



CTHRC1 Induces Pancreatic Stellate Cells (PSCs) into Myofibroblast-like Cancer-Associated Fibroblasts (myCAFs)

 ,
, 
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Reagents
2.3. Analyses of mRNA Data Using Public Databases
2.4. RNA Isolation and Quantitative Real-Time PCR (RT-qPCR)
2.5. Western Blot Analyses
2.6. Immunofluorescence (IF) Assay
2.7. Oil Red O Staining
2.8. Migration and Invasion Assay
2.9. Sandwich Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. Cell Proliferation
2.11. siRNA Transfection
2.12. Animal Study
2.13. Immunohistochemistry Staining
2.14. Statistical Analyses
3. Results
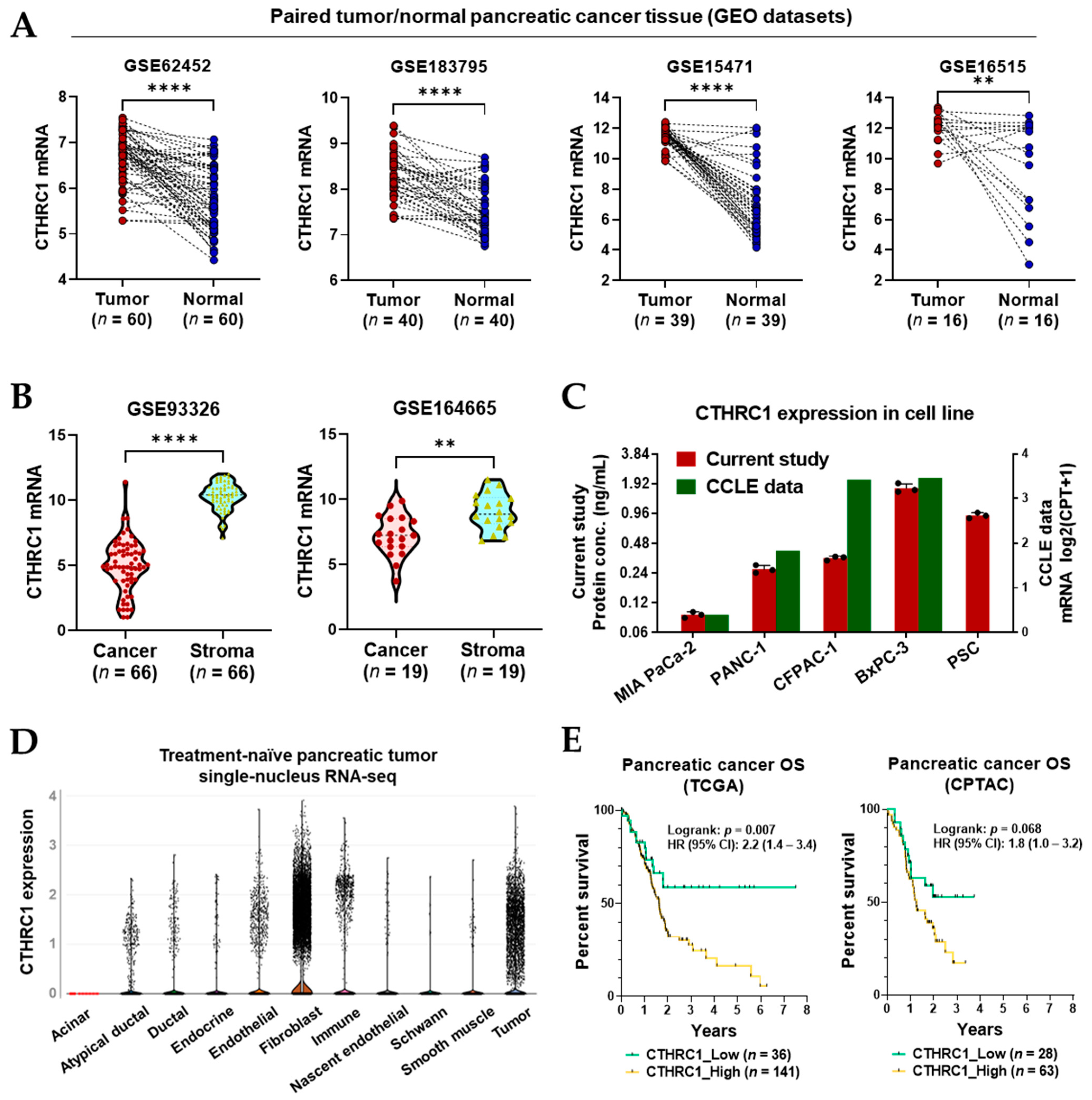
3.1. CTHRC1 Expression in Pancreatic Tumor Tissue
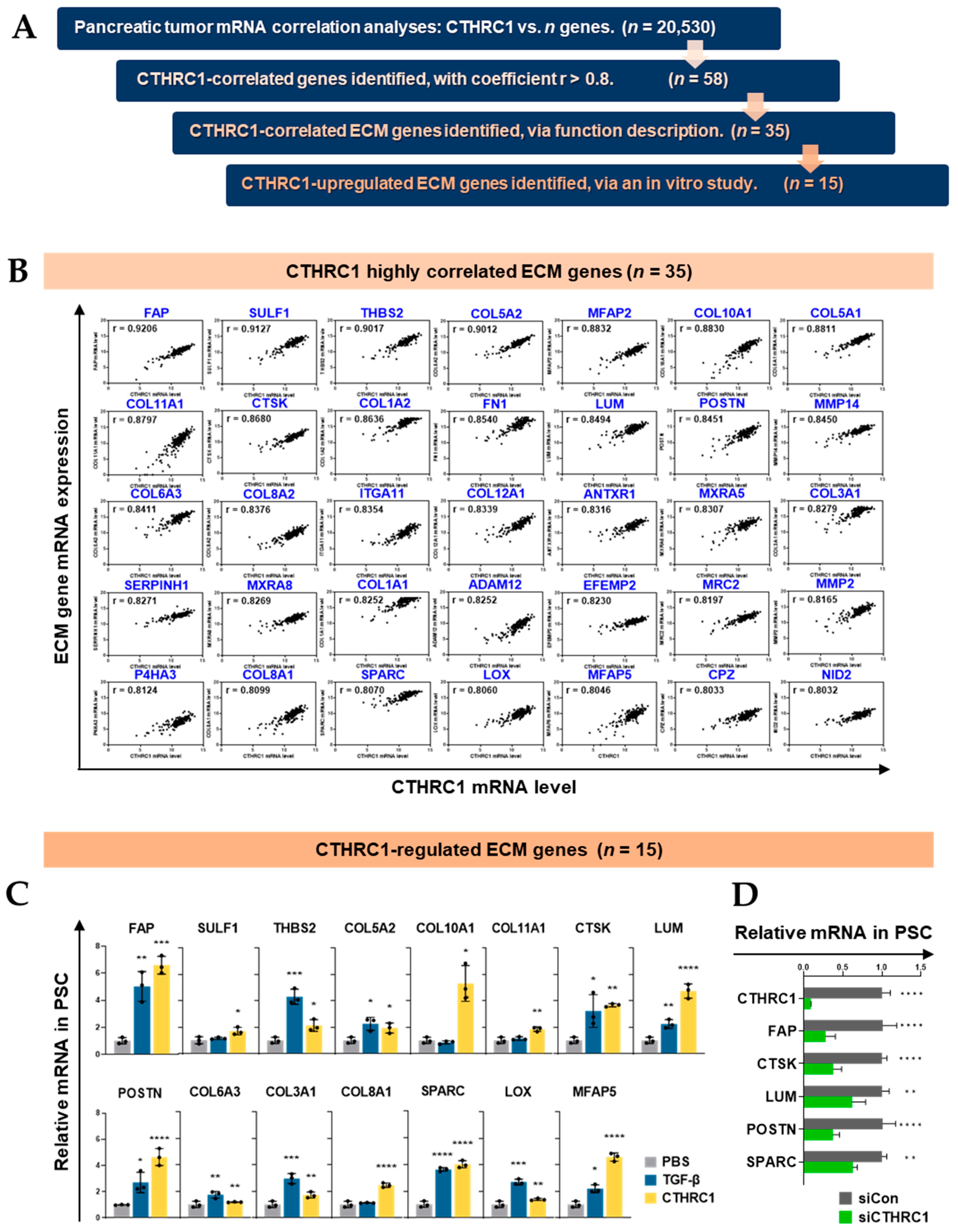
3.2. CTHRC1 Expression in Pancreatic Tumor Tissue Correlated with the Expression of ECM-Related Genes
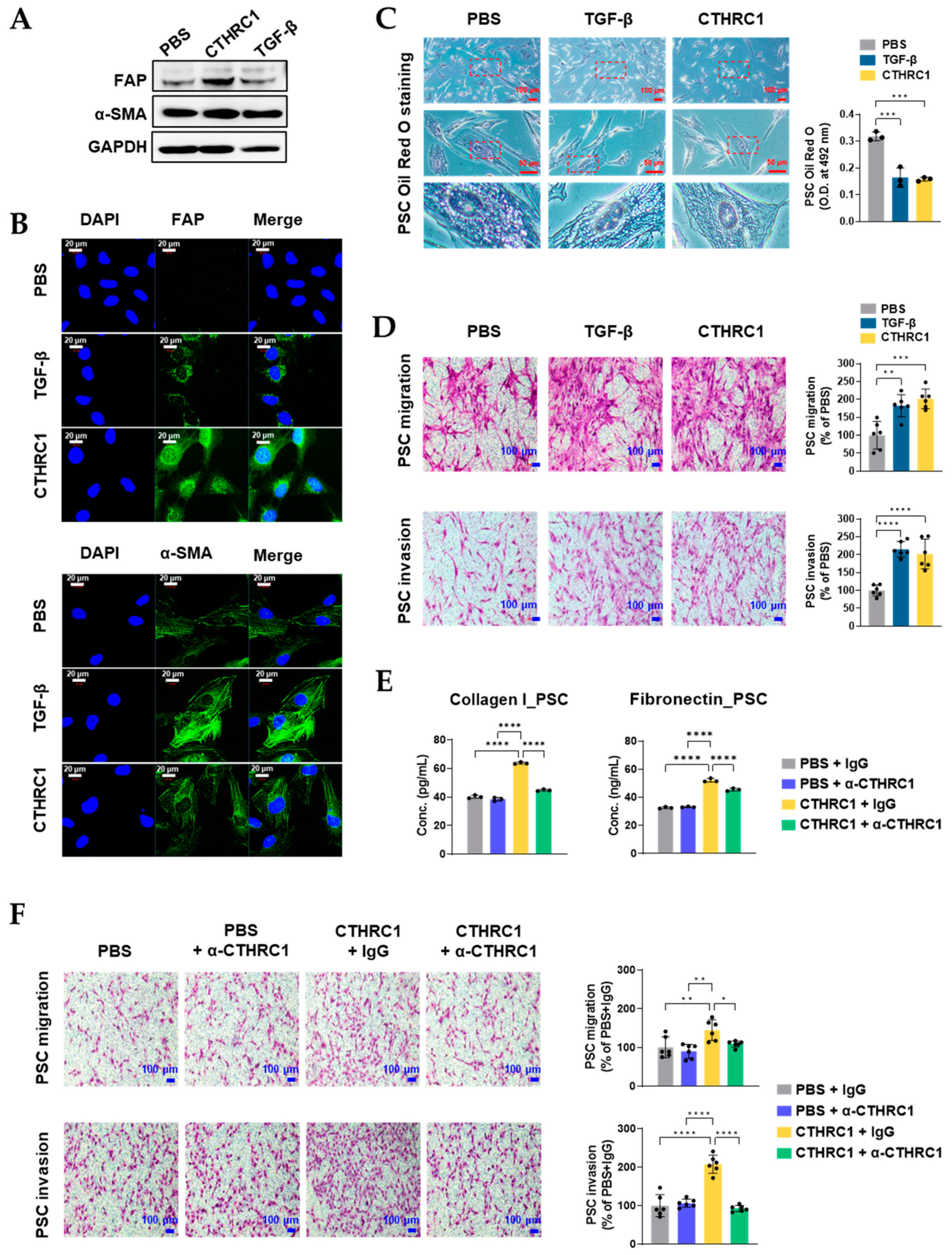
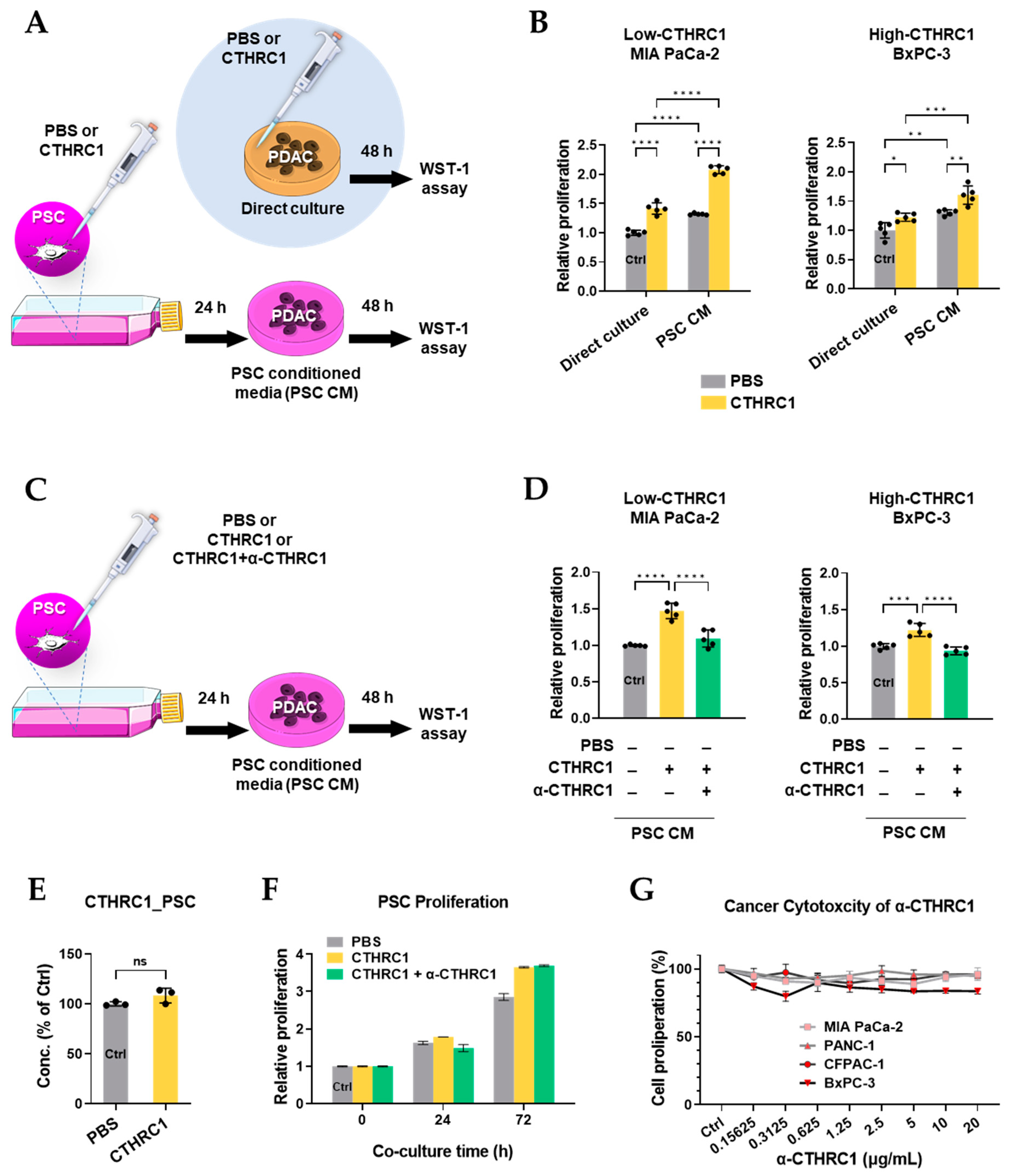
3.3. CTHRC1 Activated PSCs In Vitro
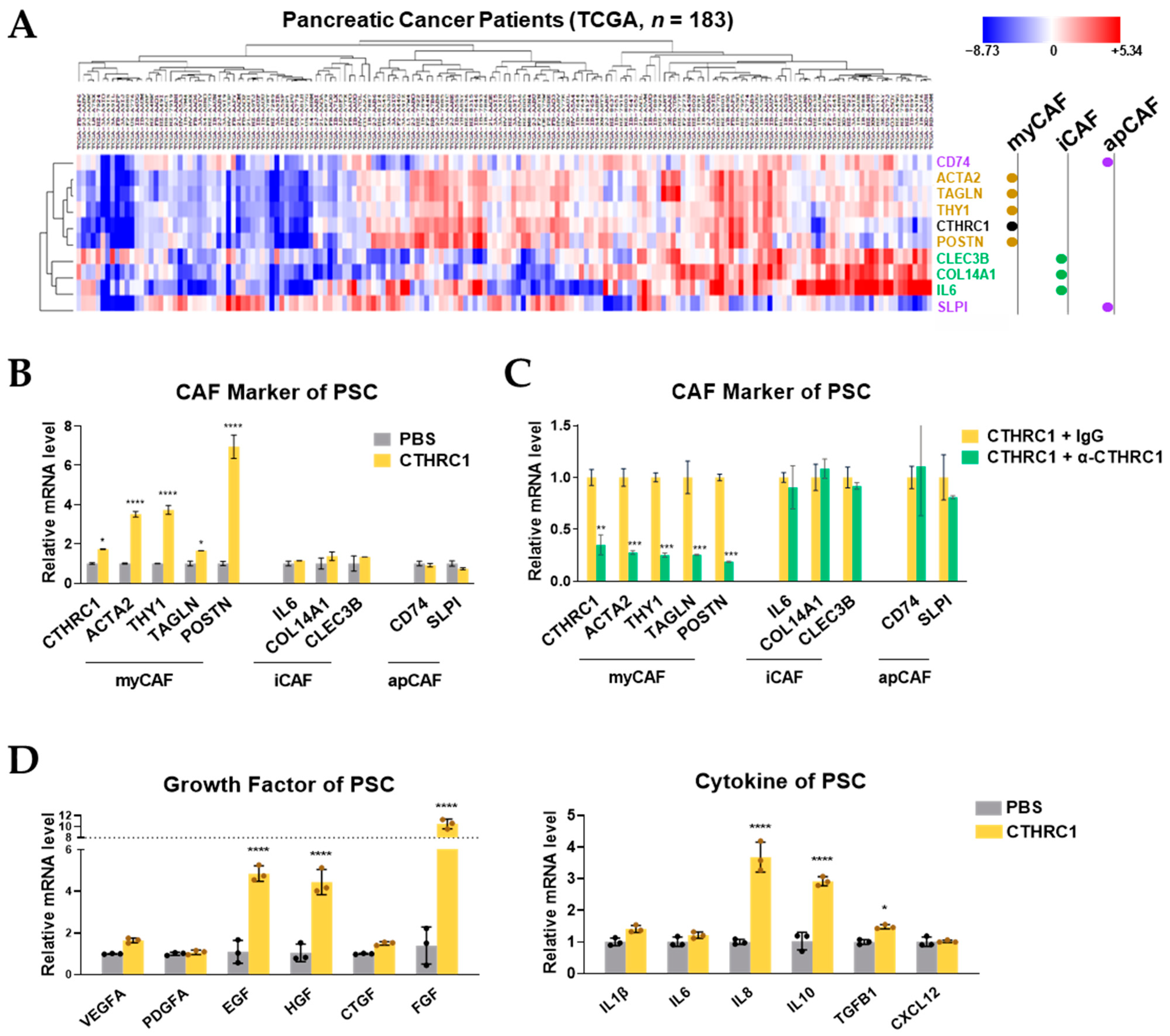
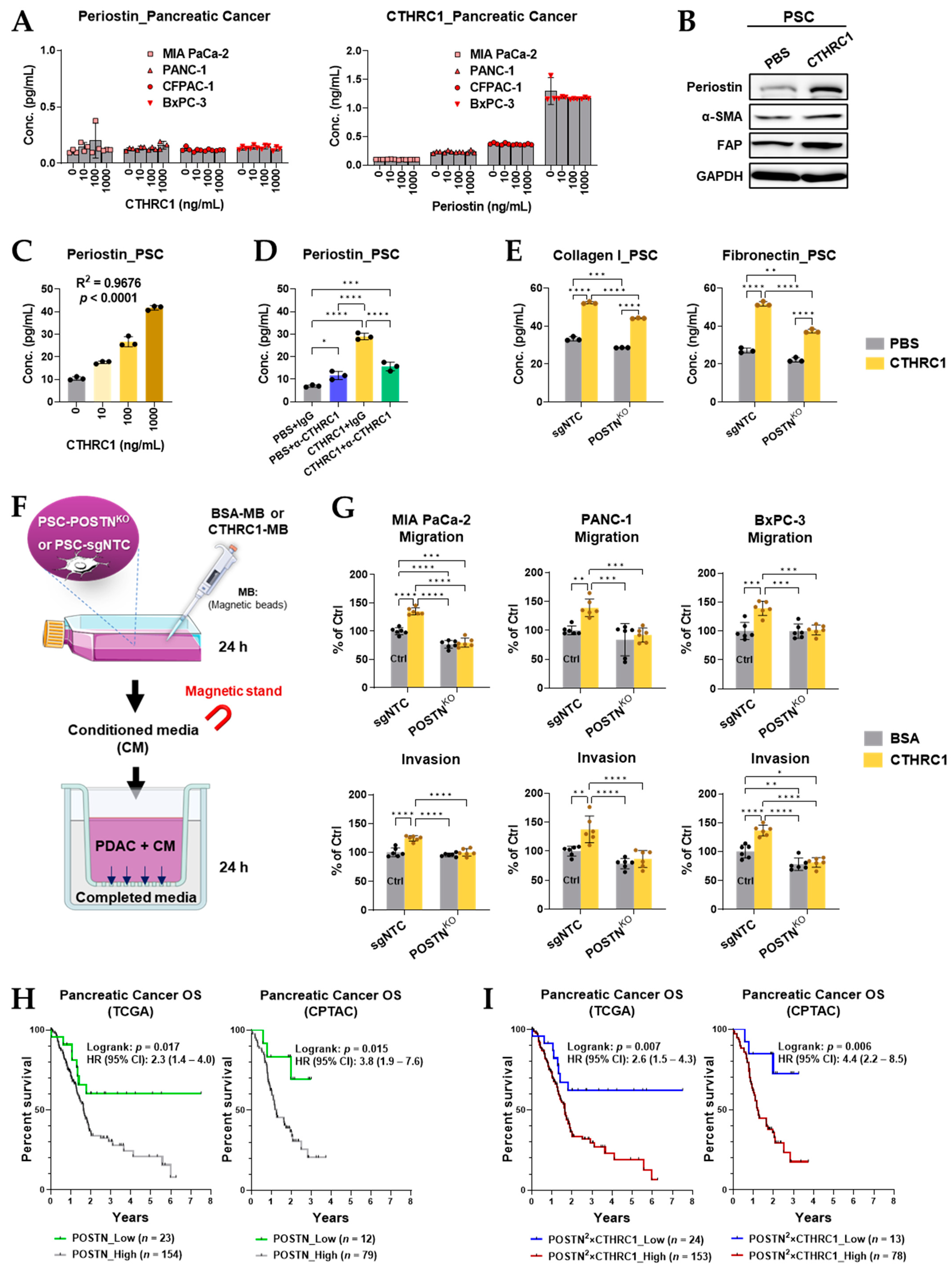
3.4. CTHRC1 Differentiated PSCs into myCAFs In Vitro
3.5. CTHRC1 Promoted Pancreatic Cancer Growth via Activated PSCs
3.6. Periostin Was Essential in the CTHRC1–PSCs–Cancer Metastasis Axis
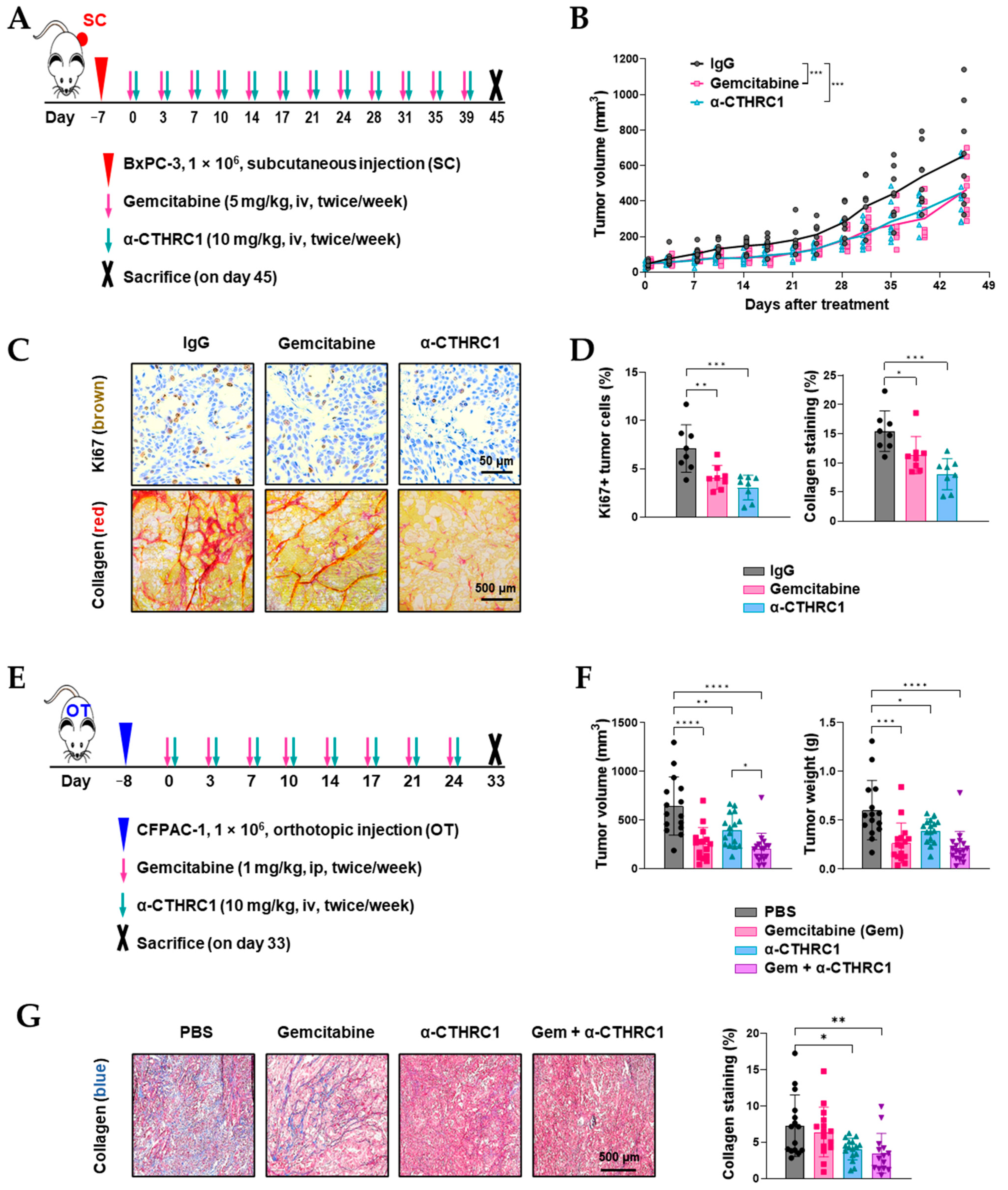
3.7. Inhibition of CTHRC1 Suppressed Tumor Growth and Induced Tumor myCAF Differentiation into iCAFs In Vivo
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wood, L.D.; Canto, M.I.; Jaffee, E.M.; Simeone, D.M. Pancreatic Cancer: Pathogenesis, Screening, Diagnosis, and Treatment. Gastroenterology 2022, 163, 386–402.e1. [Google Scholar] [CrossRef]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; et al. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Miyai, Y.; Esaki, N.; Takahashi, M.; Enomoto, A. Cancer-associated fibroblasts that restrain cancer progression: Hypotheses and perspectives. Cancer Sci. 2020, 111, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, H.; Liu, T.; Chen, T.; Wang, D.; Tang, D. Heterogeneous Pancreatic Stellate Cells Are Powerful Contributors to the Malignant Progression of Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 783617. [Google Scholar] [CrossRef] [PubMed]
- Pyagay, P.; Heroult, M.; Wang, Q.; Lehnert, W.; Belden, J.; Liaw, L.; Friesel, R.E.; Lindner, V. Collagen triple helix repeat containing 1, a novel secreted protein in injured and diseased arteries, inhibits collagen expression and promotes cell migration. Circ. Res. 2005, 96, 261–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, D.; Wei, C.; Zhang, X.; Li, S.; Liang, H.; Zheng, X.; Jiang, S.; Han, L. Pan-cancer analysis combined with experiments predicts CTHRC1 as a therapeutic target for human cancers. Cancer Cell Int. 2021, 21, 566. [Google Scholar] [CrossRef] [PubMed]
- Park, E.H.; Kim, S.; Jo, J.Y.; Kim, S.J.; Hwang, Y.; Kim, J.M.; Song, S.Y.; Lee, D.K.; Koh, S.S. Collagen triple helix repeat containing-1 promotes pancreatic cancer progression by regulating migration and adhesion of tumor cells. Carcinogenesis 2013, 34, 694–702. [Google Scholar] [CrossRef]
- Tameda, M.; Sugimoto, K.; Shiraki, K.; Yamamoto, N.; Okamoto, R.; Usui, M.; Ito, M.; Takei, Y.; Nobori, T.; Kojima, T.; et al. Collagen triple helix repeat containing 1 is overexpressed in hepatocellular carcinoma and promotes cell proliferation and motility. Int. J. Oncol. 2014, 45, 541–548. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Song, J.; Kwon, E.S.; Jo, S.; Kang, M.K.; Kim, Y.J.; Hwang, Y.; Bae, H.; Kang, T.H.; Chang, S.; et al. CTHRC1 promotes angiogenesis by recruiting Tie2-expressing monocytes to pancreatic tumors. Exp. Mol. Med. 2016, 48, e261. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.L.; Wang, T.H.; Hsu, H.C.; Yuan, R.H.; Jeng, Y.M. Overexpression of CTHRC1 in hepatocellular carcinoma promotes tumor invasion and predicts poor prognosis. PLoS ONE 2013, 8, e70324. [Google Scholar] [CrossRef]
- Li, J.; Wang, Y.; Ma, M.; Jiang, S.; Zhang, X.; Zhang, Y.; Yang, X.; Xu, C.; Tian, G.; Li, Q.; et al. Autocrine CTHRC1 activates hepatic stellate cells and promotes liver fibrosis by activating TGF-beta signaling. EBioMedicine 2019, 40, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Hamada, S.; Masamune, A.; Takikawa, T.; Suzuki, N.; Kikuta, K.; Hirota, M.; Hamada, H.; Kobune, M.; Satoh, K.; Shimosegawa, T. Pancreatic stellate cells enhance stem cell-like phenotypes in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2012, 421, 349–354. [Google Scholar] [CrossRef]
- Kim, Y.J.; Jiang, F.; Park, J.; Jeong, H.H.; Baek, J.E.; Hong, S.M.; Jeong, S.Y.; Koh, S.S. PAUF as a Target for Treatment of High PAUF-Expressing Ovarian Cancer. Front. Pharmacol. 2022, 13, 890614. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipke, J.; Brandenberger, C.; Rajces, A.; Manninger, M.; Alogna, A.; Post, H.; Muhlfeld, C. Assessment of cardiac fibrosis: A morphometric method comparison for collagen quantification. J. Appl. Physiol. 2017, 122, 1019–1030. [Google Scholar] [CrossRef]
- Lenggenhager, D.; Amrutkar, M.; Santha, P.; Aasrum, M.; Lohr, J.M.; Gladhaug, I.P.; Verbeke, C.S. Commonly Used Pancreatic Stellate Cell Cultures Differ Phenotypically and in Their Interactions with Pancreatic Cancer Cells. Cells 2019, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Kim, J.; Yang, S.; Wang, H.; Wu, C.J.; Sugimoto, H.; LeBleu, V.S.; Kalluri, R. Type I collagen deletion in alphaSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell 2021, 39, 548–565.e6. [Google Scholar] [CrossRef] [PubMed]
- Sial, N.; Ahmad, M.; Hussain, M.S.; Iqbal, M.J.; Hameed, Y.; Khan, M.; Abbas, M.; Asif, R.; Rehman, J.U.; Atif, M.; et al. CTHRC1 expression is a novel shared diagnostic and prognostic biomarker of survival in six different human cancer subtypes. Sci. Rep. 2021, 11, 19873. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Sun, B.F.; Chen, C.Y.; Zhou, J.Y.; Chen, Y.S.; Chen, H.; Liu, L.; Huang, D.; Jiang, J.; Cui, G.S.; et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res. 2019, 29, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Wang, Q.; Li, M.; Guo, H.; Liu, W.; Wang, F.; Tian, X.; Yang, Y. Single-cell RNA-seq reveals dynamic change in tumor microenvironment during pancreatic ductal adenocarcinoma malignant progression. EBioMedicine 2021, 66, 103315. [Google Scholar] [CrossRef] [PubMed]
- Sunami, Y.; Haussler, J.; Kleeff, J. Cellular Heterogeneity of Pancreatic Stellate Cells, Mesenchymal Stem Cells, and Cancer-Associated Fibroblasts in Pancreatic Cancer. Cancers 2020, 12, 3770. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhang, Q.; Yang, S.; Yang, Y.; Zhang, W.; Gao, H.; Deng, X.; Zhang, Q. A 4-gene panel as a marker at chromosome 8q in Asian gastric cancer patients. Genomics 2013, 102, 323–330. [Google Scholar] [CrossRef]
- Liu, Y.; Du, L. Role of pancreatic stellate cells and periostin in pancreatic cancer progression. Tumor Biol. 2015, 36, 3171–3177. [Google Scholar] [CrossRef]
- Yang, T.; Deng, Z.; Pan, Z.; Qian, Y.; Yao, W.; Wang, J. Prognostic value of periostin in multiple solid cancers: A systematic review with meta-analysis. J. Cell. Physiol. 2020, 235, 2800–2808. [Google Scholar] [CrossRef]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Targeting HGF/c-MET Axis in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 9170. [Google Scholar] [CrossRef]
- Xu, Z.; Pang, T.C.Y.; Liu, A.C.; Pothula, S.P.; Mekapogu, A.R.; Perera, C.J.; Murakami, T.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; et al. Targeting the HGF/c-MET pathway in advanced pancreatic cancer: A key element of treatment that limits primary tumour growth and eliminates metastasis. Br. J. Cancer 2020, 122, 1486–1495. [Google Scholar] [CrossRef]
- Vaish, U.; Jain, T.; Are, A.C.; Dudeja, V. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma: An Update on Heterogeneity and Therapeutic Targeting. Int. J. Mol. Sci. 2021, 22, 13408. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, M.K.; Jiang, F.; Kim, Y.J.; Ryu, K.; Masamune, A.; Hamada, S.; Park, Y.-Y.; Koh, S.S. CTHRC1 Induces Pancreatic Stellate Cells (PSCs) into Myofibroblast-like Cancer-Associated Fibroblasts (myCAFs). Cancers 2023, 15, 3370. https://doi.org/10.3390/cancers15133370
Kang MK, Jiang F, Kim YJ, Ryu K, Masamune A, Hamada S, Park Y-Y, Koh SS. CTHRC1 Induces Pancreatic Stellate Cells (PSCs) into Myofibroblast-like Cancer-Associated Fibroblasts (myCAFs). Cancers. 2023; 15(13):3370. https://doi.org/10.3390/cancers15133370
Chicago/Turabian StyleKang, Min Kyung, Fen Jiang, Ye Ji Kim, Kyoungjin Ryu, Atsushi Masamune, Shin Hamada, Yun-Yong Park, and Sang Seok Koh. 2023. "CTHRC1 Induces Pancreatic Stellate Cells (PSCs) into Myofibroblast-like Cancer-Associated Fibroblasts (myCAFs)" Cancers 15, no. 13: 3370. https://doi.org/10.3390/cancers15133370
APA StyleKang, M. K., Jiang, F., Kim, Y. J., Ryu, K., Masamune, A., Hamada, S., Park, Y.-Y., & Koh, S. S. (2023). CTHRC1 Induces Pancreatic Stellate Cells (PSCs) into Myofibroblast-like Cancer-Associated Fibroblasts (myCAFs). Cancers, 15(13), 3370. https://doi.org/10.3390/cancers15133370