Meningeal Lymphatics Regulate Radiotherapy Efficacy through Modulating Anti-Tumor Immunity. Cell Res. 2022, 32, 543–554. [Google Scholar] [CrossRef]
- Kikuchi, T.; Abe, T.; Ohno, T. Effects of Glioma Cells on Maturation of Dendritic Cells. J. Neurooncol. 2002, 58, 125–130. [Google Scholar] [CrossRef]
- Williams, M.A.; Rangasamy, T.; Bauer, S.M.; Killedar, S.; Karp, M.; Kensler, T.W.; Yamamoto, M.; Breysse, P.; Biswal, S.; Georas, S.N. Disruption of the Transcription Factor Nrf2 Promotes Pro-Oxidative Dendritic Cells That Stimulate Th2-Like Immunoresponsiveness upon Activation by Ambient Particulate Matter. J. Immunol. 2008, 181, 4545–4559. [Google Scholar] [CrossRef]
- Al-Huseini, L.M.A.; Aw Yeang, H.X.; Sethu, S.; Alhumeed, N.; Hamdam, J.M.; Tingle, Y.; Djouhri, L.; Kitteringham, N.; Park, B.K.; Goldring, C.E.; et al. Nuclear Factor-Erythroid 2 (NF-E2) P45-Related Factor-2 (Nrf2) Modulates Dendritic Cell Immune Function through Regulation of P38 MAPK-CAMP-Responsive Element Binding Protein/Activating Transcription Factor 1 Signaling*. J. Biol. Chem. 2013, 288, 22281–22288. [Google Scholar] [CrossRef]
- Tsai, W.-C.; Hueng, D.-Y.; Lin, C.-R.; Yang, T.C.K.; Gao, H.-W. Nrf2 Expressions Correlate with WHO Grades in Gliomas and Meningiomas. Int. J. Mol. Sci. 2016, 17, 722. [Google Scholar] [CrossRef]
- Wang, J.; Liu, P.; Xin, S.; Wang, Z.; Li, J. Nrf2 Suppresses the Function of Dendritic Cells to Facilitate the Immune Escape of Glioma Cells. Exp. Cell Res. 2017, 360, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Simonds, E.F.; Lu, E.D.; Badillo, O.; Karimi, S.; Liu, E.V.; Tamaki, W.; Rancan, C.; Downey, K.M.; Stultz, J.; Sinha, M.; et al. Deep Immune Profiling Reveals Targetable Mechanisms of Immune Evasion in Immune Checkpoint Inhibitor-Refractory Glioblastoma. J. Immunother. Cancer 2021, 9, e002181. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.W.; Quail, D.F. Immunotherapy for Glioblastoma: Current Progress and Challenges. Front. Immunol. 2021, 12, 676301. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.Q.; Wainwright, D.A.; Lee-Chang, C.; Miska, J.; Sonabend, A.M.; Heimberger, A.B.; Lukas, R.V. Next Steps for Immunotherapy in Glioblastoma. Cancers 2022, 14, 4023. [Google Scholar] [CrossRef]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E.; Sampson, J.H. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297–5303. [Google Scholar] [CrossRef]
- Lee, E.Q.; Chukwueke, U.N.; Hervey-Jumper, S.L.; de Groot, J.F.; Leone, J.P.; Armstrong, T.S.; Chang, S.M.; Arons, D.; Oliver, K.; Verble, K.; et al. Barriers to Accrual and Enrollment in Brain Tumor Trials. Neuro. Oncol. 2019, 21, 1100–1117. [Google Scholar] [CrossRef]
- Bates, A.J.; Couillard, S.A.; Arons, D.F.; Yung, W.K.A.; Vogelbaum, M.; Wen, P.Y.; Kingston, A.E. HOUT-15. Brain Tumor Patient and Caregiver Survey on Clinical Trials: Identifying Attitudes and Parriers to Patient Participation. Neuro. Oncol. 2017, 19, vi109. [Google Scholar] [CrossRef]
- Authier, A.; Farrand, K.J.; Broadley, K.W.R.; Ancelet, L.R.; Hunn, M.K.; Stone, S.; McConnell, M.J.; Hermans, I.F. Enhanced Immunosuppression by Therapy-Exposed Glioblastoma Multiforme Tumor Cells. Int. J. Cancer 2015, 136, 2566–2578. [Google Scholar] [CrossRef]
- De Martino, M.; Padilla, O.; Daviaud, C.; Wu, C.-C.; Gartrell, R.D.; Vanpouille-Box, C. Exploiting Radiation Therapy to Restore Immune Reactivity of Glioblastoma. Front. Oncol. 2021, 11, 671044. [Google Scholar] [CrossRef]
- Di Ianni, N.; Maffezzini, M.; Eoli, M.; Pellegatta, S. Revisiting the Immunological Aspects of Temozolomide Considering the Genetic Landscape and the Immune Microenvironment Composition of Glioblastoma. Front. Oncol. 2021, 11, 747690. [Google Scholar] [CrossRef]
- McGirt, M.J.; Chaichana, K.L.; Gathinji, M.; Attenello, F.J.; Than, K.; Olivi, A.; Weingart, J.D.; Brem, H.; Quiñones-Hinojosa, A.R. Independent Association of Extent of Resection with Survival in Patients with Malignant Brain Astrocytoma. J. Neurosurg. 2009, 110, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liang, B.; Li, Y.I.; Liu, X.; Huang, J.; Li, Y.M. What Is the Advance of Extent of Resection in Glioblastoma Surgical Treatment—A Systematic Review. Chin. Neurosurg. J. 2019, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bolte, A.C.; Dutta, A.B.; Hurt, M.E.; Smirnov, I.; Kovacs, M.A.; McKee, C.A.; Ennerfelt, H.E.; Shapiro, D.; Nguyen, B.H.; Frost, E.L.; et al. Meningeal Lymphatic Dysfunction Exacerbates Traumatic Brain Injury Pathogenesis. Nat. Commun. 2020, 11, 4524. [Google Scholar] [CrossRef]
- Liu, C.; Sage, J.C.; Miller, M.R.; Verhaak, R.G.W.; Hippenmeyer, S.; Vogel, H.; Foreman, O.; Bronson, R.T.; Nishiyama, A.; Luo, L.; et al. Mosaic Analysis with Double Markers Reveals Tumor Cell of Origin in Glioma. Cell 2011, 146, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant Anti-PD-1 Immunotherapy Promotes a Survival Benefit with Intratumoral and Systemic Immune Responses in Recurrent Glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Lee, A.H.; Sun, L.; Mochizuki, A.Y.; Reynoso, J.G.; Orpilla, J.; Chow, F.; Kienzler, J.C.; Everson, R.G.; Nathanson, D.A.; Bensinger, S.J.; et al. Neoadjuvant PD-1 Blockade Induces T Cell and CDC1 Activation but Fails to Overcome the Immunosuppressive Tumor Associated Macrophages in Recurrent Glioblastoma. Nat. Commun. 2021, 12, 6938. [Google Scholar] [CrossRef]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E.; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased Regulatory T-Cell Fraction amidst a Diminished CD4 Compartment Explains Cellular Immune Defects in Patients with Malignant Glioma. Cancer Res. 2006, 66, 3294–3302. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas Promote Immunosuppression through Induction of B7-H1 Expression in Tumor-Associated Macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef]
- Ayasoufi, K.; Pfaller, C.K.; Evgin, L.; Khadka, R.H.; Tritz, Z.P.; Goddery, E.N.; Fain, C.E.; Yokanovich, L.T.; Himes, B.T.; Jin, F.; et al. Brain Cancer Induces Systemic Immunosuppression through Release of Non-Steroid Soluble Mediators. Brain 2020, 143, 3629–3652. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Brem, S.; Campian, J.L.; Trusheim, J.E.; Iwamoto, F.M.; Tran, D.D.; Ansstas, G.; Cobbs, C.S.; Heth, J.A.; et al. Association of Autologous Tumor Lysate-Loaded Dendritic Cell Vaccination with Extension of Survival among Patients with Newly Diagnosed and Recurrent Glioblastoma: A Phase 3 Prospective Externally Controlled Cohort Trial. JAMA Oncol. 2023, 9, 112–121. [Google Scholar] [CrossRef]
- Eoli, M.; Corbetta, C.; Anghileri, E.; Di Ianni, N.; Milani, M.; Cuccarini, V.; Musio, S.; Paterra, R.; Frigerio, S.; Nava, S.; et al. Expansion of Effector and Memory T Cells Is Associated with Increased Survival in Recurrent Glioblastomas Treated with Dendritic Cell Immunotherapy. Neurooncol. Adv. 2019, 1, vdz022. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.-N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus Toxoid and CCL3 Improve Dendritic Cell Vaccines in Mice and Glioblastoma Patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Lowenstein, P.R.; Castro, M.G. Evolutionary Basis of a New Gene- and Immune-Therapeutic Approach for the Treatment of Malignant Brain Tumors: From Mice to Clinical Trials for Glioma Patients. Clin. Immunol. 2018, 189, 43–51. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conarroe, C.A.; Bullock, T.N.J. Ready for Prime Time? Dendritic Cells in High-Grade Gliomas. Cancers 2023, 15, 2902. https://doi.org/10.3390/cancers15112902
Conarroe CA, Bullock TNJ. Ready for Prime Time? Dendritic Cells in High-Grade Gliomas. Cancers. 2023; 15(11):2902. https://doi.org/10.3390/cancers15112902
Chicago/Turabian StyleConarroe, Claire A., and Timothy N. J. Bullock. 2023. "Ready for Prime Time? Dendritic Cells in High-Grade Gliomas" Cancers 15, no. 11: 2902. https://doi.org/10.3390/cancers15112902
APA StyleConarroe, C. A., & Bullock, T. N. J. (2023). Ready for Prime Time? Dendritic Cells in High-Grade Gliomas. Cancers, 15(11), 2902. https://doi.org/10.3390/cancers15112902