



B7-H3 Associates with IMPDH2 and Regulates Cancer Cell Survival
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Antibodies and Reagents
2.2. Plasmids, siRNA, and Primers
2.3. Cell Culture
2.4. Immunoprecipitation
2.5. Western Blotting
2.6. Metabolite Extraction
2.7. UHPLC-MS Metabolic Profiling
2.7.1. Materials and Reagents
2.7.2. Sample Preparation for HILIC Analysis
2.7.3. UHPLC-MS Analysis
2.7.4. Run Order and Quality Control
2.7.5. Metabolite Annotation and Data Processing
2.8. Immunofluorescence
2.9. Confocal Microscopy
2.10. Measurement of Membrane and Cytoplasmic IMPDH2 Intensity
2.11. Oxidative Stress Detection
2.12. Chemoresistance and Survival Assay
2.13. Hanging Drop Spheroid Assay and Immunostaining
2.14. Human Lung PCLS Sectioning
2.15. Immunostaining of Human Lung PCLS
2.16. Statistical Analysis
3. Results
3.1. B7-H3 Localisation Differs between Lung Epithelial and Cancer Cells
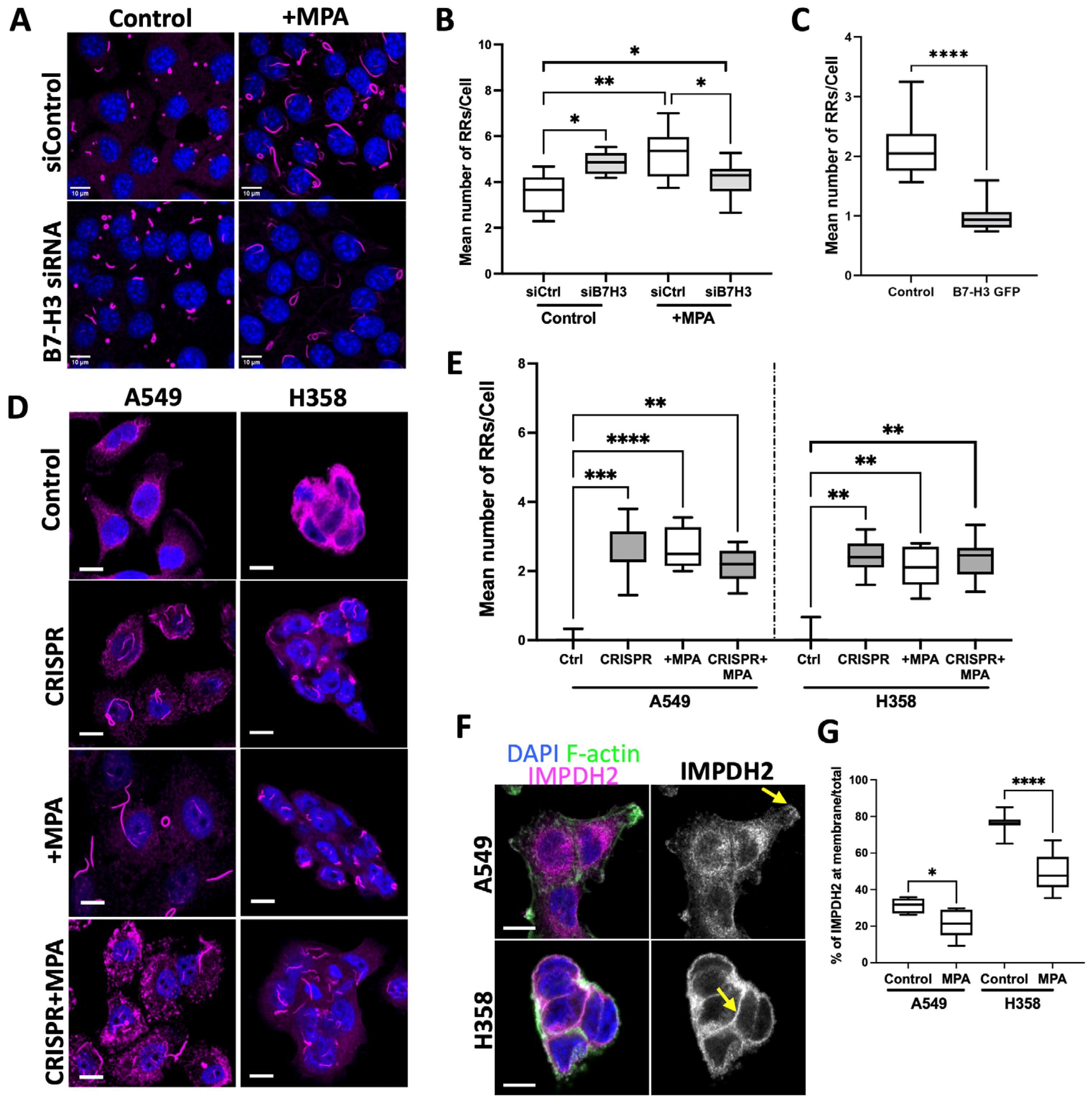
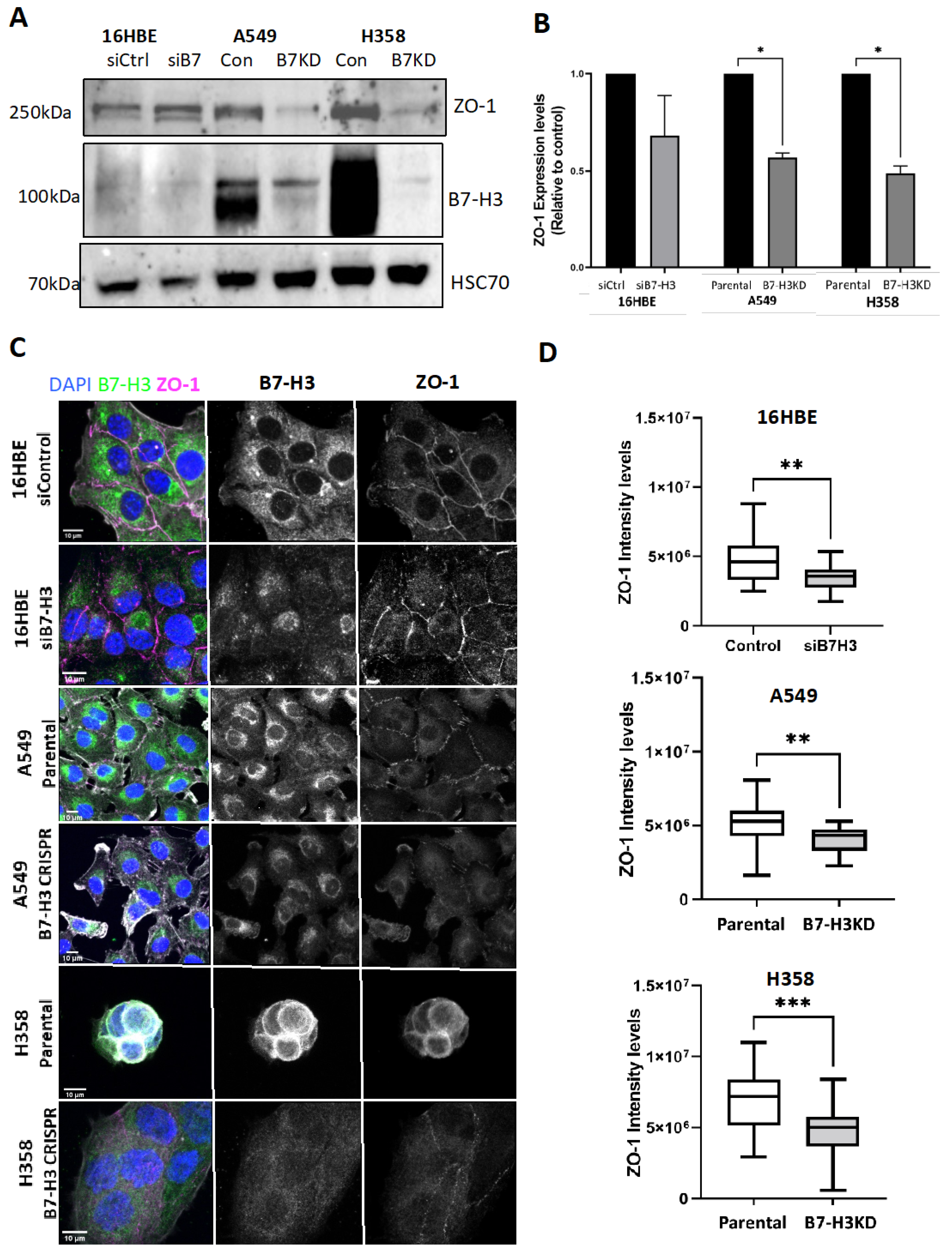
3.2. B7-H3 Negatively Regulates Rod and Ring Assembly in Cancer Cells
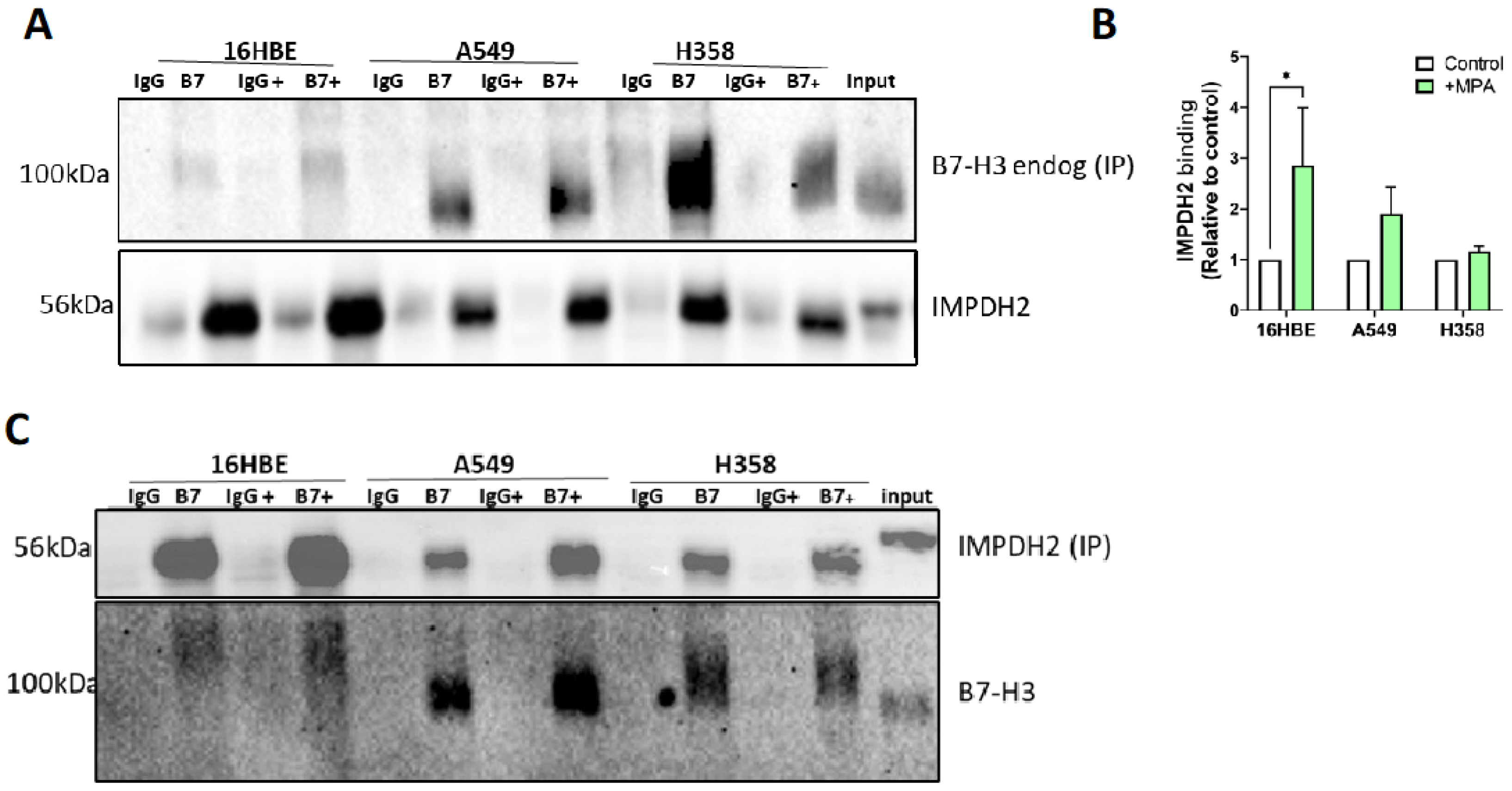
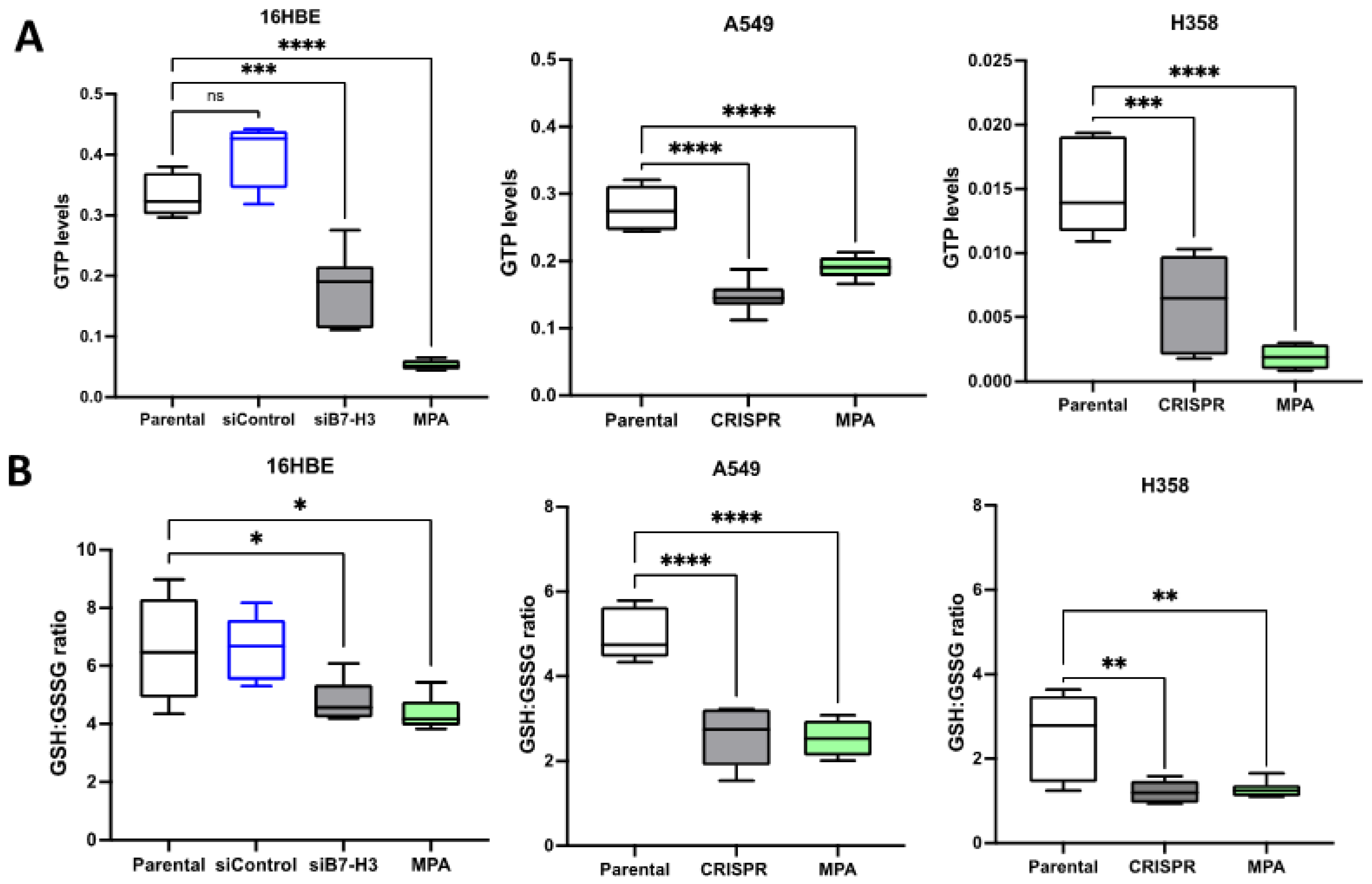
3.3. B7-H3 Forms a Complex with IMPDH2 and Regulates Downstream Metabolic Targets
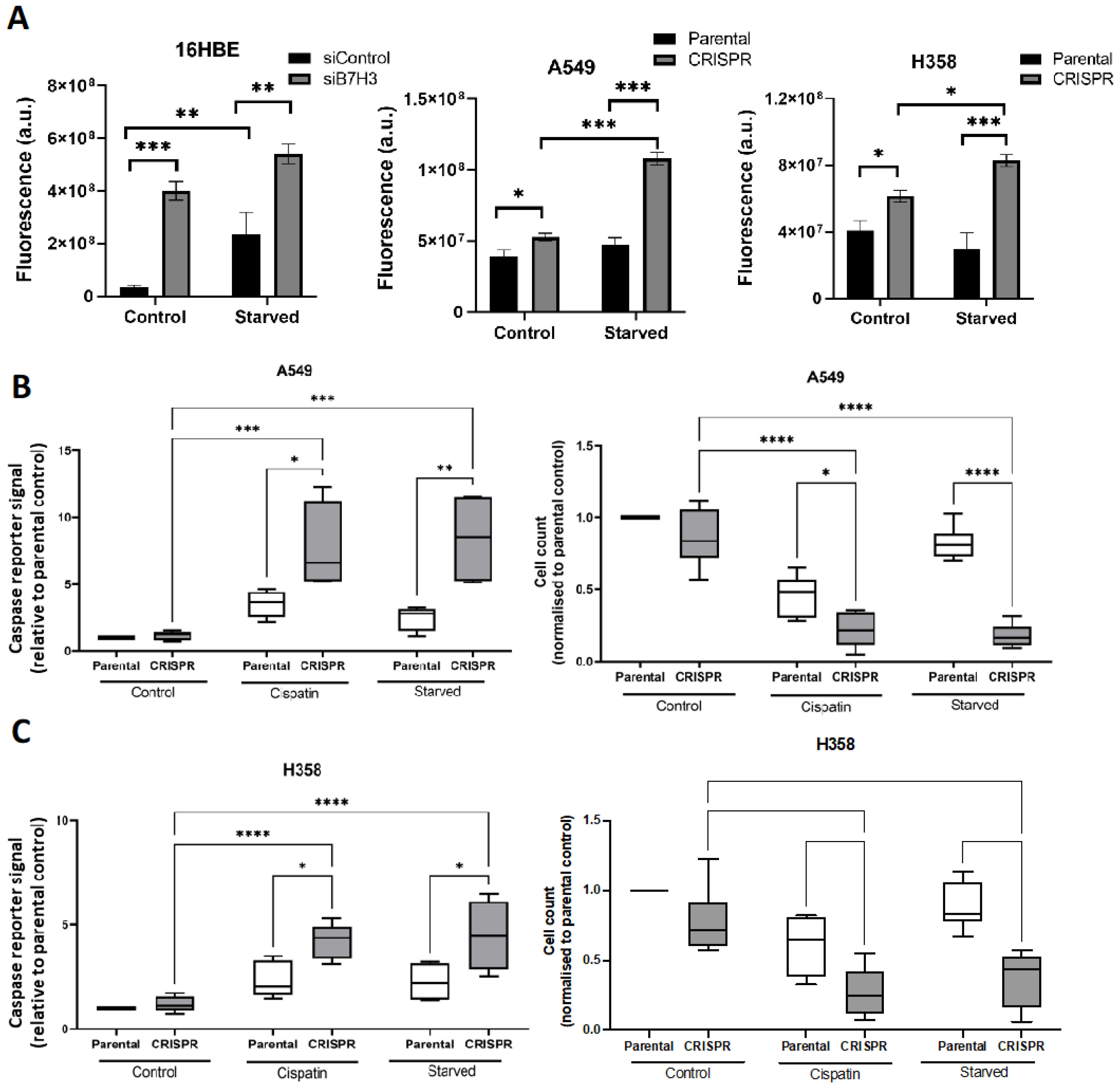
3.4. B7-H3 Suppresses Oxidative Stress and Promotes Chemoresistance and Cell Survival
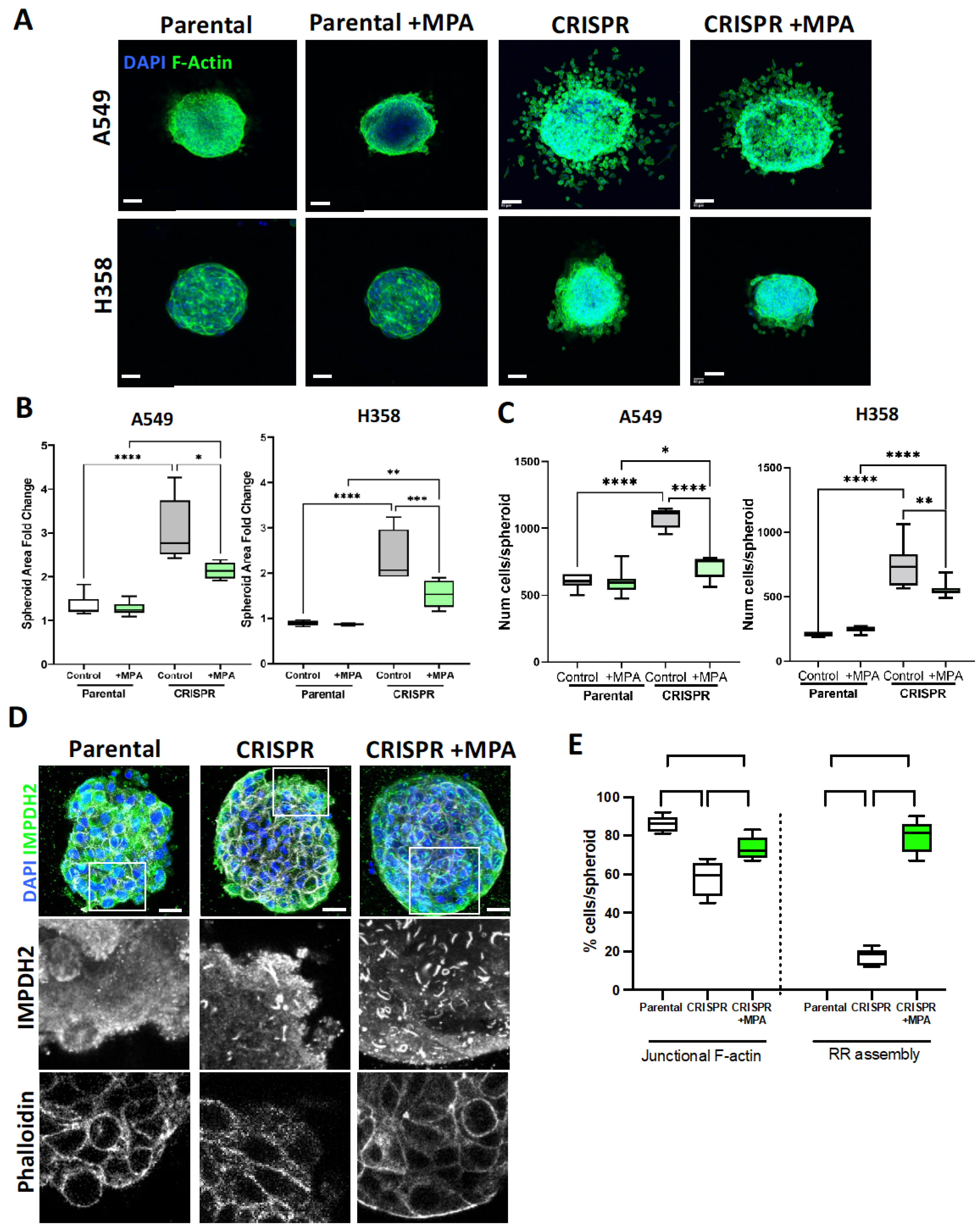
3.5. IMPDH2 Supports B7-H3-Dependent Cancer Cell 3D Invasion and Proliferation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- de Groot, P.M.; Wu, C.C.; Carter, B.W.; Munden, R.F. The epidemiology of lung cancer. Transl. Lung Cancer Res. 2018, 7, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Walser, T.; Cui, X.; Yanagawa, J.; Lee, J.M.; Heinrich, E.; Lee, G.; Sharma, S.; Dubinett, S.M. Smoking and Lung Cancer: The Role of Inflammation. Proc. Am. Thorac. Soc. 2008, 5, 811–815. [Google Scholar] [CrossRef]
- Li, J.; Zhang, X.; Hou, Z.; Cai, S.; Guo, Y.; Sun, L.; Li, A.; Li, Q.; Wang, E.; Miao, Y. P130cas-FAK interaction is essential for YAP-mediated radioresistance of non-small cell lung cancer. Cell Death Dis. 2022, 13, 783. [Google Scholar] [CrossRef] [PubMed]
- Zappa, C.; Mousa, S.A. Non-small cell lung cancer: Current treatment and future advances. Transl. Lung Cancer Res. 2016, 5, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahid, K.; Khalife, M.; Dabney, R.; Phan, A.T. Immunotherapy and targeted therapy—The new roadmap in cancer treatment. Ann. Transl. Med. 2019, 7, 595. [Google Scholar] [CrossRef]
- Slattum, G.M.; Rosenblatt, J. Tumour cell invasion: An emerging role for basal epithelial cell extrusion. Nat. Rev. Cancer 2014, 14, 495–501. [Google Scholar] [CrossRef]
- Lolo, F.-N.; Casas-Tintó, S.; Moreno, E. Cell Competition Time Line: Winners Kill Losers, which Are Extruded and Engulfed by Hemocytes. Cell Rep. 2012, 2, 526–539. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Läsche, M.; Emons, G.; Gründker, C. Shedding New Light on Cancer Metabolism: A Metabolic Tightrope Between Life and Death. Front. Oncol. 2020, 10, 409. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef] [Green Version]
- An, S.; Kumar, R.; Sheets, E.D.; Benkovic, S.J. Reversible Compartmentalization of de Novo Purine Biosynthetic Complexes in Living Cells. Science 2008, 320, 103–106. [Google Scholar] [CrossRef]
- Barfeld, S.J.; Fazli, L.; Persson, M.; Marjavaara, L.; Urbanucci, A.; Kaukoniemi, K.M.; Rennie, P.S.; Ceder, Y.; Chabes, A.; Visakorpi, T.; et al. Myc-dependent purine biosynthesis affects nucleolar stress and therapy response in prostate cancer. Oncotarget 2015, 6, 12587–12602. [Google Scholar] [CrossRef] [Green Version]
- Dostalek, M.; Gohh, R.Y.; Akhlaghi, F. Inosine Monophosphate Dehydrogenase Expression and Activity Are Significantly Lower in Kidney Transplant Recipients with Diabetes Mellitus. Ther. Drug Monit. 2013, 35, 374–383. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Zheng, Z.; Xu, Y.; Weng, H.; Gao, Y.; Qin, K.; Rong, J.; Chen, C.; Yun, M.; Zhang, J.; et al. Over-expression of IMPDH2 is associated with tumor progression and poor prognosis in hepatocellular carcinoma. Am. J. Cancer Res. 2018, 8, 1604–1614. [Google Scholar]
- Floryk, D.; Tollaksen, S.L.; Giometti, C.S.; Huberman, E. Differentiation of Human Prostate Cancer PC-3 Cells Induced by Inhibitors of Inosine 5′-Monophosphate Dehydrogenase. Cancer Res. 2004, 64, 9049–9056. [Google Scholar] [CrossRef] [Green Version]
- Li, H.-X.; Meng, Q.-P.; Liu, W.; Li, Y.-G.; Zhang, H.-M.; Bao, F.-C.; Song, L.-L.; Li, H.J. IMPDH2 mediate radioresistance and chemoresistance in osteosarcoma cells. Eur. Rev. Med Pharmacol. Sci. 2014, 18, 3038–3044. [Google Scholar]
- Hedstrom, L. IMP Dehydrogenase: Structure, Mechanism, and Inhibition. Chem. Rev. 2009, 109, 2903–2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, J.; Almquist, S.J.; Ford, P.J.; Shlyakhter, D.; Wang, Y.; Nimmesgern, E.; Germann, U.A. Regulation of inosine monophosphate dehydrogenase type I and type II isoforms in human lymphocytes. Biochem. Pharmacol. 2004, 67, 767–776. [Google Scholar] [CrossRef]
- Carcamo, W.C.; Satoh, M.; Kasahara, H.; Terada, N.; Hamazaki, T.; Chan, J.Y.F.; Yao, B.; Tamayo, S.; Covini, G.; von Mühlen, C.A.; et al. Induction of Cytoplasmic Rods and Rings Structures by Inhibition of the CTP and GTP Synthetic Pathway in Mammalian Cells. PLoS ONE 2011, 6, e29690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunter, J.H.; Thomas, E.C.; Lengefeld, N.; Kruger, S.J.; Worton, L.; Gardiner, E.M.; Jones, A.; Barnett, N.L.; Whitehead, J.P. Characterisation of inosine monophosphate dehydrogenase expression during retinal development: Differences between variants and isoforms. Int. J. Biochem. Cell Biol. 2008, 40, 1716–1728. [Google Scholar] [CrossRef]
- Keppeke, G.D.; Prado, M.S.; Nunes, E.; Perazzio, S.F.; Rodrigues, S.H.; Ferraz, M.L.G.; Chan, E.K.L.; Andrade, L.E.C. Differential capacity of therapeutic drugs to induce Rods/Rings structures in vitro and in vivo and generation of anti-Rods/Rings autoantibodies. Clin. Immunol. 2016, 173, 149–156. [Google Scholar] [CrossRef]
- Ji, Y.; Gu, J.; Makhov, A.M.; Griffith, J.D.; Mitchell, B.S. Regulation of the Interaction of Inosine Monophosphate Dehydrogenase with Mycophenolic Acid by GTP. J. Biol. Chem. 2006, 281, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Anthony, S.A.; Burrell, A.L.; Johnson, M.C.; Duong-Ly, K.C.; Kuo, Y.-M.; Simonet, J.C.; Michener, P.; Andrews, A.; Kollman, J.M.; Peterson, J.R. Reconstituted IMPDH polymers accommodate both catalytically active and inactive conformations. Mol. Biol. Cell 2017, 28, 2600–2608. [Google Scholar] [CrossRef]
- Carcamo, W.C.; Calise, S.J.; von Mühlen, C.A.; Satoh, M.; Chan, E.K. Molecular Cell Biology and Immunobiology of Mammalian Rod/Ring Structures. Int. Rev. Cell Mol. Biol. 2014, 308, 35–74. [Google Scholar] [CrossRef]
- Keppeke, G.D.; Chang, C.C.; Peng, M.; Chen, L.-Y.; Lin, W.-C.; Pai, L.-M.; Andrade, L.E.C.; Sung, L.-Y.; Liu, J.-L. IMP/GTP balance modulates cytoophidium assembly and IMPDH activity. Cell Div. 2018, 13, 5. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-C.; Lin, W.-C.; Pai, L.-M.; Lee, H.-S.; Wu, S.-C.; Ding, S.-T.; Liu, J.-L.; Sung, L.-Y. Cytoophidium assembly reflects upregulation of IMPDH activity. J. Cell Sci. 2015, 128, 3550–3555. [Google Scholar] [CrossRef] [Green Version]
- Aboelella, N.S.; Brandle, C.; Kim, T.; Ding, Z.-C.; Zhou, G. Oxidative Stress in the Tumor Microenvironment and Its Relevance to Cancer Immunotherapy. Cancers 2021, 13, 986. [Google Scholar] [CrossRef]
- Kennel, K.B.; Greten, F.R. Immune cell—Produced ROS and their impact on tumor growth and metastasis. Redox Biol. 2021, 42, 101891. [Google Scholar] [CrossRef]
- Valacchi, G.; Virgili, F.; Cervellati, C.; Pecorelli, A. OxInflammation: From Subclinical Condition to Pathological Biomarker. Front. Physiol. 2018, 9, 858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive Oxygen Species Regulate T Cell Immune Response in the Tumor Microenvironment. Oxid. Med. Cell. Longev. 2016, 2016, 1580967. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef] [Green Version]
- Picarda, E.; Ohaegbulam, K.C.; Zang, X. Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 3425–3431. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Xiang, S.; Shen, J.; Xiao, M.; Zhao, Y.; Wu, X.; Du, F.; Ji, H.; Li, M.; Zhao, Q.; et al. Comprehensive understanding of B7 family in gastric cancer: Expression profile, association with clinicopathological parameters and downstream targets. Int. J. Biol. Sci. 2020, 16, 568–582. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Wang, Y.; Zhao, J.; Gu, M.; Giscombe, R.; Lefvert, A.K.; Wang, X. B7-H3 and B7-H4 expression in non-small-cell lung cancer. Lung Cancer 2006, 53, 143–151. [Google Scholar] [CrossRef]
- Sun, X.; Vale, M.; Leung, E.; Kanwar, J.R.; Gupta, R.; Krissansen, G.W. Mouse B7-H3 induces antitumor immunity. Gene Ther. 2003, 10, 1728–1734. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Chapoval, A.I.; Flies, D.B.; Zhu, G.; Hirano, F.; Wang, S.; Lau, J.S.; Dong, H.; Tamada, K.; Flies, A.S.; et al. B7-H3 Enhances Tumor Immunity In Vivo by Costimulating Rapid Clonal Expansion of Antigen-Specific CD8+ Cytolytic T Cells. J. Immunol. 2004, 173, 5445–5450. [Google Scholar] [CrossRef] [Green Version]
- Lupu, C.M.; Eisenbach, C.; Kuefner, M.A.; Schmidt, J.; Lupu, A.D.; Stremmel, W.; Encke, J. An orthotopic colon cancer model for studying the B7-H3 antitumor effect in vivo. J. Gastrointest. Surg. 2006, 10, 635–645. [Google Scholar] [CrossRef]
- Katayama, A.; Takahara, M.; Kishibe, K.; Nagato, T.; Kunibe, I.; Katada, A.; Hayashi, T.; Harabuchi, Y. Expression of B7-H3 in hypopharyngeal squamous cell carcinoma as a predictive indicator for tumor metastasis and prognosis. Int. J. Oncol. 2011, 38, 1219–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, Q.; Chen, W.; Shan, B.; Ding, Y.; Zhang, G.; Cao, N.; Liu, L.; Zhang, Y. B7-H3 is Overexpressed in Patients Suffering Osteosarcoma and Associated with Tumor Aggressiveness and Metastasis. PLoS ONE 2013, 8, e70689. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K.; Amori, G.; Yuasa, T.; Yamamoto, S.; Yonese, J.; Ishikawa, Y. Relationship of B7-H3 expression in tumor cells and tumor vasculature with FOXP3+ regulatory T cells in renal cell carcinoma. Cancer Manag. Res. 2019, 11, 7021–7030. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Zhang, P.; Li, J.; Zhao, J.; Liu, C.; Yang, F.; Yang, D.; Gao, A.; Lin, W.; Ma, X.; et al. B7-H3 in combination with regulatory T cell is associated with tumor progression in primary human non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 13987–13995. [Google Scholar]
- Prasad, D.V.R.; Nguyen, T.; Li, Z.; Yang, Y.; Duong, J.; Wang, Y.; Dong, C. Murine B7-H3 is a negative regulator of T cells. J. Immunol. 2004, 173, 2500–2506. [Google Scholar] [CrossRef] [Green Version]
- Kang, F.-B.; Wang, L.; Jia, H.-C.; Li, D.; Li, H.-J.; Zhang, Y.-G.; Sun, D.-X. B7-H3 promotes aggression and invasion of hepatocellular carcinoma by targeting epithelial-to-mesenchymal transition via JAK2/STAT3/Slug signaling pathway. Cancer Cell Int. 2015, 15, 45. [Google Scholar] [CrossRef] [Green Version]
- Nunes-Xavier, C.E.; Karlsen, K.F.; Tekle, C.; Pedersen, C.; Øyjord, T.; Hongisto, V.; Nesland, J.M.; Tan, M.; Sahlberg, K.K.; Fodstad, O. Decreased expression of B7-H3 reduces the glycolytic capacity and sensitizes breast cancer cells to AKT/mTOR inhibitors. Oncotarget 2016, 7, 6891–6901. [Google Scholar] [CrossRef] [Green Version]
- Flem-Karlsen, K.; Tekle, C.; Øyjord, T.; Flørenes, V.A.; Mælandsmo, G.M.; Fodstad, O.; Nunes-Xavier, C.E. p38 MAPK activation through B7-H3-mediated DUSP10 repression promotes chemoresistance. Sci. Rep. 2019, 9, 5839. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Zapater, E.; Bagley, D.C.; Hernandez, V.L.; Roberts, L.B.; Maguire, T.J.A.; Voss, F.; Mertins, P.; Kirchner, M.; Peset-Martin, I.; Woszczek, G.; et al. Epithelial coxsackievirus adenovirus receptor promotes house dust mite-induced lung inflammation. Nat. Commun. 2022, 13, 6407. [Google Scholar] [CrossRef]
- Chen, J.-T.; Chen, C.-H.; Ku, K.-L.; Hsiao, M.; Chiang, C.-P.; Hsu, T.-L.; Chen, M.-H.; Wong, C.-H. Glycoprotein B7-H3 overexpression and aberrant glycosylation in oral cancer and immune response. Proc. Natl. Acad. Sci. USA 2015, 112, 13057–13062. [Google Scholar] [CrossRef]
- Schiavon, C.R.; Griffin, M.E.; Pirozzi, M.; Parashuraman, R.; Zhou, W.; Jinnah, H.A.; Reines, D.; Kahn, R.A.; Pizzirusso, M.; Chang, A.; et al. Compositional complexity of rods and rings. Mol. Biol. Cell 2018, 29, 2303–2316. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.C.; Gunter, J.H.; Webster, J.A.; Schieber, N.L.; Oorschot, V.; Parton, R.G.; Whitehead, J.P. Different Characteristics and Nucleotide Binding Properties of Inosine Monophosphate Dehydrogenase (IMPDH) Isoforms. PLoS ONE 2012, 7, e51096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitka, O.; Skalickova, S.; Gumulec, J.; Masarik, M.; Adam, V.; Hubalek, J.; Trnkova, L.; Kruseova, J.; Eckschlager, T.; Kizek, R. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol. Lett. 2012, 4, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Flem-Karlsen, K.; Fodstad, O.; Tan, M.; Nunes-Xavier, C.E. B7-H3 in Cancer—Beyond Immune Regulation. Trends Cancer 2018, 4, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Ma, H.; Zha, L.; Li, Q.; Yang, G.; Pan, H.; Fei, X.; Xu, X.; Xing, C.; Zhang, L. IMPDH2 promotes cell proliferation and epithelial-mesenchymal transition of non-small cell lung cancer by activating the Wnt/β-catenin signaling pathway. Oncol. Lett. 2020, 20, 219. [Google Scholar] [CrossRef]
- Wolfe, K.; Kofuji, S.; Yoshino, H.; Sasaki, M.; Okumura, K.; Sasaki, A.T. Dynamic compartmentalization of purine nucleotide metabolic enzymes at leading edge in highly motile renal cell carcinoma. Biochem. Biophys. Res. Commun. 2019, 516, 50–56. [Google Scholar] [CrossRef]
- Lagies, S.; Schlimpert, M.; Neumann, S.; Wäldin, A.; Kammerer, B.; Borner, C.; Peintner, L. Cells grown in three-dimensional spheroids mirror in vivo metabolic response of epithelial cells. Commun. Biol. 2020, 3, 246. [Google Scholar] [CrossRef]
- Tekle, C.; Nygren, M.K.; Chen, Y.-W.; Dybsjord, I.; Nesland, J.M.; Maelandsmo, G.M.; Fodstad, O. B7-H3 contributes to the metastatic capacity of melanoma cells by modulation of known metastasis-associated genes. Int. J. Cancer 2012, 130, 2282–2290. [Google Scholar] [CrossRef]
- Castellanos, J.R.; Purvis, I.J.; Labak, C.M.; Guda, M.R.; Tsung, A.J.; Velpula, K.K.; Asuthkar, S. B7-H3 role in the immune landscape of cancer. Am. J. Clin. Exp. Immunol. 2017, 6, 66–75. [Google Scholar]
- Zhao, X.; Li, D.-C.; Zhu, X.-G.; Gan, W.-J.; Li, Z.; Xiong, F.; Zhang, Z.-X.; Zhang, G.-B.; Zhang, X.-G.; Zhao, H. B7-H3 overexpression in pancreatic cancer promotes tumor progression. Int. J. Mol. Med. 2013, 31, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wang, Y.; Wang, J.; Dong, F.; Zhu, M.; Wan, W.; Li, H.; Wu, F.; Yan, X.; Ke, X. B7-H3 silencing inhibits tumor progression of mantle cell lymphoma and enhances chemosensitivity. Int. J. Oncol. 2015, 46, 2562–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wang, L.; Zhang, H.; Tu, F.; Qiang, Y.; Nie, C. Decreased expression of ZO-1 is associated with tumor metastases in liver cancer. Oncol. Lett. 2019, 17, 1859–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.; Liu, F.; Liu, Z.; Zhang, T.; Hua, D. B7-H3 increases thymidylate synthase expression via the PI3k-Akt pathway. Tumor Biol. 2016, 37, 9465–9472. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, G.-B.; Gan, W.-J.; Xiong, F.; Li, Z.; Zhao, H.; Zhu, D.-M.; Zhang, B.; Zhang, X.-G.; Li, D.-C. Silencing of B7-H3 increases gemcitabine sensitivity by promoting apoptosis in pancreatic carcinoma. Oncol. Lett. 2013, 5, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Rudolph, J.L.; Harrison, S.M.W.; Jin, L.; Frantz, A.L.; Harrison, D.A.; Andres, D.A.; Dávila, D.; Torres-Aleman, I.; Newmeyer, M.E.D.; et al. An evolutionarily conserved Rit GTPase–p38 MAPK signaling pathway mediates oxidative stress resistance. Mol. Biol. Cell 2011, 22, 3231–3241. [Google Scholar] [CrossRef]
- Karigane, D.; Kobayashi, H.; Morikawa, T.; Ootomo, Y.; Sakai, M.; Nagamatsu, G.; Kubota, Y.; Goda, N.; Matsumoto, M.; Nishimura, E.K.; et al. p38α Activates Purine Metabolism to Initiate Hematopoietic Stem/Progenitor Cell Cycling in Response to Stress. Cell Stem Cell 2016, 19, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Li, Y.; Guo, G.; Song, J.; Cai, Z.; Yang, J.; Chen, Z.; Wang, Y.; Huang, Y.; Gao, Q. B7-H3 Promotes the Migration and Invasion of Human Bladder Cancer Cells via the PI3K/Akt/STAT3 Signaling Pathway. J. Cancer 2017, 8, 816–824. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Liu, J.; Que, L.; Tang, X. The immunoregulatory protein B7-H3 promotes aerobic glycolysis in oral squamous carcinoma via PI3K/Akt/mTOR pathway. J. Cancer 2019, 10, 5770–5784. [Google Scholar] [CrossRef]
- Cucchi, D.; Gibson, A.; A Martin, S. The emerging relationship between metabolism and DNA repair. Cell Cycle 2021, 20, 943–959. [Google Scholar] [CrossRef]
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.-E.; Malki, M.I. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhamad, S.; Elmasry, Y.; Uwagboe, I.; Chekmeneva, E.; Sands, C.; Cooper, B.W.; Camuzeaux, S.; Salam, A.; Parsons, M. B7-H3 Associates with IMPDH2 and Regulates Cancer Cell Survival. Cancers 2023, 15, 3530. https://doi.org/10.3390/cancers15133530
Alhamad S, Elmasry Y, Uwagboe I, Chekmeneva E, Sands C, Cooper BW, Camuzeaux S, Salam A, Parsons M. B7-H3 Associates with IMPDH2 and Regulates Cancer Cell Survival. Cancers. 2023; 15(13):3530. https://doi.org/10.3390/cancers15133530
Chicago/Turabian StyleAlhamad, Salwa, Yassmin Elmasry, Isabel Uwagboe, Elena Chekmeneva, Caroline Sands, Benjamin W. Cooper, Stephane Camuzeaux, Ash Salam, and Maddy Parsons. 2023. "B7-H3 Associates with IMPDH2 and Regulates Cancer Cell Survival" Cancers 15, no. 13: 3530. https://doi.org/10.3390/cancers15133530
APA StyleAlhamad, S., Elmasry, Y., Uwagboe, I., Chekmeneva, E., Sands, C., Cooper, B. W., Camuzeaux, S., Salam, A., & Parsons, M. (2023). B7-H3 Associates with IMPDH2 and Regulates Cancer Cell Survival. Cancers, 15(13), 3530. https://doi.org/10.3390/cancers15133530