
Precision Medicine: Disease Subtyping and Tailored Treatment



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
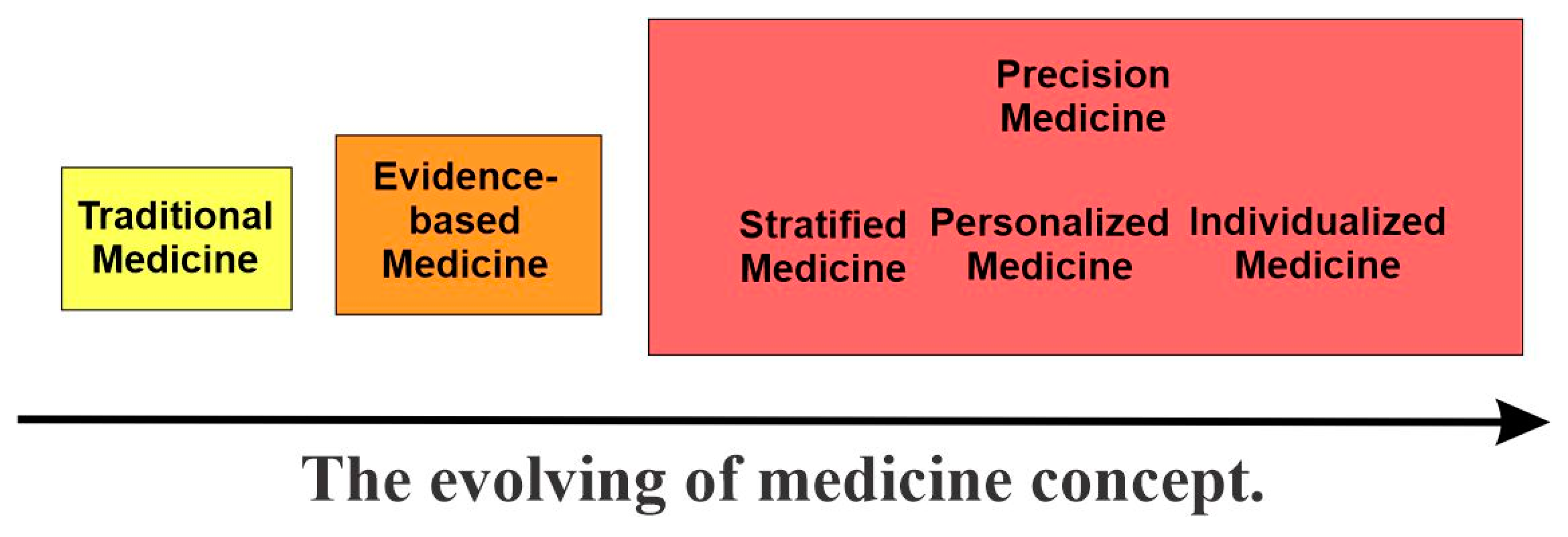
2. The Evolving of Medicine Concepts and the Emerging of Precision Medicine
2.1. Traditional Medicine and Evidence-Based Medicine
2.2. The Emergence of Precision Medicine
2.2.1. Overview
2.2.2. Stratified Medicine
2.2.3. Personalized Medicine
2.2.4. Individualized Medicine
2.2.5. P4 Medicine
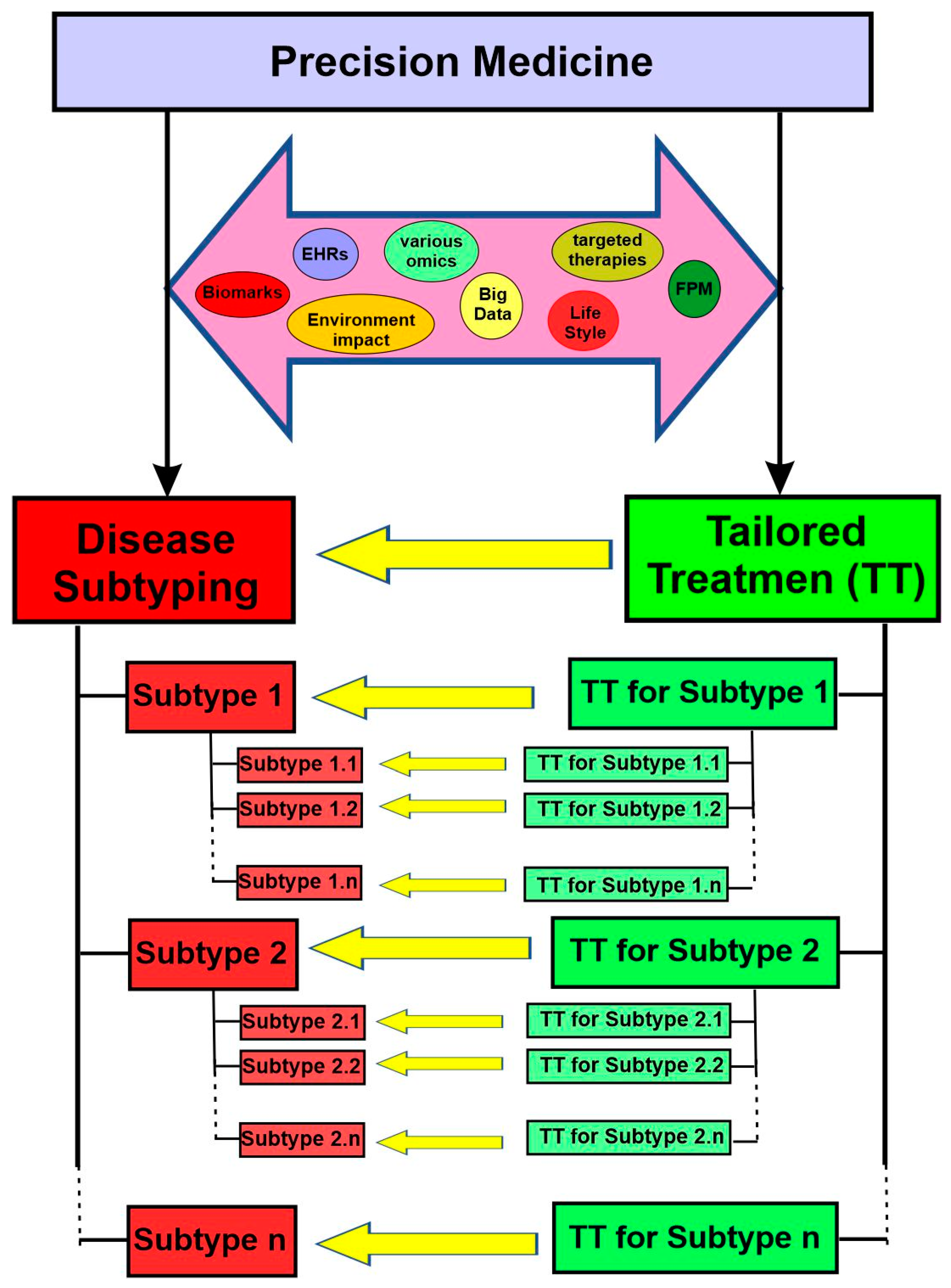
3. Central Tenets and Two Essential Objectives of Precision Medicine
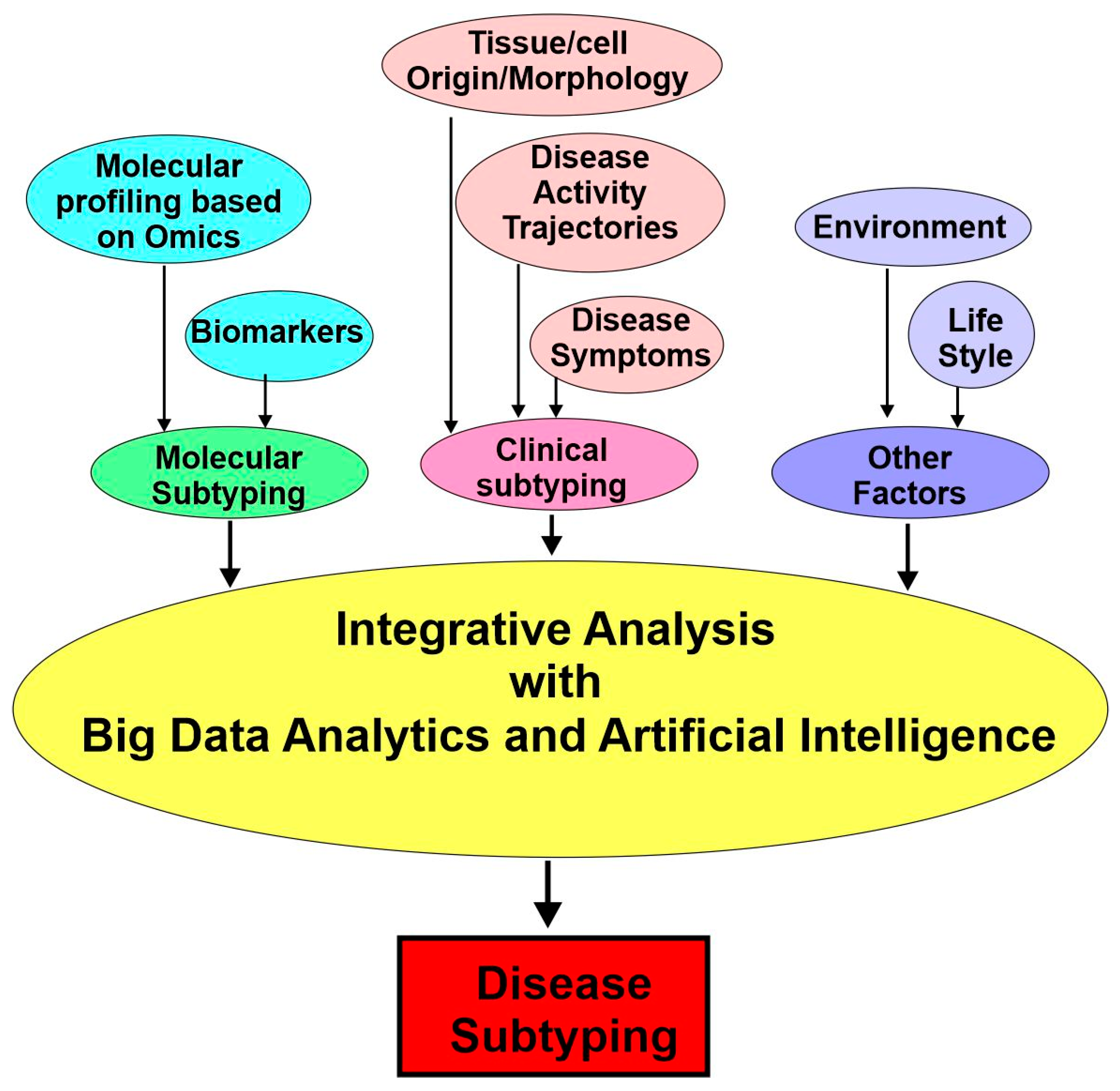
4. Disease Subtyping
4.1. Overview
4.2. Approaches for Disease Subtyping in Precision Medicine
4.3. Molecular Subtyping: Biomarkers
4.3.1. Diagnostic Biomarkers
4.3.2. Prognostic Biomarkers
4.3.3. Predictive Biomarkers
4.3.4. Other Biomarkers
4.3.5. Biomarker Combinations
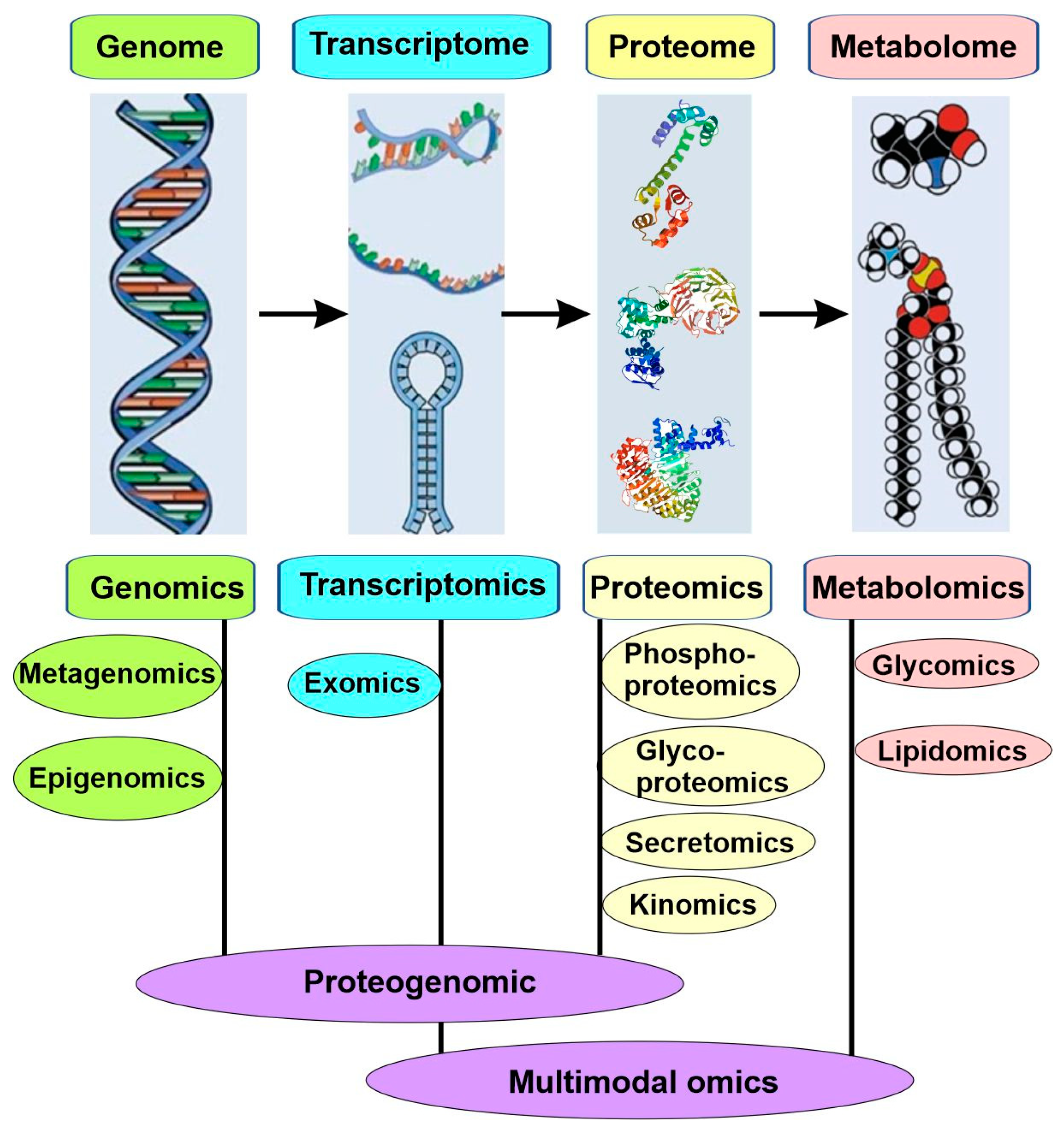
4.4. Molecular Subtyping: -Omics
4.4.1. Genomics
4.4.2. Epigenomics
4.4.3. Transcriptomics
4.4.4. Proteomics
4.4.5. Metabolomics
4.4.6. Microbiomics and Metagenomics
4.4.7. Proteogenomics and Multimodal Omics
4.5. Clinically Enriched Subtypes and Deep Phenotyping
4.6. Integrative Analysis
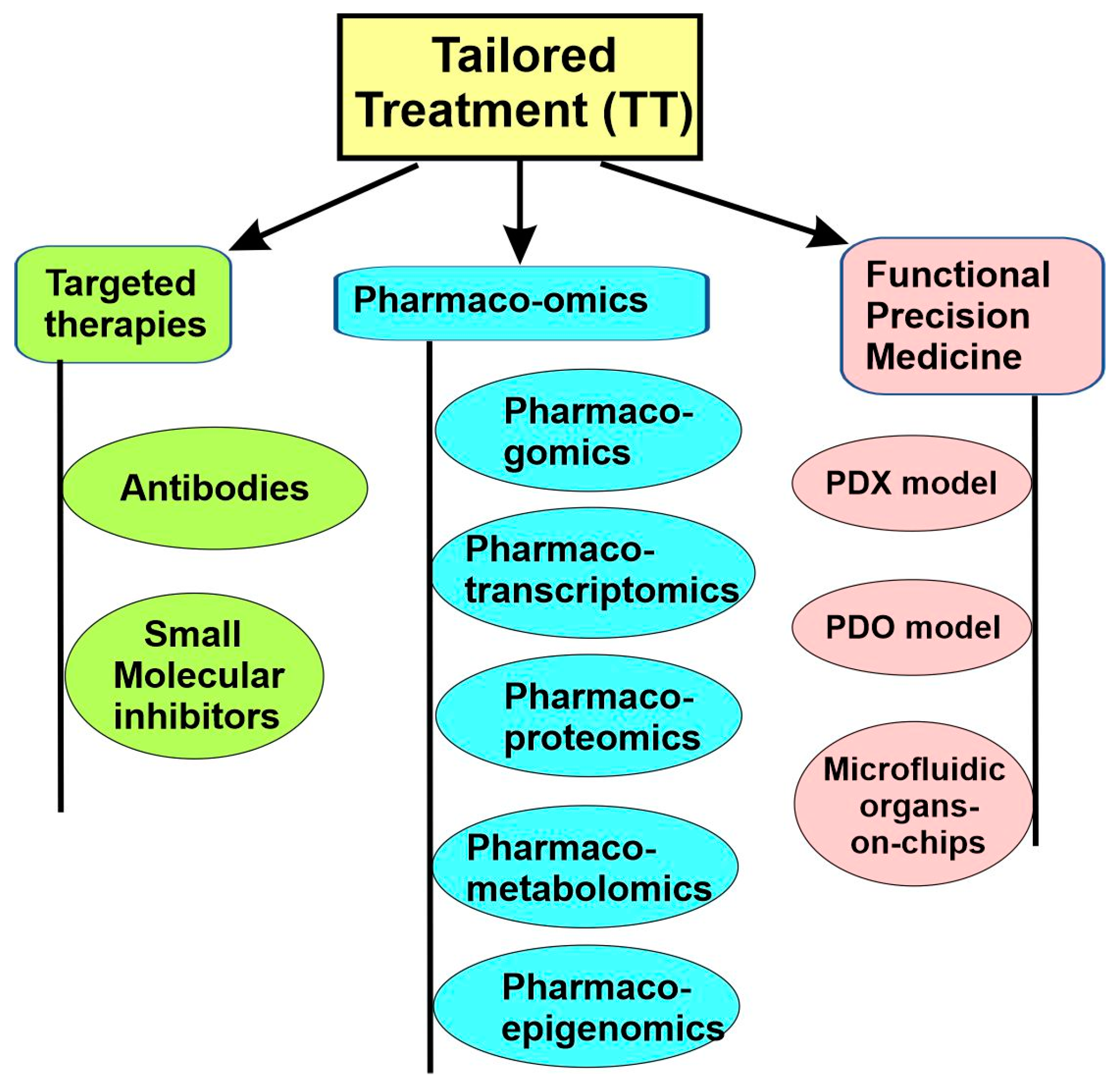
5. Tailored Treatment for the Disease Subtypes
5.1. Targeted Therapies
5.2. Targeted Therapy for Cancers
5.2.1. Antibodies
Overview
Therapeutic Antibodies Based on Natural Properties
Antibody–Drug Conjugates and Antibody–Radionuclide Conjugates
Engineered Antibodies Targeting Cytotoxic T Cells
5.2.2. Small Molecule Inhibitors
5.3. Pharmaco-Omics
5.3.1. Pharmacogenomics
5.3.2. Pharmacotranscriptomics
5.3.3. Pharmacoepigenetics
5.3.4. Pharmacoproteomics
5.3.5. Pharmacometabolomics
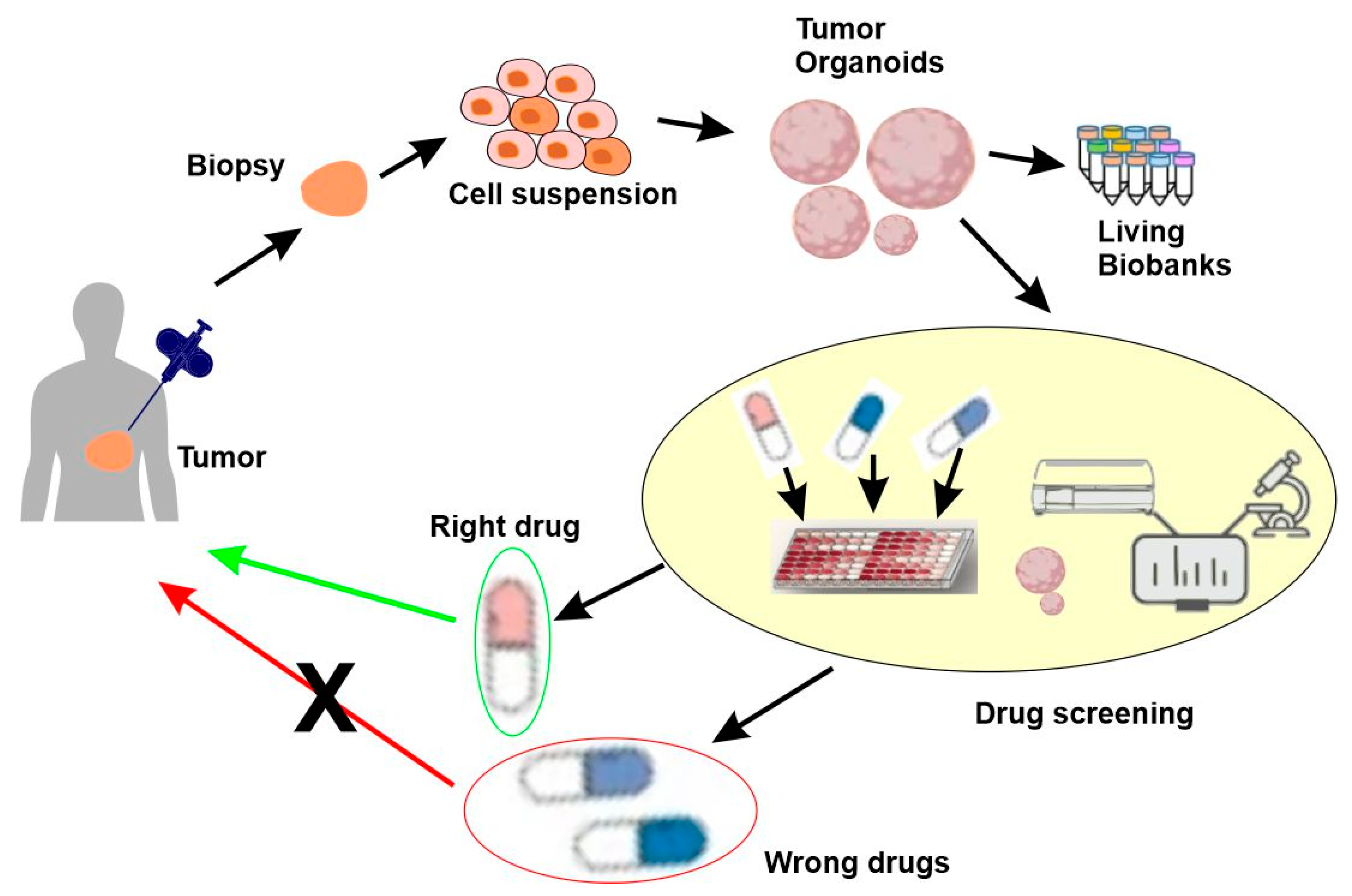
5.4. Functional Precision Medicine
5.4.1. Overview
5.4.2. Patient-Derived Xenograft (PDX) Models
5.4.3. Patient-Derived Organoids (PDOs)
5.4.4. Microfluidic Organs-on-Chips
6. Other Aspects of Precision Medicine
6.1. Environmental, Social, and Behavioral Factors
6.2. Electronic Health Records
6.3. Digital Health
6.4. Big Data Analytics and Artificial Intelligence
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Misale, S.; Bozic, I.; Tong, J.; Peraza-Penton, A.; Lallo, A.; Baldi, F.; Lin, K.H.; Truini, M.; Trusolino, L.; Bertotti, A.; et al. Vertical suppression of the EGFR pathway prevents onset of resistance in colorectal cancers. Nat. Commun. 2015, 6, 8305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litman, T. Personalized medicine-concepts, technologies, and applications in inflammatory skin diseases. APMIS 2019, 127, 386–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sackett, D.L. Evidence-based medicine. Semin. Perinatol. 1997, 21, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Djulbegovic, B.; Guyatt, G.H. Progress in evidence-based medicine: A quarter century on. Lancet 2017, 390, 415–423. [Google Scholar] [CrossRef]
- Masic, I.; Miokovic, M.; Muhamedagic, B. Evidence based medicine—New approaches and challenges. Acta. Inform. Med. 2008, 16, 219–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckmann, J.S.; Lew, D. Reconciling evidence-based medicine and precision medicine in the era of big data: Challenges and opportunities. Genome Med. 2016, 8, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannidis, J.P.A.; Khoury, M.J. Evidence-based medicine and big genomic data. Hum. Mol. Genet. 2018, 27, R2–R7. [Google Scholar] [CrossRef] [Green Version]
- Gordon, E.; Koslow, S. Integrative Neuroscience and Personalized Medicine; Oxford University Press: Oxford, UK, 2010. [Google Scholar]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.Y. Scientific advance in acupuncture. Am. J. Chin. Med. 1979, 7, 53–75. [Google Scholar] [CrossRef] [PubMed]
- National Research Council (US) Committee on a Framework for Developing a New Taxonomy of Disease. The National Academies Collection: Reports funded by National Institutes of Health. In Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Bell, J. Stratified medicines: Towards better treatment for disease. Lancet 2014, 383 (Suppl. 1), S3–S5. [Google Scholar] [CrossRef] [PubMed]
- Trusheim, M.R.; Berndt, E.R.; Douglas, F.L. Stratified medicine: Strategic and economic implications of combining drugs and clinical biomarkers. Nat. Rev. Drug Discov. 2007, 6, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W.M. Can personalized medicine survive? Can. Fam. Physician 1971, 17, 29–88. [Google Scholar] [PubMed]
- Langreth, R.; Waldholz, M. New era of personalized medicine: Targeting drugs for each unique genetic profile. Oncologist 1999, 4, 426–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jørgensen, J.T. Twenty Years with Personalized Medicine: Past, Present, and Future of Individualized Pharmacotherapy. Oncologist 2019, 24, e432–e440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topol, E.J. Individualized medicine from prewomb to tomb. Cell 2014, 157, 241–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischel-Ghodsian, N. Mitochondrial DNA mutations and diabetes: Another step toward individualized medicine. Ann. Intern. Med. 2001, 134, 777–779. [Google Scholar] [CrossRef] [PubMed]
- Esslinger, S. Primary nursing care: Individualized patient care. OH. Osteopath. Hosp. 1979, 23, 8–10. [Google Scholar]
- Ghia, J.N.; Gregg, J.M. The University of North Carolina Pain Center. I. Organization and function. Anesth. Prog. 1982, 29, 41–46. [Google Scholar]
- Auffray, C.; Charron, D.; Hood, L. Predictive, preventive, personalized and participatory medicine: Back to the future. Genome Med. 2010, 2, 57. [Google Scholar] [CrossRef]
- Hood, L.; Friend, S.H. Predictive, personalized, preventive, participatory (P4) cancer medicine. Nat. Rev.Clin. Oncol. 2011, 8, 184–187. [Google Scholar] [CrossRef]
- Wu, P.Y.; Cheng, C.W.; Kaddi, C.D.; Venugopalan, J.; Hoffman, R.; Wang, M.D. -Omic and Electronic Health Record Big Data Analytics for Precision Medicine. IEEE Trans. Bio.-Med. Eng. 2017, 64, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Saria, S.; Goldenberg, A. Subtyping: What It is and Its Role in Precision Medicine. IEEE Intell. Syst. 2015, 30, 70–75. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Wei, S.; Tao, J.; Xu, J.; Chen, X.; Wang, Z.; Zhang, N.; Zuo, L.; Jia, Z.; Chen, H.; Sun, H.; et al. Ten Years of EWAS. Adv. Sci. 2021, 8, e2100727. [Google Scholar] [CrossRef] [PubMed]
- de Giambattista, C.; Ventura, P.; Trerotoli, P.; Margari, M.; Palumbi, R.; Margari, L. Subtyping the Autism Spectrum Disorder: Comparison of Children with High Functioning Autism and Asperger Syndrome. J. Autism. Dev. Disord. 2019, 49, 138–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, R.; Chupp, G. Phenotypes and endotypes of adult asthma: Moving toward precision medicine. J. Allergy Clin. Immunol. 2019, 144, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D. Precision medicine and management of rheumatoid arthritis. J. Autoimmun. 2020, 110, 102405. [Google Scholar] [CrossRef]
- Toro-Domínguez, D.; Alarcón-Riquelme, M.E. Precision medicine in autoimmune diseases: Fact or fiction. Rheumatology 2021, 60, 3977–3985. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.A.; Loscalzo, J. Emerging Role of Precision Medicine in Cardiovascular Disease. Circ. Res. 2018, 122, 1302–1315. [Google Scholar] [CrossRef]
- Eggers, C.; Pedrosa, D.J.; Kahraman, D.; Maier, F.; Lewis, C.J.; Fink, G.R.; Schmidt, M.; Timmermann, L. Parkinson subtypes progress differently in clinical course and imaging pattern. PLoS ONE 2012, 7, e46813. [Google Scholar] [CrossRef] [Green Version]
- Lötvall, J.; Akdis, C.A.; Bacharier, L.B.; Bjermer, L.; Casale, T.B.; Custovic, A.; Lemanske, R.F., Jr.; Wardlaw, A.J.; Wenzel, S.E.; Greenberger, P.A. Asthma endotypes: A new approach to classification of disease entities within the asthma syndrome. J. Allergy Clin. Immunol. 2011, 127, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.R.; Hripcsak, G.; Shen, Y.; Chung, W.K.; Weng, C. Defining a comprehensive verotype using electronic health records for personalized medicine. J. Am. Med. Inform. Assoc. 2013, 20, e232–e238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, F.S. Exceptional opportunities in medical science: A view from the National Institutes of Health. JAMA 2015, 313, 131–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyawali, B. Point: The Imprecise Pursuit of Precision Medicine: Are Biomarkers to Blame? J. Natl. Compr. Cancer Netw. 2017, 15, 859–862. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Subbiah, V.; Kurzrock, R. Counterpoint: Successes in the Pursuit of Precision Medicine: Biomarkers Take Credit. J. Natl. Compr. Cancer Netw. 2017, 15, 863–866. [Google Scholar] [CrossRef] [Green Version]
- Thompson, I.M.; Ankerst, D.P.; Chi, C.; Goodman, P.J.; Tangen, C.M.; Lucia, M.S.; Feng, Z.; Parnes, H.L.; Coltman, C.A., Jr. Assessing prostate cancer risk: Results from the Prostate Cancer Prevention Trial. J. Natl. Cancer Inst. 2006, 98, 529–534. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z. Personalized medicine: The future of breast cancer management. Breast Cancer Manag. 2015, 4, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Ballman, K.V. Biomarker: Predictive or Prognostic? J. Clin. Oncol. 2015, 33, 3968–3971. [Google Scholar] [CrossRef]
- Baselga, J.; Cortés, J.; Im, S.A.; Clark, E.; Ross, G.; Kiermaier, A.; Swain, S.M. Biomarker analyses in CLEOPATRA: A phase III, placebo-controlled study of pertuzumab in human epidermal growth factor receptor 2-positive, first-line metastatic breast cancer. J. Clin. Oncol. Oncol. 2014, 32, 3753–3761. [Google Scholar] [CrossRef]
- Yin, L.; Duan, J.J.; Bian, X.W.; Yu, S.C. Triple-negative breast cancer molecular subtyping and treatment progress. Breast Cancer Res. 2020, 22, 61. [Google Scholar] [CrossRef]
- Bianchini, G.; De Angelis, C.; Licata, L.; Gianni, L. Treatment landscape of triple-negative breast cancer-expanded options, evolving needs. Nat. Rev. Clin. Oncol. 2022, 19, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Lluch, A.; Eroles, P.; Perez-Fidalgo, J.A. Emerging EGFR antagonists for breast cancer. Expert Opin. Emerg. Drugs 2014, 19, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Epigenomics: Technologies and Applications. Circ. Res. 2018, 122, 1191–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, R.; Shirley, N.; Bleackley, M.; Dolan, S.; Shafee, T. Transcriptomics technologies. PLoS Comput. Biol. 2017, 13, e1005457. [Google Scholar] [CrossRef] [Green Version]
- Rao, A.; Barkley, D.; França, G.S.; Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature 2021, 596, 211–220. [Google Scholar] [CrossRef]
- Al-Amrani, S.; Al-Jabri, Z.; Al-Zaabi, A.; Alshekaili, J.; Al-Khabori, M. Proteomics: Concepts and applications in human medicine. World J. Biol. Chem. 2021, 12, 57–69. [Google Scholar] [CrossRef]
- Clish, C.B. Metabolomics: An emerging but powerful tool for precision medicine. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000588. [Google Scholar] [CrossRef] [Green Version]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e310. [Google Scholar] [CrossRef] [Green Version]
- Jacob, M.; Lopata, A.L.; Dasouki, M.; Abdel Rahman, A.M. Metabolomics toward personalized medicine. Mass Spectrom. Rev. 2019, 38, 221–238. [Google Scholar] [CrossRef]
- Naithani, N.; Sinha, S.; Misra, P.; Vasudevan, B.; Sahu, R. Precision medicine: Concept and tools. Med. J. Armed Forces India 2021, 77, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.S. Microbiomics: Were we all wrong before? Periodontol. 2000 2021, 85, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Athanasopoulou, K.; Adamopoulos, P.G.; Scorilas, A. Unveiling the Human Gastrointestinal Tract Microbiome: The Past, Present, and Future of Metagenomics. Biomedicines 2023, 11, 827. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I. Proteogenomics: Concepts, applications and computational strategies. Nat. Methods 2014, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, J.D.; Berg, H.C.; Church, G.M. Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics 2004, 4, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Bantscheff, M.; Lemeer, S.; Savitski, M.M.; Kuster, B. Quantitative mass spectrometry in proteomics: Critical review update from 2007 to the present. Anal. Bioanal. Chem. 2012, 404, 939–965. [Google Scholar] [CrossRef]
- Mann, M.; Kulak, N.A.; Nagaraj, N.; Cox, J. The coming age of complete, accurate, and ubiquitous proteomes. Mol. Cell 2013, 49, 583–590. [Google Scholar] [CrossRef] [Green Version]
- Albright, J.C.; Goering, A.W.; Doroghazi, J.R.; Metcalf, W.W.; Kelleher, N.L. Strain-specific proteogenomics accelerates the discovery of natural products via their biosynthetic pathways. J. Ind. Microbiol. Biotechnol. 2014, 41, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef] [Green Version]
- Method of the Year 2019: Single-cell multimodal omics. Nat. Methods 2020, 17, 2020. [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Li, G.; Liu, Y.; Zhang, Y.; Kubo, N.; Yu, M.; Fang, R.; Kellis, M.; Ren, B. Joint profiling of DNA methylation and chromatin architecture in single cells. Nat. Methods 2019, 16, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Preissl, S.; Ren, B. Single-cell multimodal omics: The power of many. Nat. Methods 2020, 17, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Robinson, P.N. Deep phenotyping for precision medicine. Hum. Mutat. 2012, 33, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Delude, C.M. Deep phenotyping: The details of disease. Nature 2015, 527, S14–S15. [Google Scholar] [CrossRef]
- Doshi-Velez, F.; Ge, Y.; Kohane, I. Comorbidity clusters in autism spectrum disorders: An electronic health record time-series analysis. Pediatrics 2014, 133, e54–e63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rooden, S.M.; Colas, F.; Martínez-Martín, P.; Visser, M.; Verbaan, D.; Marinus, J.; Chaudhuri, R.K.; Kok, J.N.; van Hilten, J.J. Clinical subtypes of Parkinson’s disease. Mov. Disord. 2011, 26, 51–58. [Google Scholar] [CrossRef]
- Xu, M.; Song, J. Targeted Therapy in Cardiovascular Disease: A Precision Therapy Era. Front. Pharmacol. 2021, 12, 623674. [Google Scholar] [CrossRef]
- Ehrlich, P. Experimental Researches on Specific Therapy: On Immunity with special Reference to the Relationship between Distribution and Action of Antigens**[Reprinted (with amendments by the Ed.) from The Harben Lectures for 1907 of the Royal Institute of Public Health, London: Lewis, 1908; cf. Bibl. 200 and 181.]: FIRST HARBEN LECTURE††[Delivered in London, 5 June 1907.]. In The Collected Papers of Paul Ehrlich; Himmelweit, F., Ed.; Pergamon: Oxford, UK, 1960; pp. 106–117. [Google Scholar]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef]
- Nicolazzi, M.A.; Carnicelli, A.; Fuorlo, M.; Scaldaferri, A.; Masetti, R.; Landolfi, R.; Favuzzi, A.M.R. Anthracycline and trastuzumab-induced cardiotoxicity in breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- Barnas, J.L.; Looney, R.J.; Anolik, J.H. B cell targeted therapies in autoimmune disease. Curr. Opin. Immunol. 2019, 61, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Quon, B.S.; Rowe, S.M. New and emerging targeted therapies for cystic fibrosis. BMJ 2016, 352, i859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Badger, C.C.; Anasetti, C.; Davis, J.; Bernstein, I.D. Treatment of malignancy with unmodified antibody. Pathol. Immunopathol. Res. 1987, 6, 419–434. [Google Scholar] [CrossRef]
- Khazaeli, M.B.; Conry, R.M.; LoBuglio, A.F. Human immune response to monoclonal antibodies. J. Immunother. Emphas. Tumor Immunol. 1994, 15, 42–52. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Goydel, R.S.; Rader, C. Antibody-based cancer therapy. Oncogene 2021, 40, 3655–3664. [Google Scholar] [CrossRef]
- Franzin, R.; Netti, G.S.; Spadaccino, F.; Porta, C.; Gesualdo, L.; Stallone, G.; Castellano, G.; Ranieri, E. The Use of Immune Checkpoint Inhibitors in Oncology and the Occurrence of AKI: Where Do We Stand? Front. Immunol. 2020, 11, 574271. [Google Scholar] [CrossRef]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct. Target. Ther. 2022, 7, 39. [Google Scholar] [CrossRef]
- Zahavi, D.; Weiner, L. Monoclonal Antibodies in Cancer Therapy. Antibodies 2020, 9, 34. [Google Scholar] [CrossRef]
- Lambert, J.M.; Berkenblit, A. Antibody-Drug Conjugates for Cancer Treatment. Annu. Rev. Med. 2018, 69, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Steiner, M.; Neri, D. Antibody-radionuclide conjugates for cancer therapy: Historical considerations and new trends. Clin. Cancer Res. 2011, 17, 6406–6416. [Google Scholar] [CrossRef] [Green Version]
- Aghanejad, A.; Bonab, S.F.; Sepehri, M.; Haghighi, F.S.; Tarighatnia, A.; Kreiter, C.; Nader, N.D.; Tohidkia, M.R. A review on targeting tumor microenvironment: The main paradigm shift in the mAb-based immunotherapy of solid tumors. Int. J. Biol. Macromol. 2022, 207, 592–610. [Google Scholar] [CrossRef]
- Elzahhar, P.; Belal, A.S.F.; Elamrawy, F.; Helal, N.A.; Nounou, M.I. Bioconjugation in Drug Delivery: Practical Perspectives and Future Perceptions. Methods Mol. Biol. 2019, 2000, 125–182. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Ledford, H. Melanoma drug wins US approval. Nature 2011, 471, 561. [Google Scholar] [CrossRef] [Green Version]
- Dougan, M.; Pietropaolo, M. Time to dissect the autoimmune etiology of cancer antibody immunotherapy. J. Clin. Investig. 2020, 130, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Turanli, B.; Karagoz, K.; Gulfidan, G.; Sinha, R.; Mardinoglu, A.; Arga, K.Y. A Network-Based Cancer Drug Discovery: From Integrated Multi-Omics Approaches to Precision Medicine. Curr. Pharm. Des. 2018, 24, 3778–3790. [Google Scholar] [CrossRef] [PubMed]
- Weinshilboum, R.M.; Wang, L. Pharmacogenetics and pharmacogenomics: Development, science, and translation. Annu. Rev. Genom. Hum. Genet. 2006, 7, 223–245. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [Green Version]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.A.; Manley, K.A.; Mc, K.V. Genetic control of isoniazid metabolism in man. Br. Med. J. 1960, 2, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Kalow, W.; Gunn, D.R. The relation between dose of succinylcholine and duration of apnea in man. J. Pharmacol. Exp. Ther. 1957, 120, 203–214. [Google Scholar]
- Kalow, W.; Gunn, D.R. Some statistical data on atypical cholinesterase of human serum. Ann. Hum. Genet. 1959, 23, 239–250. [Google Scholar] [CrossRef]
- Evans, F.T.; Gray, P.W.; Lehmann, H.; Silk, E. Sensitivity to succinylcholine in relation to serum-cholinesterase. Lancet 1952, 1, 1229–1230. [Google Scholar] [CrossRef]
- Bartels, C.F.; van der Spek, A.F.; La Du, B.N. Two polymorphisms in the non-coding regions of the BCHE gene. Nucleic Acids Res. 1990, 18, 6171. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.F.; Oliveira, T.G.; Molfetta, G.A.; Garcia, L.V.; Ferreira, C.A.; Marques, A.A.; Silva, W.A., Jr. Biochemical and genetic analysis of butyrylcholinesterase (BChE) in a family, due to prolonged neuromuscular blockade after the use of succinylcholine. Genet. Mol. Biol. 2011, 34, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Rieder, M.J.; Reiner, A.P.; Gage, B.F.; Nickerson, D.A.; Eby, C.S.; McLeod, H.L.; Blough, D.K.; Thummel, K.E.; Veenstra, D.L.; Rettie, A.E. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N. Engl. J. Med. 2005, 352, 2285–2293. [Google Scholar] [CrossRef] [Green Version]
- Daly, A.K.; King, B.P. Pharmacogenetics of oral anticoagulants. Pharmacogenetics 2003, 13, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Aithal, G.P.; Day, C.P.; Kesteven, P.J.; Daly, A.K. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet 1999, 353, 717–719. [Google Scholar] [CrossRef]
- Xicota, L.; De Toma, I.; Maffioletti, E.; Pisanu, C.; Squassina, A.; Baune, B.T.; Potier, M.C.; Stacey, D.; Dierssen, M. Recommendations for pharmacotranscriptomic profiling of drug response in CNS disorders. Eur. Neuropsychopharmacol. 2022, 54, 41–53. [Google Scholar] [CrossRef]
- Jabbarzadeh Kaboli, P.; Luo, S.; Chen, Y.; Jomhori, M.; Imani, S.; Xiang, S.; Wu, Z.; Li, M.; Shen, J.; Zhao, Y.; et al. Pharmacotranscriptomic profiling of resistant triple-negative breast cancer cells treated with lapatinib and berberine shows upregulation of PI3K/Akt signaling under cytotoxic stress. Gene 2022, 816, 146171. [Google Scholar] [CrossRef]
- Pan, Z.; Wang, K.; Wang, X.; Jia, Z.; Yang, Y.; Duan, Y.; Huang, L.; Wu, Z.X.; Zhang, J.Y.; Ding, X. Cholesterol promotes EGFR-TKIs resistance in NSCLC by inducing EGFR/Src/Erk/SP1 signaling-mediated ERRα re-expression. Mol. Cancer 2022, 21, 77. [Google Scholar] [CrossRef] [PubMed]
- Majchrzak-Celińska, A.; Baer-Dubowska, W. Pharmacoepigenetics: An element of personalized therapy? Expert Opin. Drug Metab. Toxicol. 2017, 13, 387–398. [Google Scholar] [CrossRef]
- Peedicayil, J. Pharmacoepigenetics and Pharmacoepigenomics: An Overview. Curr. Drug Discov. Technol. 2019, 16, 392–399. [Google Scholar] [CrossRef]
- Schrader, S.; Perfilyev, A.; Martinell, M.; García-Calzón, S.; Ling, C. Statin therapy is associated with epigenetic modifications in individuals with Type 2 diabetes. Epigenomics 2021, 13, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Nuotio, M.L.; Sánez Tähtisalo, H.; Lahtinen, A.; Donner, K.; Fyhrquist, F.; Perola, M.; Kontula, K.K.; Hiltunen, T.P. Pharmacoepigenetics of hypertension: Genome-wide methylation analysis of responsiveness to four classes of antihypertensive drugs using a double-blind crossover study design. Epigenetics 2022, 17, 1432–1445. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Zolla, L. Pharmacoproteomics: A chess game on a protein field. Drug Discov. Today 2010, 15, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.A.J.; Sawicka, K.; Sawicka, J.; Franczak, A.; Cohen, A.; Bil-Lula, I.; Sawicki, G. Protection of the transplant kidney during cold perfusion with doxycycline: Proteomic analysis in a rat model. Proteome Sci. 2020, 18, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, H.; Jia, W.; Hu, Z. Emerging Applications of Metabolomics in Clinical Pharmacology. Clin. Pharmacol. Ther. 2019, 106, 544–556. [Google Scholar] [CrossRef]
- Mussap, M.; Loddo, C.; Fanni, C.; Fanos, V. Metabolomics in pharmacology—A delve into the novel field of pharmacometabolomics. Expert Rev. Clin. Pharmacol. 2020, 13, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Sreekumar, A.; Poisson, L.M.; Rajendiran, T.M.; Khan, A.P.; Cao, Q.; Yu, J.; Laxman, B.; Mehra, R.; Lonigro, R.J.; Li, Y.; et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 2009, 457, 910–914. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Kachroo, P.; Sordillo, J.E.; Lutz, S.M.; Weiss, S.T.; Kelly, R.S.; McGeachie, M.J.; Wu, A.C.; Lasky-Su, J.A. Pharmaco-Metabolomics of Inhaled Corticosteroid Response in Individuals with Asthma. J. Pers. Med. 2021, 11, 1148. [Google Scholar] [CrossRef]
- Du, J.; Su, Y.; Qian, C.; Yuan, D.; Miao, K.; Lee, D.; Ng, A.H.C.; Wijker, R.S.; Ribas, A.; Levine, R.D.; et al. Raman-guided subcellular pharmaco-metabolomics for metastatic melanoma cells. Nat. Commun. 2020, 11, 4830. [Google Scholar] [CrossRef]
- Letai, A.; Bhola, P.; Welm, A.L. Functional precision oncology: Testing tumors with drugs to identify vulnerabilities and novel combinations. Cancer Cell 2022, 40, 26–35. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirschbaum, A.; Geisse, N.C.; Sister, T.J.; Meyer, L.M. Effect of certain folic acid antagonists on transplanted myeloid and lymphoid leukemias of the F strain of mice. Cancer Res. 1950, 10, 762–768. [Google Scholar] [PubMed]
- Yoshida, G.J. Applications of patient-derived tumor xenograft models and tumor organoids. J. Hematol. Oncol. 2020, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- DeRose, Y.S.; Wang, G.; Lin, Y.C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Corà, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef] [Green Version]
- Guillen, K.P.; Fujita, M.; Butterfield, A.J.; Scherer, S.D.; Bailey, M.H.; Chu, Z.; DeRose, Y.S.; Zhao, L.; Cortes-Sanchez, E.; Yang, C.H.; et al. A human breast cancer-derived xenograft and organoid platform for drug discovery and precision oncology. Nat. Cancer 2022, 3, 232–250. [Google Scholar] [CrossRef]
- Fior, R.; Póvoa, V.; Mendes, R.V.; Carvalho, T.; Gomes, A.; Figueiredo, N.; Ferreira, M.G. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc. Natl. Acad. Sci. USA 2017, 114, E8234–E8243. [Google Scholar] [CrossRef]
- Zou, T.; Mao, X.; Yin, J.; Li, X.; Chen, J.; Zhu, T.; Li, Q.; Zhou, H.; Liu, Z. Emerging roles of RAC1 in treating lung cancer patients. Clin. Genet. 2017, 91, 520–528. [Google Scholar] [CrossRef]
- Rajeshkumar, N.V.; Yabuuchi, S.; Pai, S.G.; De Oliveira, E.; Kamphorst, J.J.; Rabinowitz, J.D.; Tejero, H.; Al-Shahrour, F.; Hidalgo, M.; Maitra, A.; et al. Treatment of Pancreatic Cancer Patient-Derived Xenograft Panel with Metabolic Inhibitors Reveals Efficacy of Phenformin. Clin. Cancer Res. 2017, 23, 5639–5647. [Google Scholar] [CrossRef] [Green Version]
- Garcia, P.L.; Miller, A.L.; Gamblin, T.L.; Council, L.N.; Christein, J.D.; Arnoletti, J.P.; Heslin, M.J.; Reddy, S.; Richardson, J.H.; Cui, X.; et al. JQ1 Induces DNA Damage and Apoptosis, and Inhibits Tumor Growth in a Patient-Derived Xenograft Model of Cholangiocarcinoma. Mol. Cancer Ther. 2018, 17, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; Auffinger, B.; Guo, D.; Hasan, T.; Deheeger, M.; Tobias, A.L.; Kim, J.Y.; Atashi, F.; Zhang, L.; Lesniak, M.S.; et al. Dedifferentiation of Glioma Cells to Glioma Stem-like Cells By Therapeutic Stress-induced HIF Signaling in the Recurrent GBM Model. Mol. Cancer Ther. 2016, 15, 3064–3076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fack, F.; Tardito, S.; Hochart, G.; Oudin, A.; Zheng, L.; Fritah, S.; Golebiewska, A.; Nazarov, P.V.; Bernard, A.; Hau, A.C.; et al. Altered metabolic landscape in IDH-mutant gliomas affects phospholipid, energy, and oxidative stress pathways. EMBO Mol. Med. 2017, 9, 1681–1695. [Google Scholar] [CrossRef] [PubMed]
- Garner, E.F.; Williams, A.P.; Stafman, L.L.; Aye, J.M.; Mroczek-Musulman, E.; Moore, B.P.; Stewart, J.E.; Friedman, G.K.; Beierle, E.A. FTY720 Decreases Tumorigenesis in Group 3 Medulloblastoma Patient-Derived Xenografts. Sci. Rep. 2018, 8, 6913. [Google Scholar] [CrossRef] [Green Version]
- Lawson, D.A.; Bhakta, N.R.; Kessenbrock, K.; Prummel, K.D.; Yu, Y.; Takai, K.; Zhou, A.; Eyob, H.; Balakrishnan, S.; Wang, C.Y.; et al. Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 2015, 526, 131–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Ayachi, I.; Fatima, I.; Wend, P.; Alva-Ornelas, J.A.; Runke, S.; Kuenzinger, W.L.; Silva, J.; Silva, W.; Gray, J.K.; Lehr, S.; et al. The WNT10B Network Is Associated with Survival and Metastases in Chemoresistant Triple-Negative Breast Cancer. Cancer Res. 2019, 79, 982–993. [Google Scholar] [CrossRef] [Green Version]
- Weeden, C.E.; Holik, A.Z.; Young, R.J.; Ma, S.B.; Garnier, J.M.; Fox, S.B.; Antippa, P.; Irving, L.B.; Steinfort, D.P.; Wright, G.M.; et al. Cisplatin Increases Sensitivity to FGFR Inhibition in Patient-Derived Xenograft Models of Lung Squamous Cell Carcinoma. Mol. Cancer Ther. 2017, 16, 1610–1622. [Google Scholar] [CrossRef] [Green Version]
- Talebi, A.; Dehairs, J.; Rambow, F.; Rogiers, A.; Nittner, D.; Derua, R.; Vanderhoydonc, F.; Duarte, J.A.G.; Bosisio, F.; Van den Eynde, K.; et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat. Commun. 2018, 9, 2500. [Google Scholar] [CrossRef] [Green Version]
- Grasset, E.M.; Bertero, T.; Bozec, A.; Friard, J.; Bourget, I.; Pisano, S.; Lecacheur, M.; Maiel, M.; Bailleux, C.; Emelyanov, A.; et al. Matrix Stiffening and EGFR Cooperate to Promote the Collective Invasion of Cancer Cells. Cancer Res. 2018, 78, 5229–5242. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.F.; Palakurthi, S.; Zeng, Q.; Zhou, S.; Ivanova, E.; Huang, W.; Zervantonakis, I.K.; Selfors, L.M.; Shen, Y.; Pritchard, C.C.; et al. Establishment of Patient-Derived Tumor Xenograft Models of Epithelial Ovarian Cancer for Preclinical Evaluation of Novel Therapeutics. Clin. Cancer Res. 2017, 23, 1263–1273. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.J.; Kandoth, C.; Williams, A.B.; et al. Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 2018, 173, 515–528.e517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef] [Green Version]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-derived organoids model treatment response of metastatic gastrointestinal cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Bruun, J.; Kryeziu, K.; Eide, P.W.; Moosavi, S.H.; Eilertsen, I.A.; Langerud, J.; Røsok, B.; Totland, M.Z.; Brunsell, T.H.; Pellinen, T.; et al. Patient-Derived Organoids from Multiple Colorectal Cancer Liver Metastases Reveal Moderate Intra-patient Pharmacotranscriptomic Heterogeneity. Clin. Cancer Res. 2020, 26, 4107–4119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e122. [Google Scholar] [CrossRef]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid cultures derived from patients with advanced prostate cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Driehuis, E.; Kolders, S.; Spelier, S.; Lõhmussaar, K.; Willems, S.M.; Devriese, L.A.; de Bree, R.; de Ruiter, E.J.; Korving, J.; Begthel, H.; et al. Oral Mucosal Organoids as a Potential Platform for Personalized Cancer Therapy. Cancer Discov. 2019, 9, 852–871. [Google Scholar] [CrossRef]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.H.N.; Siu, H.C.; Law, S.; Ho, S.L.; Yue, S.S.K.; Tsui, W.Y.; Chan, D.; Chan, A.S.; Ma, S.; Lam, K.O.; et al. A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 2018, 23, 882–897.e811. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Francies, H.E.; Secrier, M.; Perner, J.; Miremadi, A.; Galeano-Dalmau, N.; Barendt, W.J.; Letchford, L.; Leyden, G.M.; Goffin, E.K.; et al. Organoid cultures recapitulate esophageal adenocarcinoma heterogeneity providing a model for clonality studies and precision therapeutics. Nat. Commun. 2018, 9, 2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boretto, M.; Maenhoudt, N.; Luo, X.; Hennes, A.; Boeckx, B.; Bui, B.; Heremans, R.; Perneel, L.; Kobayashi, H.; Van Zundert, I.; et al. Patient-derived organoids from endometrial disease capture clinical heterogeneity and are amenable to drug screening. Nat. Cell Biol. 2019, 21, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Radulovich, N.; Ng, C.; Liu, N.; Notsuda, H.; Cabanero, M.; Martins-Filho, S.N.; Raghavan, V.; Li, Q.; Mer, A.S.; et al. Organoid Cultures as Preclinical Models of Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 1162–1174. [Google Scholar] [CrossRef] [Green Version]
- Papaccio, F.; Cabeza-Segura, M.; Garcia-Micò, B.; Tarazona, N.; Roda, D.; Castillo, J.; Cervantes, A. Will Organoids Fill the Gap towards Functional Precision Medicine? J. Pers. Med. 2022, 12, 1939. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, J.; Wang, X.; Feng, L.; Wu, J.; Zhu, X.; Wen, W.; Gong, X. Organ-on-a-chip: Recent breakthroughs and future prospects. BioMedical Eng. OnLine 2020, 19, 9. [Google Scholar] [CrossRef] [Green Version]
- Doherty, E.L.; Aw, W.Y.; Hickey, A.J.; Polacheck, W.J. Microfluidic and Organ-on-a-Chip Approaches to Investigate Cellular and Microenvironmental Contributions to Cardiovascular Function and Pathology. Front. Bioeng. Biotechnol. 2021, 9, 624435. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Sontheimer-Phelps, A.; Hassell, B.A.; Ingber, D.E. Modelling cancer in microfluidic human organs-on-chips. Nat. Rev. Cancer 2019, 19, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Mackenbach, J.P. Genetics and health inequalities: Hypotheses and controversies. J. Epidemiol. Community Health 2005, 59, 268–273. [Google Scholar] [CrossRef]
- Joyner, M.J.; Paneth, N. Promises, promises, and precision medicine. J. Clin. Investig. 2019, 129, 946–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.M.; Shanley, T.P. The Missing -Omes: Proposing Social and Environmental Nomenclature in Precision Medicine. Clin. Transl. Sci. 2017, 10, 64–66. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, S.A. Shattuck Lecture. We can do better--improving the health of the American people. N. Engl. J. Med. 2007, 357, 1221–1228. [Google Scholar] [CrossRef] [Green Version]
- Pendergrass, S.A.; Crawford, D.C. Using Electronic Health Records To Generate Phenotypes For Research. Curr. Protoc. Hum. Genet. 2019, 100, e80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abul-Husn, N.S.; Kenny, E.E. Personalized Medicine and the Power of Electronic Health Records. Cell 2019, 177, 58–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, M.K.; Sauer, S.J.; Nath, S.; Robinson, T.J.; Morse, M.A.; Devi, G.R. X-linked inhibitor of apoptosis protein mediates tumor cell resistance to antibody-dependent cellular cytotoxicity. Cell Death Dis. 2016, 7, e2073. [Google Scholar] [CrossRef]
- Wolford, B.N.; Willer, C.J.; Surakka, I. Electronic health records: The next wave of complex disease genetics. Hum. Mol. Genet. 2018, 27, R14–R21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO Guidelines Approved by the Guidelines Review Committee. In WHO Guideline Recommendations on Digital Interventions for Health System Strengthening; World Health Organization: Geneva, Switzerland, 2019.
- Fadahunsi, K.P.; O’Connor, S.; Akinlua, J.T.; Wark, P.A.; Gallagher, J.; Carroll, C.; Car, J.; Majeed, A.; O’Donoghue, J. Information Quality Frameworks for Digital Health Technologies: Systematic Review. J. Med. Internet Res. 2021, 23, e23479. [Google Scholar] [CrossRef]
- Bhavnani, S.P.; Narula, J.; Sengupta, P.P. Mobile technology and the digitization of healthcare. Eur. Heart J. 2016, 37, 1428–1438. [Google Scholar] [CrossRef]
- Ristevski, B.; Chen, M. Big data analytics in medicine and healthcare. J. Integr. Bioinform. 2018, 15, 20170030. [Google Scholar] [CrossRef]
- Raghupathi, W.; Raghupathi, V. Big data analytics in healthcare: Promise and potential. Health Inf. Sci. Syst. 2014, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Preininger, A. Ai in health: State of the art, challenges, and future directions. Yearb. Med. Inform. 2019, 28, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, R.C.; Wang, Z. Precision Medicine: Disease Subtyping and Tailored Treatment. Cancers 2023, 15, 3837. https://doi.org/10.3390/cancers15153837
Wang RC, Wang Z. Precision Medicine: Disease Subtyping and Tailored Treatment. Cancers. 2023; 15(15):3837. https://doi.org/10.3390/cancers15153837
Chicago/Turabian StyleWang, Richard C., and Zhixiang Wang. 2023. "Precision Medicine: Disease Subtyping and Tailored Treatment" Cancers 15, no. 15: 3837. https://doi.org/10.3390/cancers15153837
APA StyleWang, R. C., & Wang, Z. (2023). Precision Medicine: Disease Subtyping and Tailored Treatment. Cancers, 15(15), 3837. https://doi.org/10.3390/cancers15153837