
SAM-Competitive EZH2-Inhibitors Induce Platinum Resistance by EZH2-Independent Induction of ABC-Transporters

 , , , , , , ,
, , , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Patient Samples
2.2. EZH2 Expression Analysis
2.3. Cell Lines
2.4. Cell Viability Assay
2.5. Apoptosis Assay
2.6. Cell Cycle Assay
2.7. Mass Spectrometry with Inductive Coupled Plasma (ICP-MS)
2.8. Transduction
2.9. Cell Treatment for Expression Analysis
2.10. Western Blot
2.11. cDNA Synthesis and RT-qPCR
2.12. RNA-Sequencing of Treated Cell Culture Samples
2.13. Statistics
3. Results
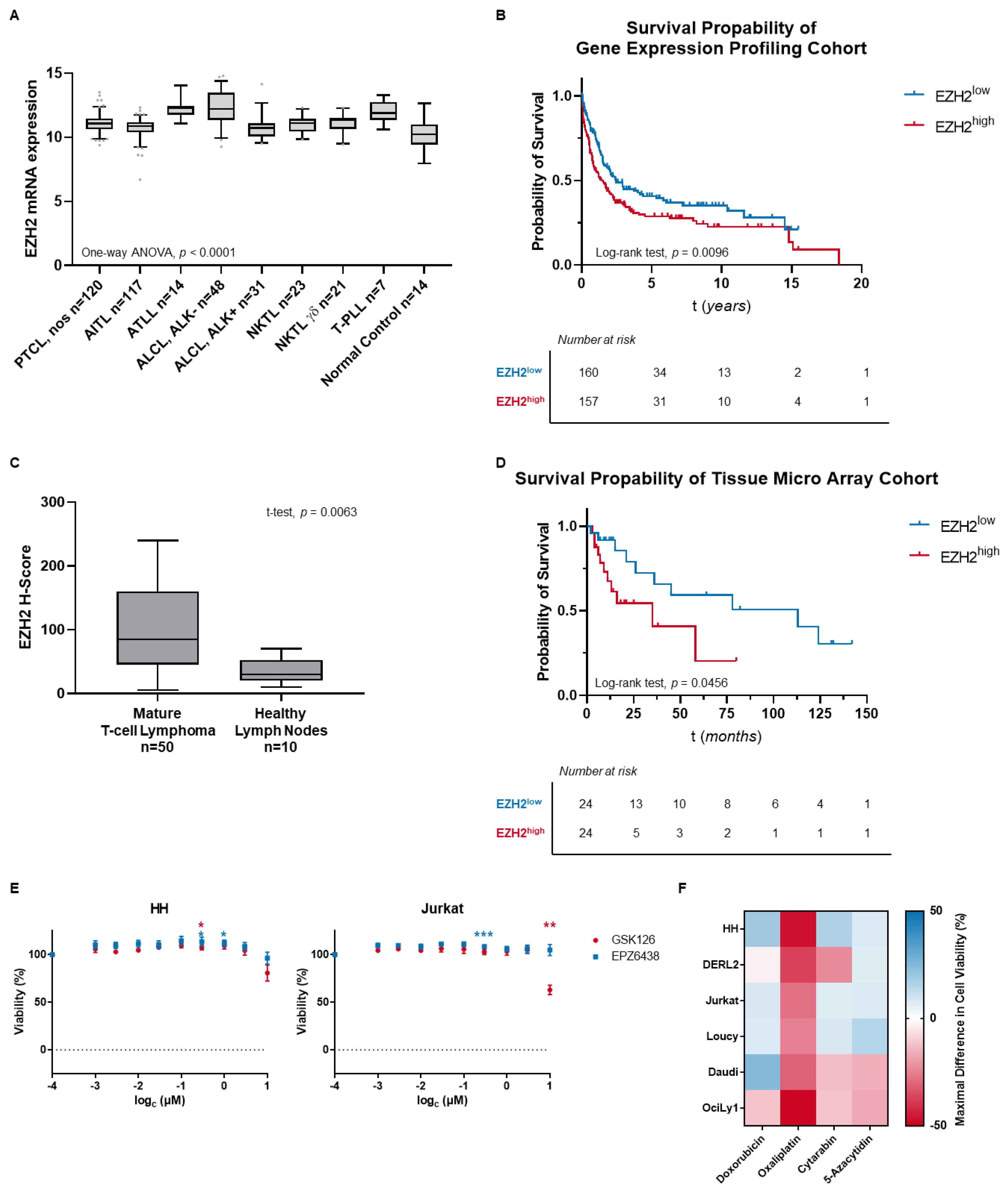
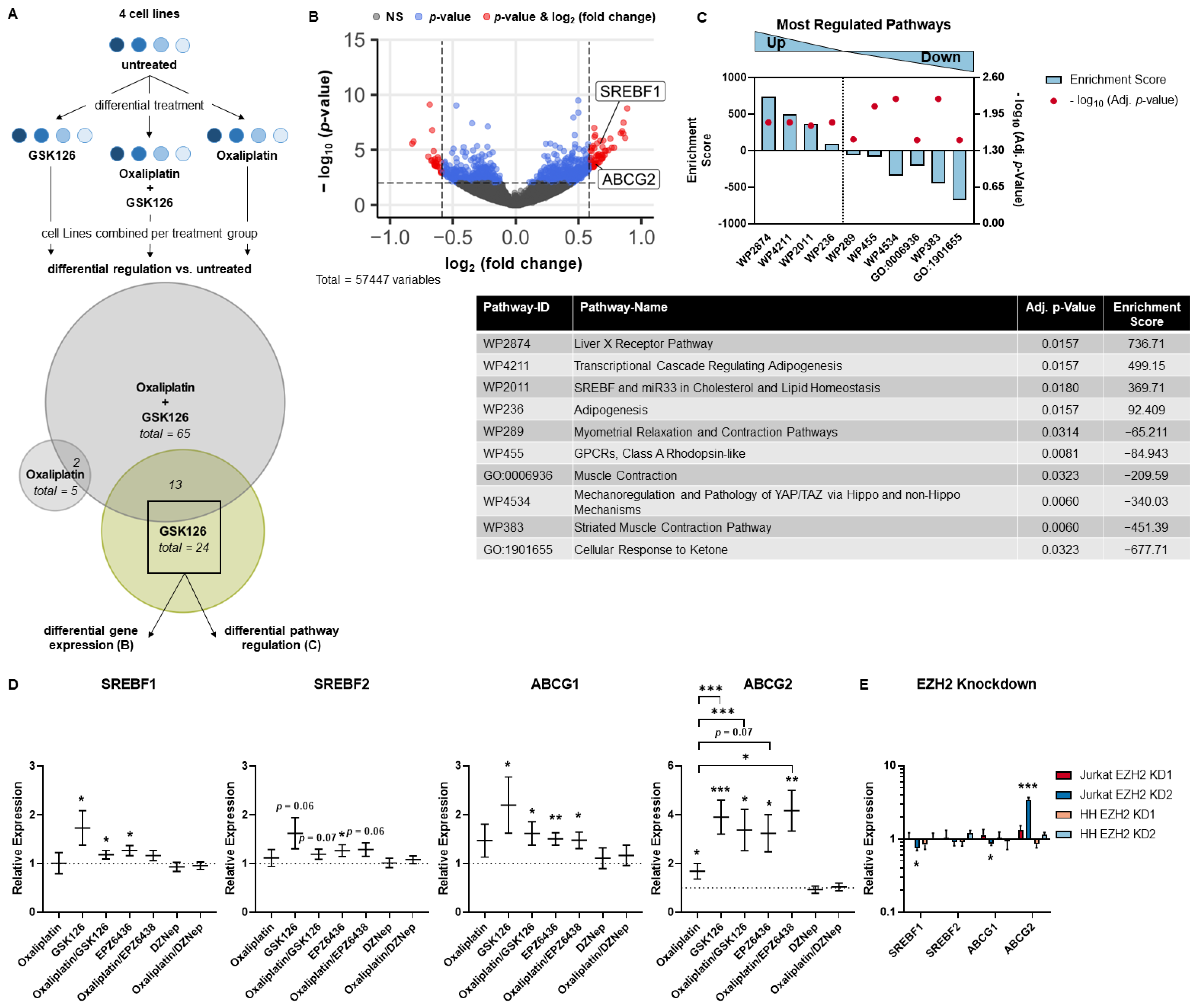
3.1. MTCL Overexpress EZH2, but Selective EZH2 Inhibitors Show No Reduction of Cell Viability
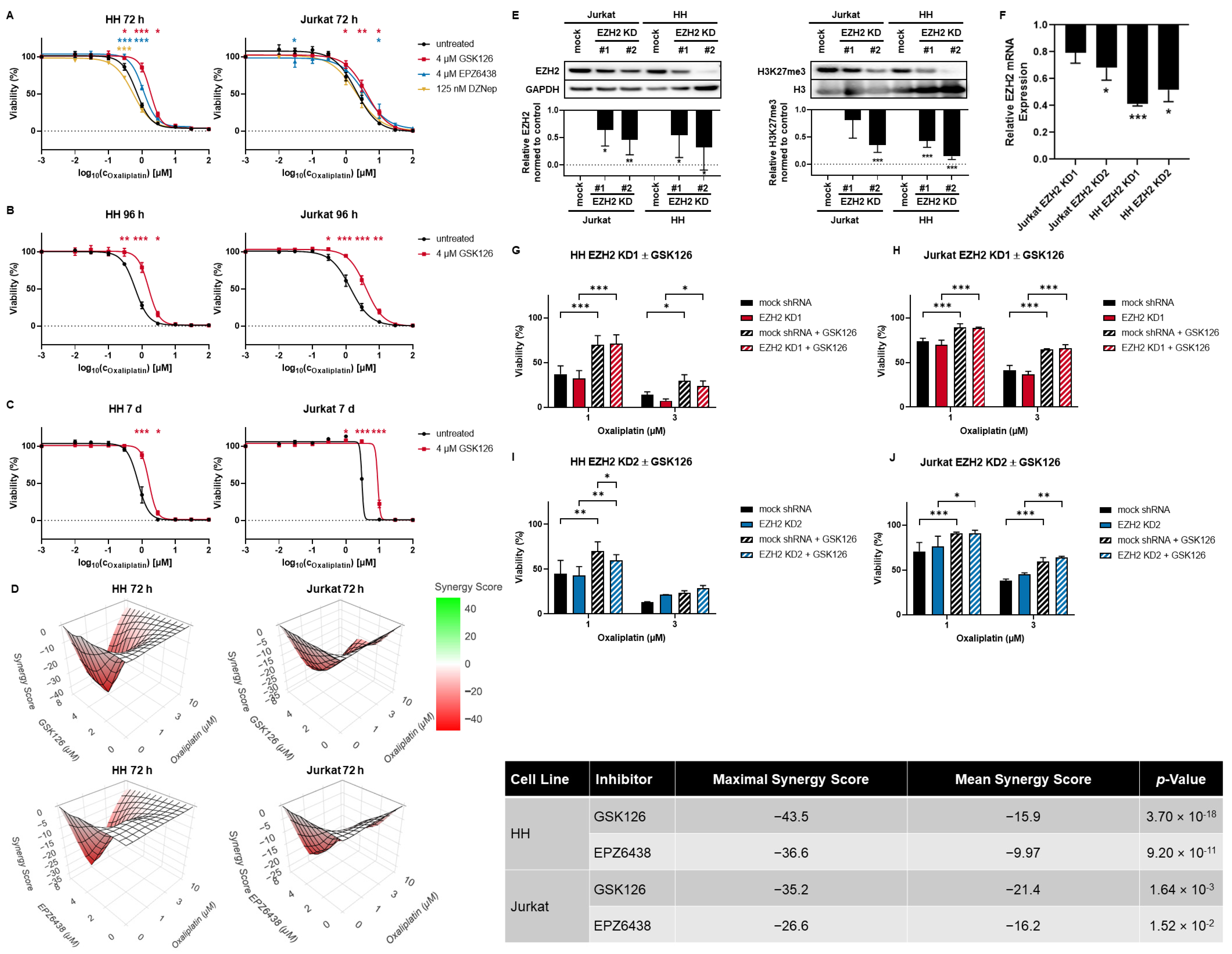
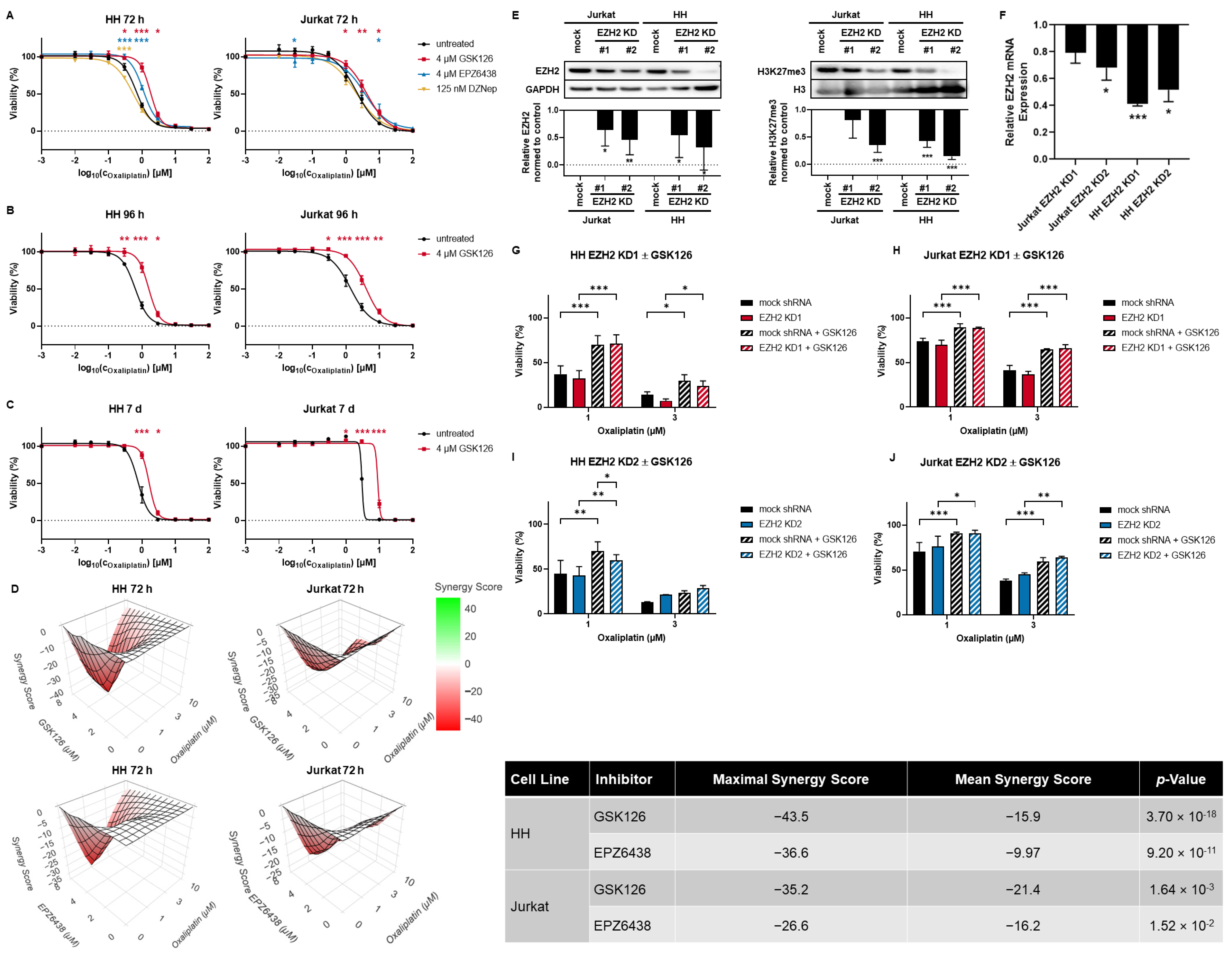
3.2. SAM-Competitive EZH2 Inhibitors Increase Oxaliplatin Resistance
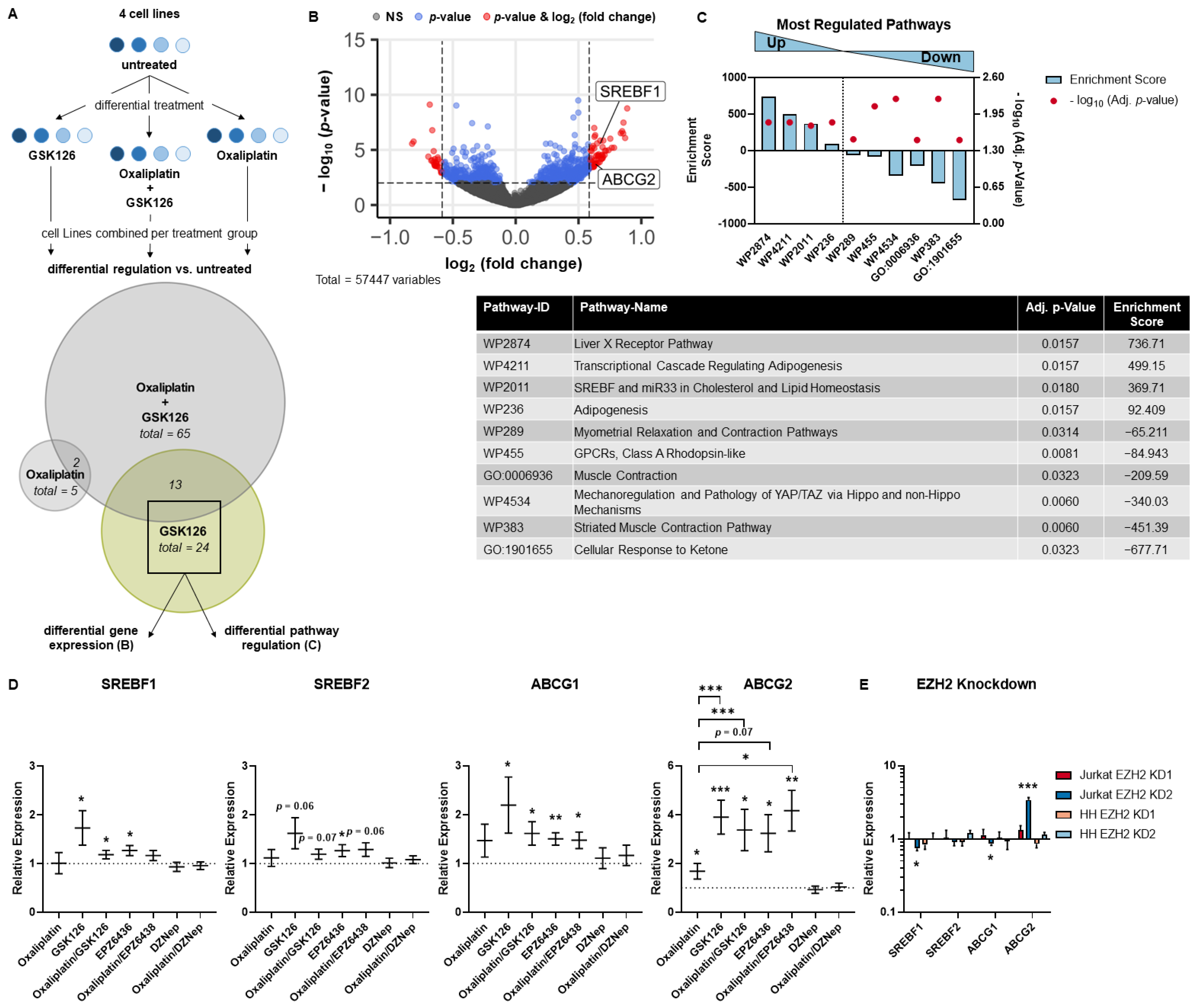
3.3. SREBP and ABC Inhibition Reduce Effects Induced by Pharmacological EZH2 Inhibition
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Iii, A.D.P.; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Li, Z.; Wu, Z.; Aau, M.; Guan, P.; Karuturi, R.M.; Liou, Y.C.; Yu, Q. Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol. Cell 2011, 43, 798–810. [Google Scholar] [CrossRef]
- Margueron, R.; Li, G.; Sarma, K.; Blais, A.; Zavadil, J.; Woodcock, C.L.; Dynlacht, B.D.; Reinberg, D. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol. Cell 2008, 32, 503–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’angelo, V.; Iannotta, A.; Ramaglia, M.; Lombardi, A.; Zarone, M.R.; Desiderio, V.; Affinita, M.C.; Pecoraro, G.; Di Martino, M.; Indolfi, P.; et al. EZH2 is increased in paediatric T-cell acute lymphoblastic leukemia and is a suitable molecular target in combination treatment approaches. J. Exp. Clin. Cancer Res. 2015, 34, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulati, N.; Béguelin, W.; Giulino-Roth, L. Enhancer of zeste homolog 2 (EZH2) inhibitors. Leuk. Lymphoma 2018, 59, 1574–1585. [Google Scholar] [CrossRef]
- Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. World Health Organization Classification of Tumours, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- Vose, J.; Armitage, J.; Weisenburger, D. International peripheral T-cell and natural killer/T-cell lymphoma study: Pathology findings and clinical outcomes. J. Clin. Oncol. 2008, 26, 4124–4130. [Google Scholar] [PubMed]
- Houot, R.; Poeschel, V.; Altmann, B.; Angel, S.; Thurner, L.; Illmer, T.; Andre, M.; Dreyling, M.; Maisonneuve, H.; Tilly, H.; et al. Prolonged Remissions After Nivolumab Plus Gemcitabine/Oxaliplatin in Relapsed/Refractory T-cell Lymphoma. Hemasphere 2022, 6, e672. [Google Scholar] [CrossRef]
- Yao, Y.Y.; Tang, Y.; Zhu, Q.; Zhuang, Y.; Cheng, Y.M.; Wang, L.; Zou, L.F. Gemcitabine, oxaliplatin and dexamethasone as salvage treatment for elderly patients with refractory and relapsed peripheral T-cell lymphoma. Leuk. Lymphoma 2013, 54, 1194–1200. [Google Scholar] [CrossRef]
- Mounier, N.; El Gnaoui, T.; Tilly, H.; Canioni, D.; Sebban, C.; Casasnovas, R.-O.; Delarue, R.; Sonet, A.; Beaussart, P.; Petrella, T.; et al. Rituximab plus gemcitabine and oxaliplatin in patients with refractory/relapsed diffuse large B-cell lymphoma who are not candidates for high-dose therapy. A phase II Lymphoma Study Association trial. Haematologica 2013, 98, 1726–1731. [Google Scholar] [CrossRef]
- Zhang, H.; Lv, H.; Jia, X.; Hu, G.; Kong, L.; Zhang, T.; Li, L.; Pan, Y.; Zhai, Q.; Meng, B.; et al. Clinical significance of enhancer of zeste homolog 2 and histone deacetylases 1 and 2 expression in peripheral T-cell lymphoma. Oncol. Lett. 2019, 18, 1415–1423. [Google Scholar] [CrossRef] [Green Version]
- Schümann, F.L.; Groß, E.; Bauer, M.; Rohde, C.; Sandmann, S.; Terziev, D.; Müller, L.P.; Posern, G.; Wienke, A.; Fend, F.; et al. Divergent Effects of EZH1 and EZH2 Protein Expression on the Prognosis of Patients with T-Cell Lymphomas. Biomedicines 2021, 9, 1842. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Y.; Gong, Y.; Zhang, T.; Huang, J.; Tan, Z.; Xue, L. Finding an easy way to harmonize: A review of advances in clinical research and combination strategies of EZH2 inhibitors. Clin. Epigenet. 2021, 13, 62. [Google Scholar] [CrossRef] [PubMed]
- Glazer, R.I.; Hartman, K.D.; Knode, M.C.; Richard, M.M.; Chiang, P.K.; Tseng, C.K.; Marquez, V.E. 3-Deazaneplanocin: A new and potent inhibitor of S-adenosylhomocysteine hydrolase and its effects on human promyelocytic leukemia cell line HL-60. Biochem. Biophys. Res. Commun. 1986, 135, 688–694. [Google Scholar] [CrossRef]
- Miranda, T.B.; Cortez, C.C.; Yoo, C.B.; Liang, G.; Abe, M.; Kelly, T.K.; Marquez, V.E.; Jones, P.A. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol. Cancer Ther. 2009, 8, 1579–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Selvaraju, S.; Curtin, M.L.; Jakob, C.G.; Zhu, H.; Comess, K.M.; Shaw, B.; The, J.; Lima-Fernandes, E.; Szewczyk, M.M.; et al. The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat. Chem. Biol. 2017, 13, 389–395. [Google Scholar] [CrossRef]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Xu, B.; Konze, K.D.; Jin, J.; Wang, G.G. Targeting EZH2 and PRC2 dependence as novel anticancer therapy. Exp. Hematol. 2015, 43, 698–712. [Google Scholar] [CrossRef] [Green Version]
- Yamagishi, M.; Hori, M.; Fujikawa, D.; Ohsugi, T.; Honma, D.; Adachi, N.; Katano, H.; Hishima, T.; Kobayashi, S.; Nakano, K.; et al. Targeting Excessive EZH1 and EZH2 Activities for Abnormal Histone Methylation and Transcription Network in Malignant Lymphomas. Cell Rep. 2019, 29, 2321–2337.e7. [Google Scholar] [CrossRef]
- Ishitsuka, K.; Izutsu, K.; Maruyama, D.; Makita, S.; Jacobsen, E.D.; Horwitz, S.; Kusumoto, S.; Allen, P.; Porcu, P.; Imaizumi, Y.; et al. First-in-Human Study of the Ezh1 and Ezh2 Dual Inhibitor Valemetostat Tosylate (DS-3201B) In Patients with Relapsed or Refractory Non-Hodgkin Lymphomas. Hematol. Oncol. 2021, 39, 38–40. [Google Scholar] [CrossRef]
- Kuser-Abali, G.; Gong, L.; Yan, J.; Liu, Q.; Zeng, W.; Williamson, A.; Lim, C.B.; Molloy, M.E.; Little, J.B.; Huang, L.; et al. An EZH2-mediated epigenetic mechanism behind p53-dependent tissue sensitivity to DNA damage. Proc. Natl. Acad. Sci. USA 2018, 115, 3452–3457. [Google Scholar] [CrossRef] [Green Version]
- Göllner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.-U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Ariës, I.M.; Bodaar, K.; Karim, S.A.; Ni Chonghaile, T.; Hinze, L.; Burns, M.A.; Pfirrmann, M.; Degar, J.; Landrigan, J.T.; Balbach, S.; et al. PRC2 loss induces chemoresistance by repressing apoptosis in T cell acute lymphoblastic leukemia. J. Exp. Med. 2018, 215, 3094–3114. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Wright, G.; Wang, C.; Rosenwald, A.; Gascoyne, R.D.; Weisenburger, D.D.; Greiner, T.C.; Smith, L.; Guo, S.; Wilcox, R.A.; et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood 2014, 123, 2915–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detre, S.; Jotti, G.S.; Dowsett, M. A “quickscore” method for immunohistochemical semiquantitation: Validation for oestrogen receptor in breast carcinomas. J. Clin. Pathol. 1995, 48, 876–878. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.; Wang, W.; Aldahdooh, J.; Malyutina, A.; Shadbahr, T.; Tanoli, Z.; Pessia, A.; Tang, J. SynergyFinder Plus: Toward Better Interpretation and Annotation of Drug Combination Screening Datasets. Genom. Proteom. Bioinform. 2022, 20, 587–596. [Google Scholar] [CrossRef]
- Chang, C.T.; Ang, J.Y.; Wong, J.M.; Tan, S.S.; Chin, S.K.; Lim, A.B.; Tan, W.H.; Yuen, K.H. Bioequivalence and pharmacokinetic comparison of two fixed dose combination of Metformin/Glibenclamide formulations in healthy subjects under fed condition. Med. J. Malaysia 2020, 75, 286–291. [Google Scholar]
- Barr, M.P.; Gray, S.G.; Hoffmann, A.C.; Hilger, R.A.; Thomale, J.; O’flaherty, J.D.; Fennell, D.A.; Richard, D.; O’leary, J.J.; O’byrne, K.J. Generation and characterisation of cisplatin-resistant non-small cell lung cancer cell lines displaying a stem-like signature. PLoS ONE 2013, 8, e54193. [Google Scholar] [CrossRef] [Green Version]
- Melnikova, M.; Wauer, U.S.; Mendus, D.; Hilger, R.A.; Oliver, T.G.; Mercer, K.; Gohlke, B.O.; Erdmann, K.; Niederacher, D.; Neubauer, H.; et al. Diphenhydramine increases the therapeutic window for platinum drugs by simultaneously sensitizing tumor cells and protecting normal cells. Mol. Oncol. 2020, 14, 686–703. [Google Scholar] [CrossRef]
- Chernyakov, D.; Groß, A.; Fischer, A.; Bornkessel, N.; Schultheiss, C.; Gerloff, D.; Edemir, B. Loss of RANBP3L leads to transformation of renal epithelial cells towards a renal clear cell carcinoma like phenotype. J. Exp. Clin. Cancer Res. 2021, 40, 226. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Durinck, S.; Spellman, P.T.; Birney, E.; Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009, 4, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genomebiol. Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Białopiotrowicz, E.; Noyszewska-Kania, M.; Kachamakova-Trojanowska, N.; Łoboda, A.; Cybulska, M.; Grochowska, A.; Kopczyński, M.; Mikula, M.; Prochorec-Sobieszek, M.; Firczuk, M.; et al. Serine Biosynthesis Pathway Supports MYC–miR-494–EZH2 Feed-Forward Circuit Necessary to Maintain Metabolic and Epigenetic Reprogramming of Burkitt Lymphoma Cells. Cancers 2020, 12, 580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Tang, H.; Wang, K.; Zheng, Y.; Feng, J.; Dong, H.; Jin, Y.; Cao, C.; Chen, X.; Gao, G. Pharmacological inhibition of EZH2 combined with DNA-damaging agents interferes with the DNA damage response in MM cells. Mol. Med. Rep. 2019, 19, 4249–4255. [Google Scholar] [CrossRef] [Green Version]
- Knutson, S.K.; Warholic, N.M.; Johnston, L.D.; Klaus, C.R.; Wigle, T.J.; Iwanowicz, D.; Littlefield, B.A.; Porter-Scott, M.; Smith, J.J.; Moyer, M.P.; et al. Synergistic Anti-Tumor Activity of EZH2 Inhibitors and Glucocorticoid Receptor Agonists in Models of Germinal Center Non-Hodgkin Lymphomas. PLoS ONE 2014, 9, e111840. [Google Scholar] [CrossRef]
- Yang, P.-M.; Hong, Y.-H.; Hsu, K.-C.; Liu, T.-P. p38α/S1P/SREBP2 activation by the SAM-competitive EZH2 inhibitor GSK343 limits its anticancer activity but creates a druggable vulnerability in hepatocellular carcinoma. Am. J. Cancer Res. 2019, 9, 2120–2139. [Google Scholar]
- Richly, H.; Aloia, L.; Di Croce, L. Roles of the Polycomb group proteins in stem cells and cancer. Cell Death Dis. 2011, 2, e204. [Google Scholar] [CrossRef] [Green Version]
- Sarkozy, C.; Morschhauser, F.; Dubois, S.; Molina, T.; Michot, J.M.; Cullières-Dartigues, P.; Suttle, B.; Karlin, L.; Le Gouill, S.; Picquenot, J.-M.; et al. A LYSA Phase Ib Study of Tazemetostat (EPZ-6438) plus R-CHOP in Patients with Newly Diagnosed Diffuse Large B-Cell Lymphoma (DLBCL) with Poor Prognosis Features. Clin. Cancer Res. 2020, 26, 3145–3153. [Google Scholar] [CrossRef] [Green Version]
- Morschhauser, F.; Tilly, H.; Chaidos, A.; McKay, P.; Phillips, T.; Assouline, S.; Batlevi, C.L.; Campbell, P.; Ribrag, V.; Damaj, G.L.; et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020, 21, 1433–1442. [Google Scholar] [CrossRef]
- Fujikawa, D.; Nakagawa, S.; Hori, M.; Kurokawa, N.; Soejima, A.; Nakano, K.; Yamochi, T.; Nakashima, M.; Kobayashi, S.; Tanaka, Y.; et al. Polycomb-dependent epigenetic landscape in adult T-cell leukemia. Blood 2016, 127, 1790–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoy, S.M. Tazemetostat: First Approval. Drugs 2020, 80, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Liu, Y.; Hsu, Y.-J.; Fujiwara, Y.; Kim, J.; Mao, X.; Yuan, G.-C.; Orkin, S.H. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol. Cell 2008, 32, 491–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ecker, J.; Langmann, T.; Moehle, C.; Schmitz, G. Isomer specific effects of Conjugated Linoleic Acid on macrophage ABCG1 transcription by a SREBP-1c dependent mechanism. Biochem. Biophys. Res. Commun. 2007, 352, 805–811. [Google Scholar] [CrossRef]
- Choi, C.-H. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int. 2005, 5, 30. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Song, G.; Xu, Z.; Bu, Y.; Chang, F.; Jia, F.; Xiao, X.; Ren, X.; Zhang, M.; Jia, Q. Oxaliplatin resistance is enhanced by saracatinib via upregulation Wnt-ABCG1 signaling in hepatocellular carcinoma. BMC Cancer 2020, 20, 31. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Zhang, J.-T. Multidrug resistance in cancer chemotherapy and xenobiotic protection mediated by the half ATP-binding cassette transporter ABCG2. Curr. Med. Chem. Anticancer Agents 2004, 4, 31–42. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [Green Version]
- Morin, R.; Mungall, K.; Pleasance, E.; Mungall, A.; Goya, R.; Huff, R.; Scott, D.W.; Ding, J.; Roth, A.; Chiu, R.; et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood 2013, 122, 1256–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568.e14. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Groß, E.; Hilger, R.-A.; Schümann, F.L.; Bauer, M.; Bouska, A.; Rohde, C.; Willscher, E.; Lützkendorf, J.; Müller, L.P.; Edemir, B.; et al. SAM-Competitive EZH2-Inhibitors Induce Platinum Resistance by EZH2-Independent Induction of ABC-Transporters. Cancers 2023, 15, 3043. https://doi.org/10.3390/cancers15113043
Groß E, Hilger R-A, Schümann FL, Bauer M, Bouska A, Rohde C, Willscher E, Lützkendorf J, Müller LP, Edemir B, et al. SAM-Competitive EZH2-Inhibitors Induce Platinum Resistance by EZH2-Independent Induction of ABC-Transporters. Cancers. 2023; 15(11):3043. https://doi.org/10.3390/cancers15113043
Chicago/Turabian StyleGroß, Elisabeth, Ralf-Axel Hilger, Franziska Lea Schümann, Marcus Bauer, Alyssa Bouska, Christian Rohde, Edith Willscher, Jana Lützkendorf, Lutz Peter Müller, Bayram Edemir, and et al. 2023. "SAM-Competitive EZH2-Inhibitors Induce Platinum Resistance by EZH2-Independent Induction of ABC-Transporters" Cancers 15, no. 11: 3043. https://doi.org/10.3390/cancers15113043
APA StyleGroß, E., Hilger, R.-A., Schümann, F. L., Bauer, M., Bouska, A., Rohde, C., Willscher, E., Lützkendorf, J., Müller, L. P., Edemir, B., Mueller, T., Herling, M., Binder, M., Wickenhauser, C., Iqbal, J., Posern, G., & Weber, T. (2023). SAM-Competitive EZH2-Inhibitors Induce Platinum Resistance by EZH2-Independent Induction of ABC-Transporters. Cancers, 15(11), 3043. https://doi.org/10.3390/cancers15113043