
Selection of tRNA Genes in Human Breast Tumours Varies Substantially between Individuals


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Breast Cancer H3K27ac ChIP-seq Data
2.1.1. Downloading ChIP-seq Datasets
2.1.2. Regions of Interest
2.1.3. Quantification of H3K27ac Signals Using Easeq
2.1.4. Data Analysis
2.2. Comparison of tRNA Isoacceptor Expression between Tumour and Normal Breast Tissue
2.3. Analysis of Correlation between tRNA Expression and Patient Survival
3. Results
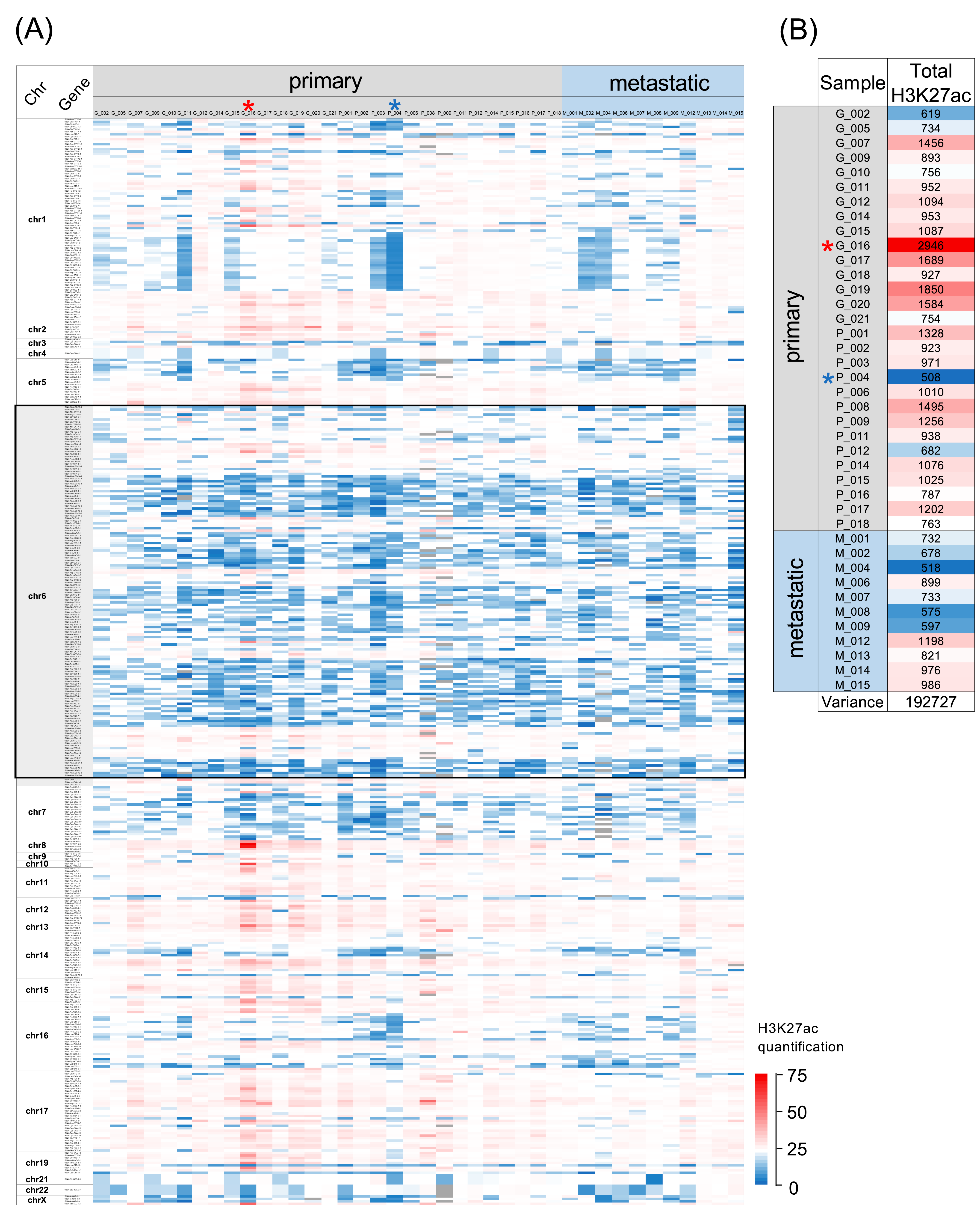
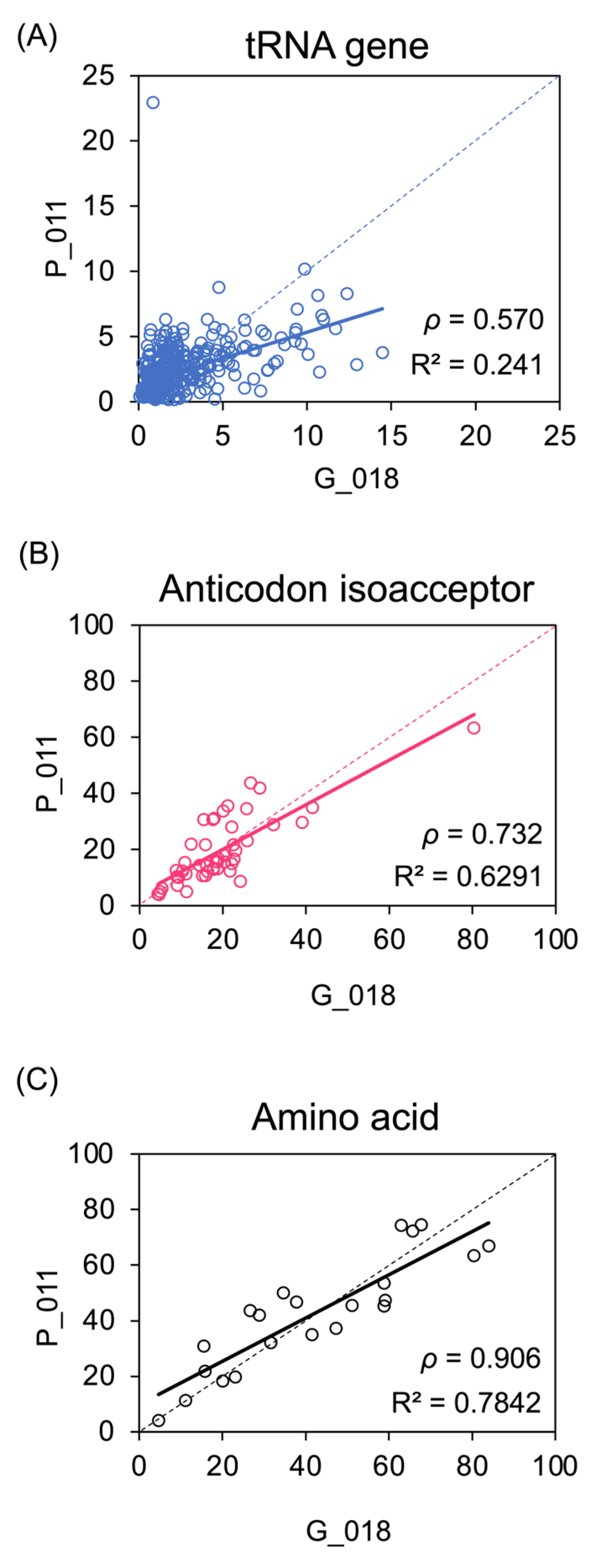
3.1. H3K27ac Enrichment Varies Considerably between tRNA Gene Loci and also between Tumours
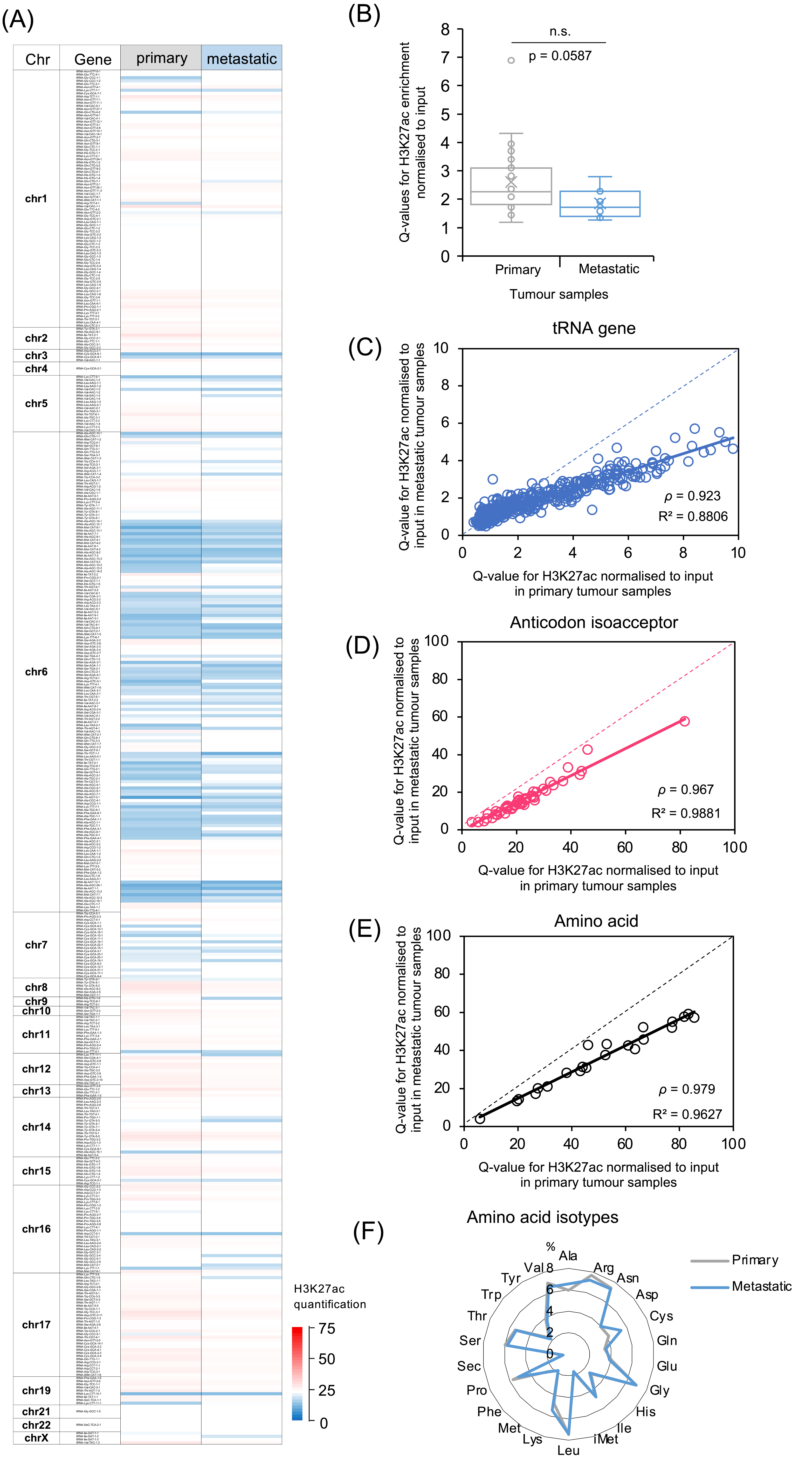
3.2. Comparison of H3K27ac Enrichment at tRNA Gene Loci between Primary and Metastatic Cancers
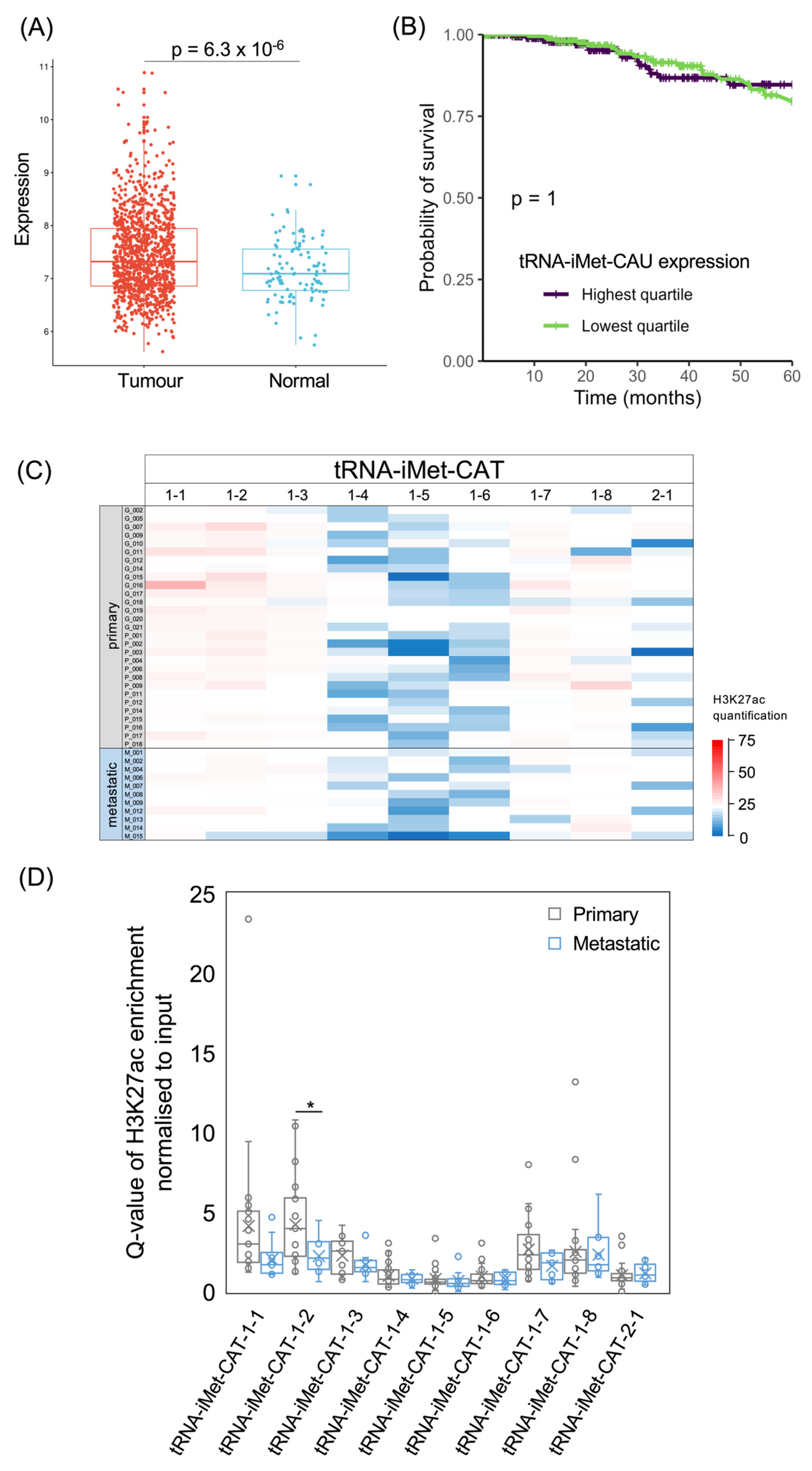
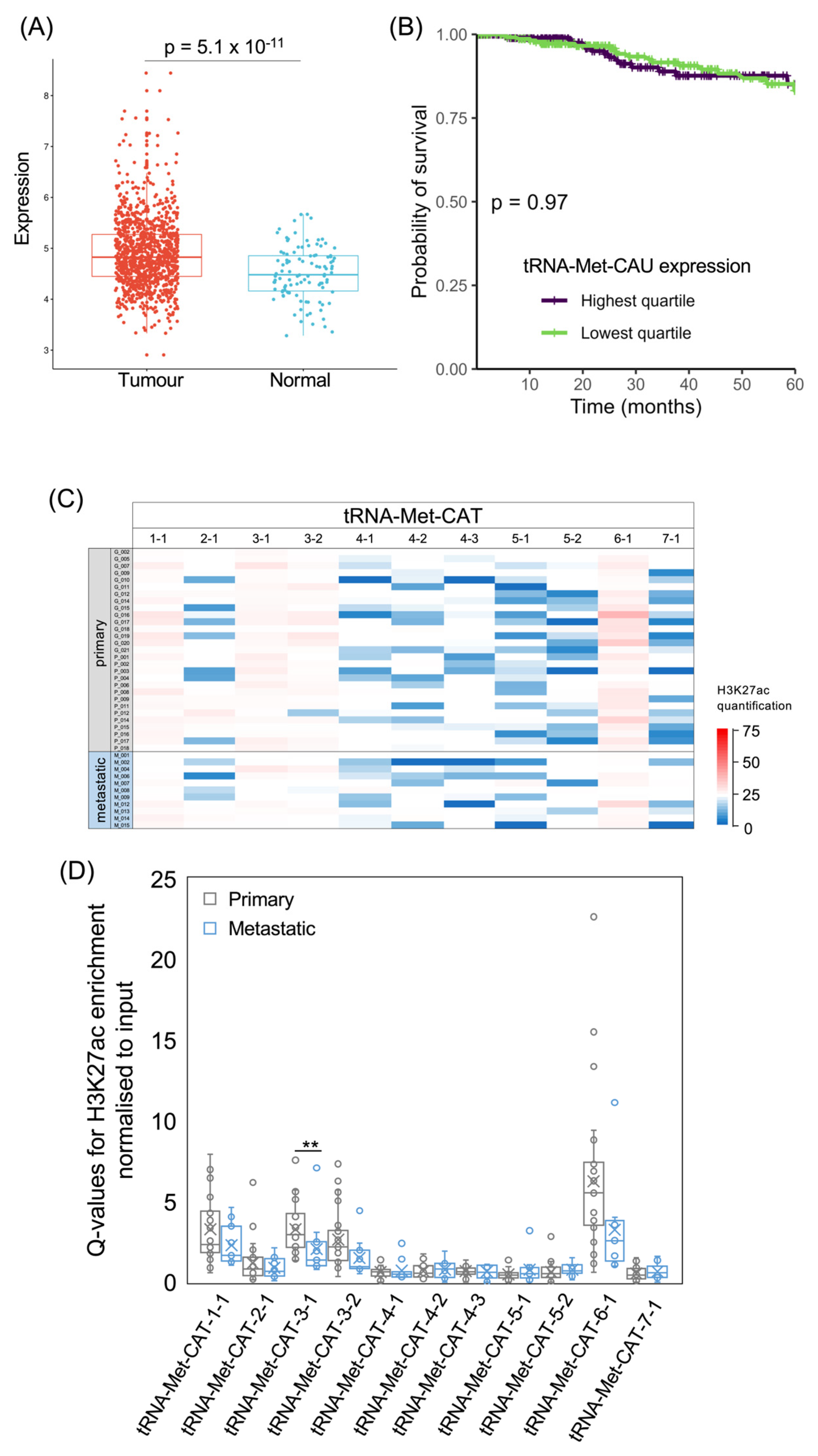
3.3. tRNA-iMet and tRNA-Met Genes
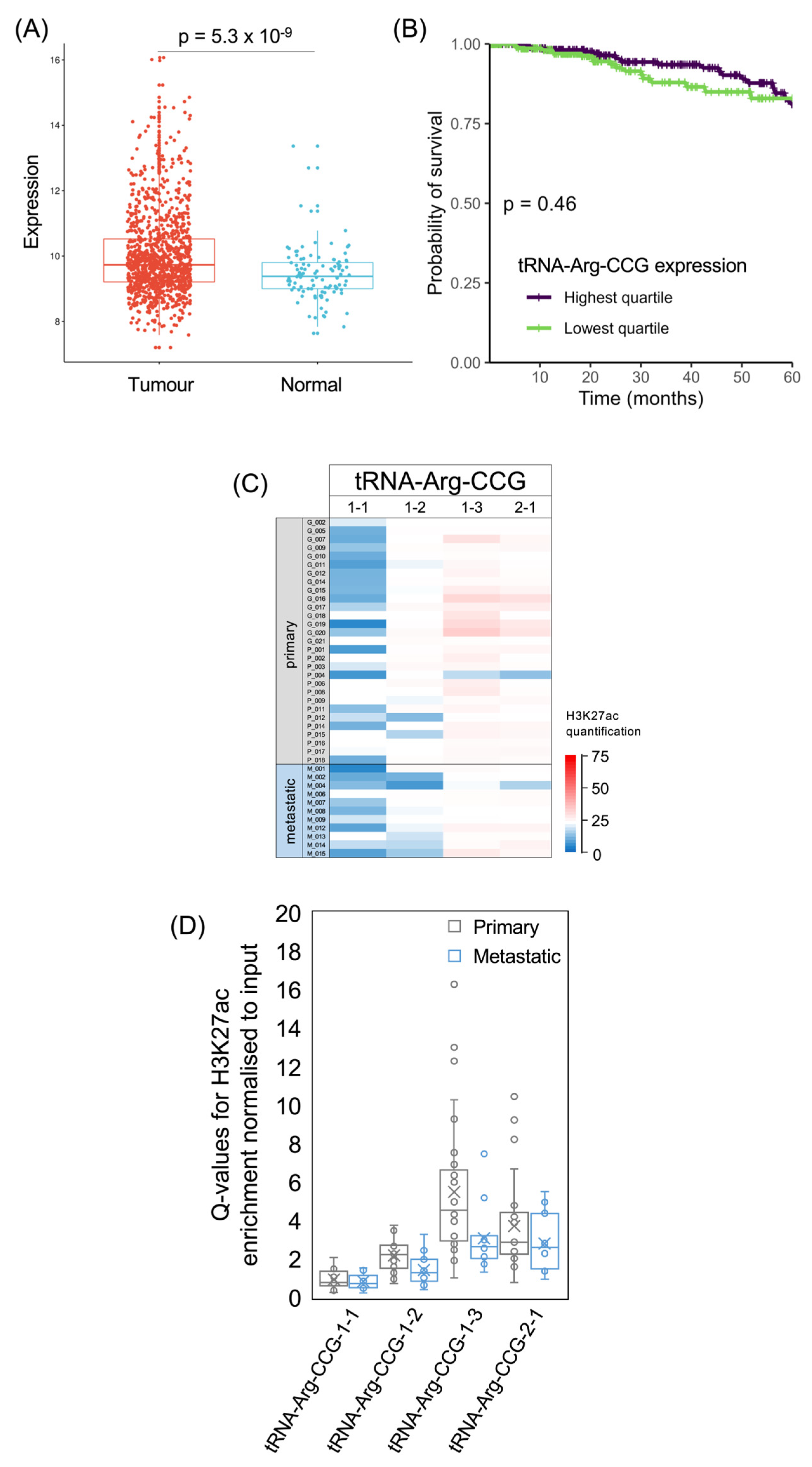
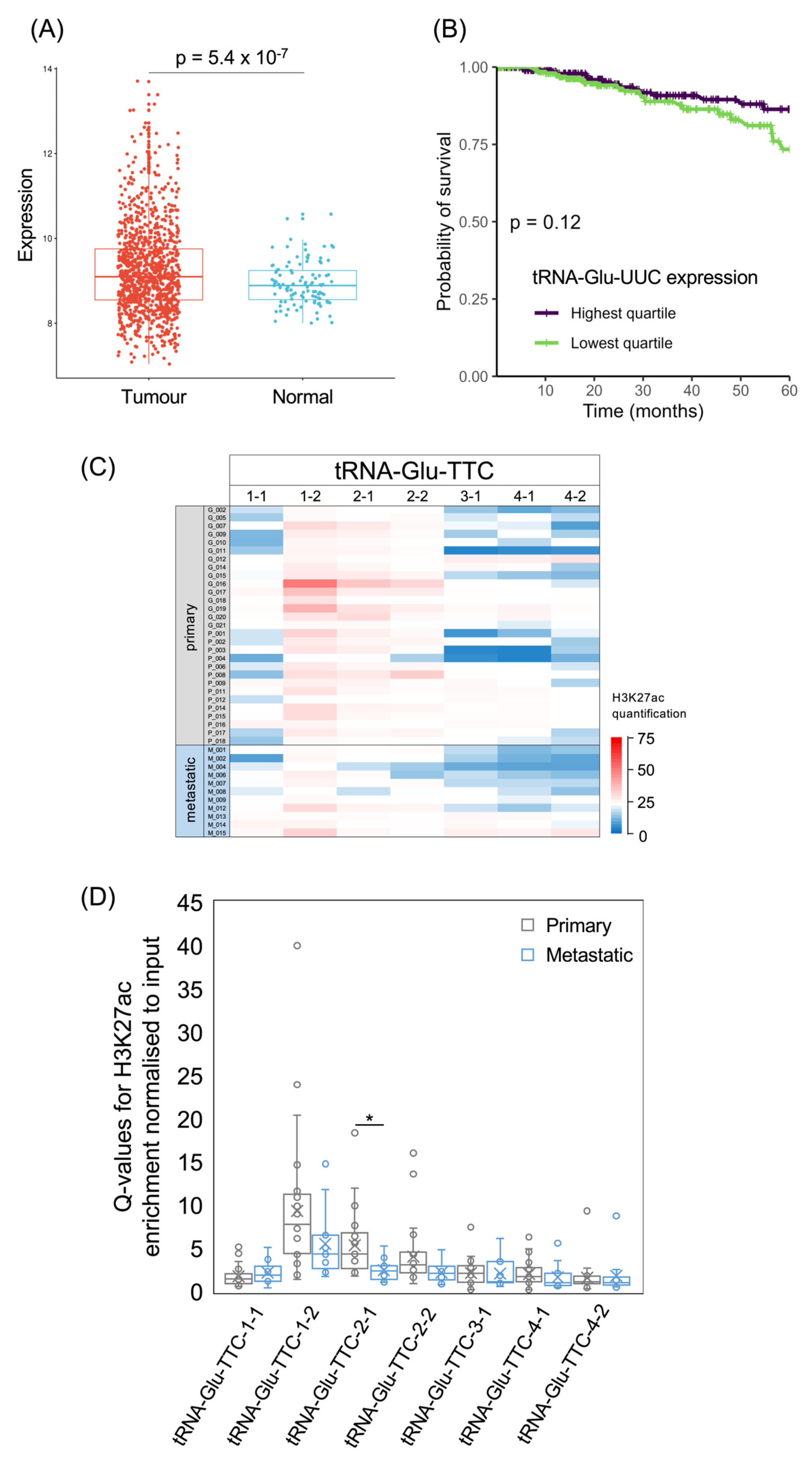
3.4. tRNA-Arg-CCG and tRNA-Glu-TTC Genes
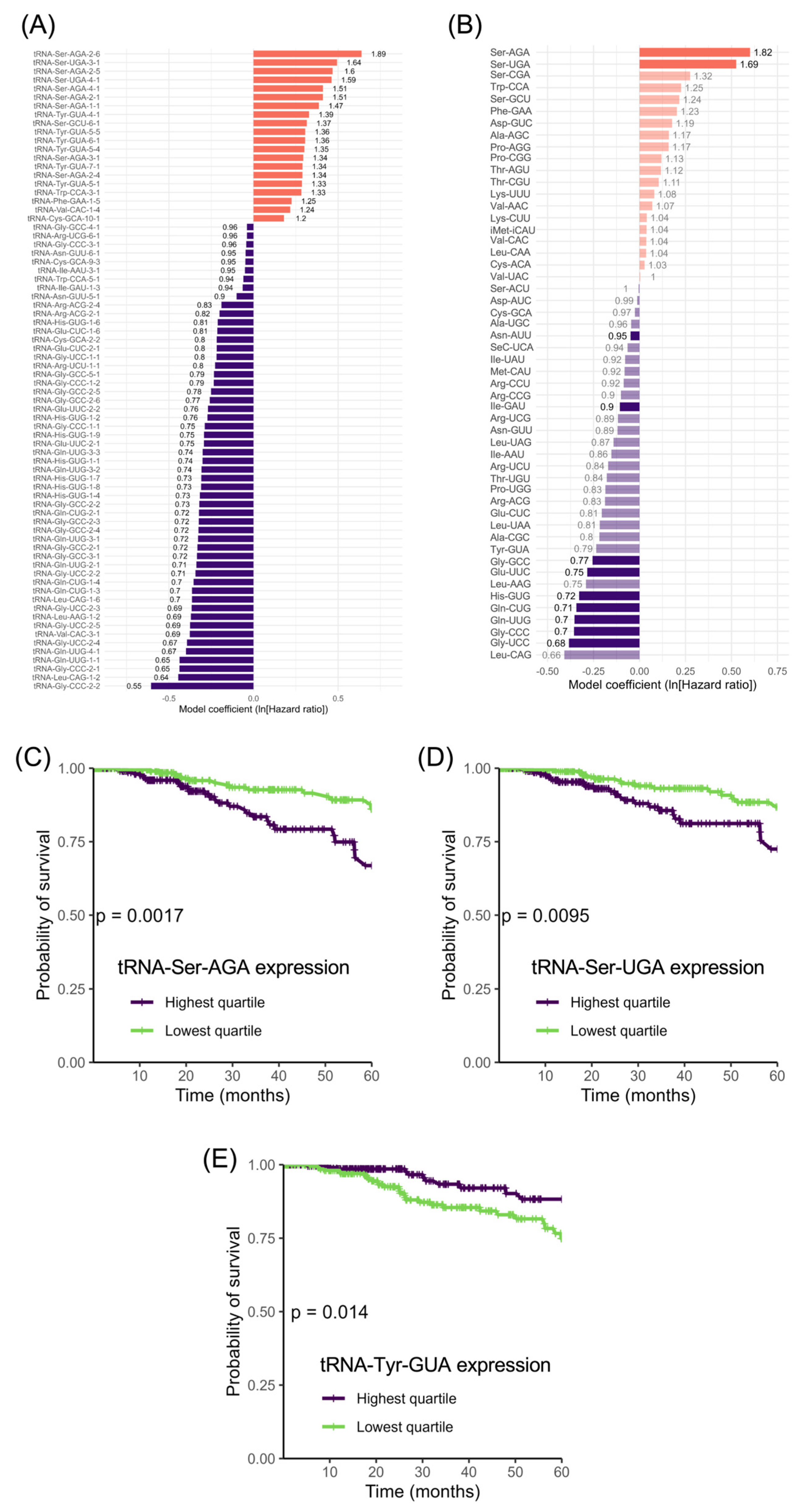
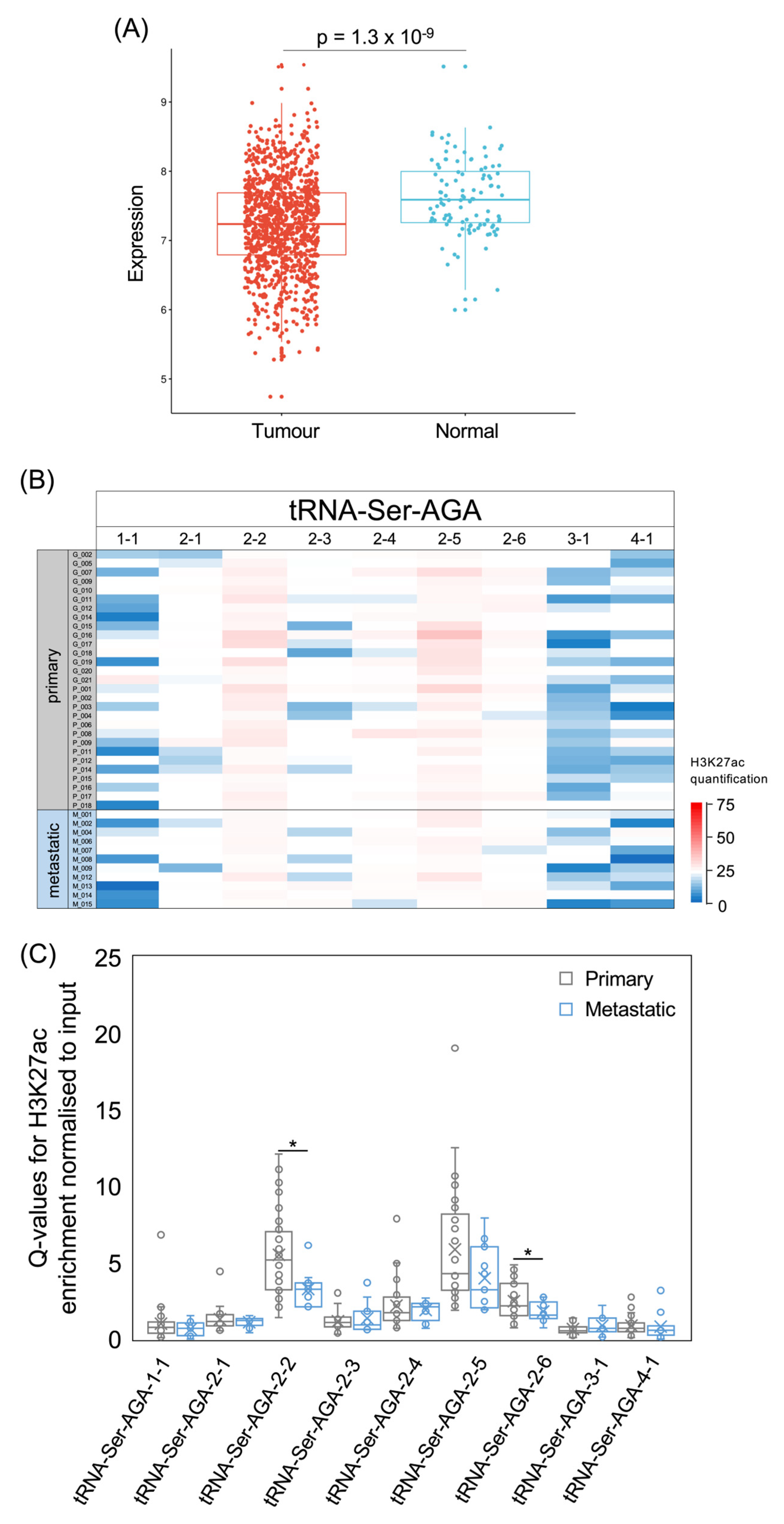
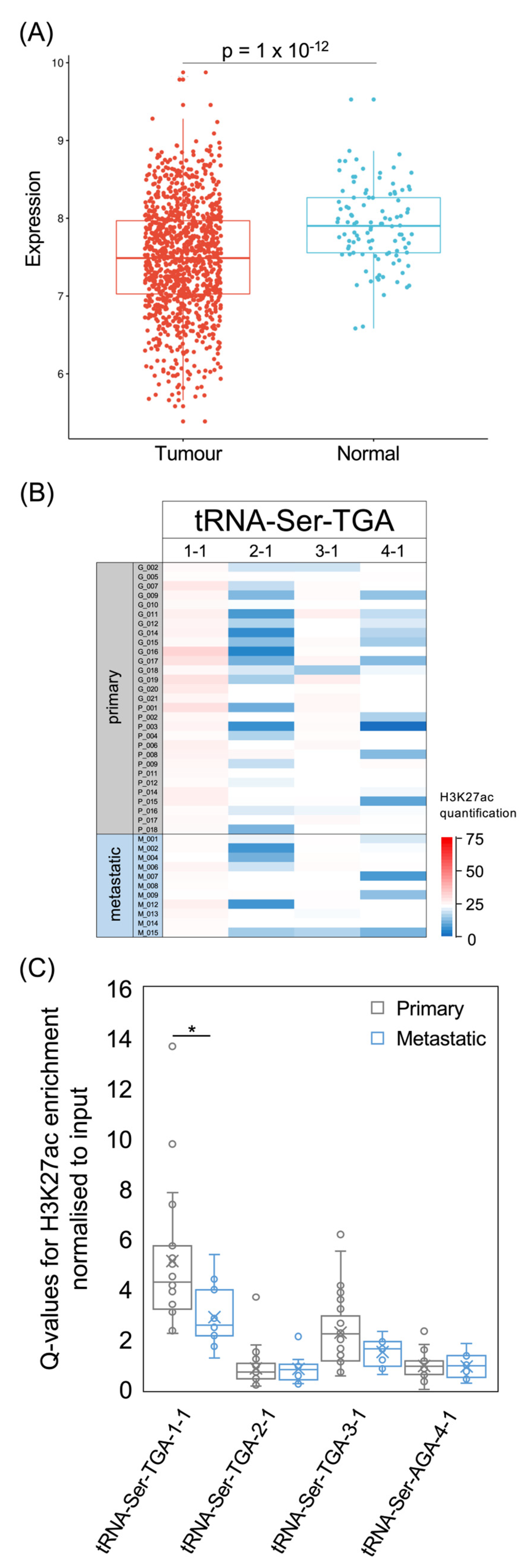
3.5. tRNA-Ser Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chan, P.P.; Lowe, T.M. GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016, 44, D184–D189. [Google Scholar] [CrossRef]
- White, R.J. Transcription by RNA polymerase III—More complex than we thought. Nat. Rev. Genet. 2011, 12, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Hughes, L.A.; Rudler, D.L.; Siira, S.; McCubbin, T.; Raven, S.A.; Browne, J.M.; Ermer, J.A.; Rientjes, J.; Rodger, J.; Marcellin, E.; et al. Copy number variation in tRNA isodecoder genes impairs mammalian development and balanced translation. Nat. Commun. 2023, 14, 2210. [Google Scholar] [CrossRef] [PubMed]
- White, R.J.; Gottlieb, T.M.; Downes, C.S.; Jackson, S.P. Cell cycle regulation of RNA polymerase III transcription. Mol. Cell Biol. 1995, 15, 6653–6662. [Google Scholar] [CrossRef]
- Gingold, H.; Tehler, D.; Christoffersen, N.R.; Nielsen, M.M.; Asmar, F.; Kooistra, S.M.; Christophersen, N.S.; Christensen, L.L.; Borre, M.; Sorensen, K.D.; et al. A dual program for translation regulation in cellular proliferation and differentiation. Cell 2014, 158, 1281–1292. [Google Scholar] [CrossRef]
- Goodarzi, H.; Nguyen, H.C.B.; Zhang, S.; Dill, B.D.; Molina, H.; Tavazoie, S.F. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell 2016, 165, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.; Coller, J. Codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018, 19, 20–30. [Google Scholar] [CrossRef] [PubMed]
- White, R.J. RNA polymerases I and III, growth control and cancer. Nat. Rev. Mol. Cell Biol. 2005, 6, 69–78. [Google Scholar] [CrossRef]
- Grewal, S.S. Why should cancer biologists care about tRNAs? tRNA synthesis, mRNA translation and the control of growth. Biochem. Biophys. Acta 2015, 1849, 898–907. [Google Scholar] [CrossRef]
- Huang, S.; Sun, B.; Xiong, Z.; Shu, Y.; Zhou, H.; Zhang, W.; Xiong, J.; Li, Q. The dysregulation of tRNAs and tRNA derivatives in cancer. J. Exp. Clin. Cancer Res. 2018, 37, 101. [Google Scholar] [CrossRef]
- Lant, J.T.; Berg, M.D.; Heinemann, I.U.; Brandl, C.J.; O’Donoghue, P. Pathways to disease from natural variations in human cytoplasmic tRNAs. J. Biol. Chem. 2019, 294, 5294–5308. [Google Scholar] [CrossRef] [PubMed]
- Orellana, E.A.; Siegal, E.; Gregory, R.I. tRNA dysregulation and disease. Nat. Rev. Genet. 2022, 23, 651–664. [Google Scholar] [CrossRef]
- Pavon-Eternod, M.; Gomes, S.; Geslain, R.; Dai, Q.; Rosner, M.R.; Pan, T. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res. 2009, 37, 7268–7280. [Google Scholar] [CrossRef]
- Krishnan, P.; Ghosh, S.; Wang, B.; Heyns, M.; Li, D.; Mackey, J.R.; Kovalchuk, O.; Damaraju, S. Genome-wide profiling of transfer RNAs and their role as novel prognostic markers for breast cancer. Sci. Rep. 2016, 6, 32843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ye, Y.; Gong, J.; Ruan, H.; Liu, C.-J.; Xiang, Y.; Cai, C.; Guo, A.-Y.; Ling, J.; Diao, L.; et al. Global analysis of tRNA and translation factor expression reveals a dynamic landscape of translational regulation in human cancers. Commun. Biol. 2018, 1, 234. [Google Scholar] [CrossRef]
- Sangha, A.K.; Kantidakis, T. The aminoacyl-tRNA synthetase and tRNA expression levels are deregulated in cancer and correlate independently with patient survival. Curr. Issues Mol. Biol. 2022, 44, 3001–3017. [Google Scholar] [CrossRef]
- Dittmar, K.A.; Goodenbour, J.M.; Pan, T. Tissue-specific differences in human transfer RNA expression. PLoS Genet. 2006, 2, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, D.; Kovalchuk, I.; Apel, I.J.; Chinnalyan, A.M.; Woycicki, R.K.; Cantor, C.R.; Kovalchuk, O. miR-34a directly targets tRNAiMet precursors and affects cellular proliferation, cell cycle, and apoptosis. Proc. Natl. Acad. Sci. USA 2018, 115, 7392–7397. [Google Scholar] [CrossRef]
- Behrens, A.; Rodschinka, G.; Nedialkova, D.D. High-resolution quantitative profiling of tRNA abundance and modification status in eukaryotes by mim-tRNAseq. Mol. Cell 2021, 81, 1802–1815. [Google Scholar] [CrossRef]
- Wiener, D.; Schwartz, S. How many tRNAs are out there? Mol. Cell 2021, 81, 1595–1597. [Google Scholar] [CrossRef]
- Cozen, A.E.; Quartley, E.; Holmes, A.D.; Hrabeta-Robinson, E.; Phizicky, E.M.; Lowe, T.M. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods 2015, 12, 879–884. [Google Scholar] [CrossRef]
- Zheng, G.; Qin, Y.; Clark, W.C.; Dai, Q.; Yi, C.; He, C.; Lambowitz, A.M.; Pan, T. Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 2015, 12, 835–837. [Google Scholar] [CrossRef]
- Pinkard, O.; McFarland, S.; Sweet, T.; Coller, J. Quantitative tRNA-sequencing uncovers metazoan tissue-specific tRNA regulation. Nat. Commun. 2020, 11, 4104. [Google Scholar] [CrossRef]
- Gogakos, T.; Brown, M.; Garzia, A.; Meyer, C.; Hafner, M.; Tuschl, T. Characterizing expression and processing of precursor and mature human tRNAs by hydro-tRNAseq and PAR-CLIP. Cell Rep. 2017, 20, 1463–1475. [Google Scholar] [CrossRef]
- Barski, A.; Chepelev, I.; Liko, D.; Cuddapah, S.; Fleming, A.B.; Birch, J.L.; Cui, K.; White, R.J.; Zhao, K. Pol II and its associated epigenetic marks are present at pol III-transcribed noncoding RNA genes. Nat. Struct. Mol. Biol. 2010, 17, 629–634. [Google Scholar] [CrossRef]
- Moqtaderi, Z.; Wang, J.; Raha, D.; White, R.J.; Snyder, M.; Weng, Z.; Struhl, K. Genomic binding profiles of functionally distinct RNA polymerase III transcription complexes in human cells. Nat. Struct. Mol. Biol. 2010, 17, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Oler, A.J.; Alla, R.K.; Roberts, D.N.; Wong, A.; Hollenhorst, P.C.; Chandler, K.J.; Cassiday, P.A.; Nelson, C.A.; Hagedorn, C.H.; Graves, B.J.; et al. Human RNA polymerase III transcriptomes and relationships to pol II promoter chromatin and enhancer-binding factors. Nat. Struct. Mol. Biol. 2010, 17, 620–628. [Google Scholar] [CrossRef]
- Raha, D.; Wang, Z.; Moqtaderi, Z.; Wu, L.; Zhong, G.; Gerstein, M.; Struhl, K.; Snyder, M. Close association of RNA polymerase II and many transcription factors with Pol III genes. Proc. Natl. Acad. Sci. USA 2010, 107, 3639–3644. [Google Scholar] [CrossRef] [PubMed]
- Kutter, C.; Brown, G.D.; Goncalves, A.; Wilson, M.D.; Watt, S.; Brazma, A.; White, R.J.; Odom, D.T. Pol III binding in six mammalian genomes shows high conservation among amino acid isotypes despite divergence in tRNA gene usage. Nat. Genet. 2011, 43, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Orioli, A.; Praz, V.; Lhote, P.; Hernandez, N. Human MAF1 targets and represses active RNA polymerase III genes by preventing recruitment rather than inducing long-term transcriptional arrest. Genome Res. 2016, 26, 624–635. [Google Scholar] [CrossRef]
- Park, J.-L.; Lee, Y.-S.; Song, M.-J.; Hong, S.-H.; Ahn, J.-H.; Seo, E.-H.; Shin, S.-P.; Lee, S.-J.; Johnson, B.-H.; Stampfer, M.R.; et al. Epigenetic regulation of RNA polymerase III transcription in early breast tumorigenesis. Oncogene 2017, 36, 6793–6804. [Google Scholar] [CrossRef]
- Van Bortle, K.; Marciano, D.P.; Liu, Q.; Chou, T.; Lipchik, A.M.; Gollapudi, S.; Geller, B.S.; Monte, E.; Kamakaka, R.T.; Snyder, M.P. A cancer-associated RNA polymerase III identity drives robust transcription and expression of snaR-A noncoding RNA. Nat. Commun. 2022, 13, 3007. [Google Scholar] [CrossRef] [PubMed]
- Van Bortle, K.; Phanstiel, D.H.; Snyder, M.P. Topological organization and dynamic regulation of human tRNA genes during macrophage differentiation. Genome Biol. 2017, 18, 180. [Google Scholar] [CrossRef] [PubMed]
- Patten, D.K.; Corleone, G.; Gyorffy, B.; Perone, Y.; Slaven, N.; Barozzi, I.; Erdos, E.; Saiakhova, A.; Goddard, K.; Vingiani, A.; et al. Enhancer mapping uncovers phenotypic heterogeneity and evolution in patients with luminal breast cancer. Nat. Med. 2018, 24, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Network, T.C.G.A.R. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC table browser data retrieval tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef]
- Lerdrup, M.; Johansen, J.V.; Agrawal-Singh, S.; Hansen, K. An interactive environment for agile analysis and visualization of ChIP-sequencing data. Nat. Struct. Mol. Biol. 2016, 23, 349–357. [Google Scholar] [CrossRef]
- Zhang, Z.; Ruan, H.; Liu, C.J.; Ye, Y.; Gong, J.; Diao, L.; Guo, A.-Y.; Han, L. tRic: A user-friendly data portal to explore the expression landscape of tRNAs in human cancers. RNA Biol. 2020, 17, 1674–1679. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000; ISBN 0-387-98784-3. [Google Scholar]
- Therneau, T.M. R Package, Version 3.3-1; A Package for Survival Analysis in R. 2022. Available online: https://CRAN.R-project.org/package=survival (accessed on 12 June 2023).
- Schramm, L.; Hernandez, N. Recruitment of RNA polymerase III to its target promoters. Genes Dev. 2002, 16, 2593–2620. [Google Scholar] [CrossRef]
- Mertens, C.; Roeder, R.G. Different functional modes of p300 in activation of RNA polymerase III transcription from chromatin templates. Mol. Cell Biol. 2008, 28, 5764–5776. [Google Scholar] [CrossRef]
- Sizer, R.E.; Chahid, N.; Butterfield, S.P.; Donze, D.; Bryant, N.J.; White, R.J. TFIIIC-based chromatin insulators through eukaryotic evolution. Gene 2022, 835, 146533. [Google Scholar] [CrossRef]
- Gomez-Roman, N.; Grandori, C.; Eisenman, R.N.; White, R.J. Direct activation of RNA polymerase III transcription by c-Myc. Nature 2003, 421, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Vervoorts, J.; Luscher-Firzlaff, J.M.; Rottmann, S.; Lilischkis, R.; Walsemann, G.; Dohmann, K.; Austen, M.; Luscher, B. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep. 2003, 4, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Malcolm, J.R.; Leese, N.K.; Lamond-Warner, P.I.; Brackenbury, W.J.; White, R.J. Widespread association of ERα with RMRP and tRNA genes in MCF-7 cells and breast cancers. Gene 2022, 821, 146280. [Google Scholar] [CrossRef] [PubMed]
- Horton, R.; Wilming, L.; Rand, V.; Lovering, R.C.; Bruford, E.A.; Khodiyar, V.K.; Lush, M.J.; Povey, S.; Talbot, C.C.; Wright, M.W.; et al. Gene map of the extended human MHC. Nat. Rev. Genet. 2004, 5, 889–899. [Google Scholar] [CrossRef]
- Pavon-Eternod, M.; Gomes, S.; Rosner, M.R.; Pan, T. Overexpression of initiator methionine tRNA leads to global reprogramming of tRNA expression and increased proliferation in human epithelial cells. RNA 2013, 19, 461–466. [Google Scholar] [CrossRef]
- Li, J.; Shen, Z.; Luo, L.; Ye, D.; Deng, H.; Gu, S.; Zhou, C. tRNAIniCAT inhibits proliferation and promotes apoptosis of laryngeal squamous cell carcinoma cells. J. Clin. Lab Anal. 2021, 35, e23821. [Google Scholar] [CrossRef]
- Macari, F.; El-houfi, Y.; Boldina, G.; Xu, H.; Khoury-Hanna, S.; Ollier, J.; Yazdani, L.; Zheng, G.; Bieche, I.; Legrand, N.; et al. TRIM6/61 connects PKCa with translational control through tRNAiMet stabilization: Impact on tumorigenesis. Oncogene 2016, 35, 1785–1796. [Google Scholar] [CrossRef]
- Birch, J.; Clarke, C.J.; Campbell, A.D.; Campbell, K.; Mitchell, L.E.; Liko, D.; Kalna, G.; Strathdee, D.; Sansom, O.J.; Neilson, M.; et al. The initiator methionine tRNA drives cell migration and invasion leading to increased metastatic potential in melanoma. Biol. Open 2016, 5, 1371–1379. [Google Scholar] [CrossRef]
- Clarke, C.J.; Berg, T.J.; Birch, J.; Ennis, D.; Mitchell, L.E.; Cloix, C.; Campbell, A.D.; Sumpton, D.; Nixon, C.; Campbell, K.; et al. The initiator methionine tRNA drives secretion of type II collagen from stromal fibroblasts to promote tumor growth and angiogenesis. Curr. Biol. 2016, 26, 755–765. [Google Scholar] [CrossRef]
- Thornlow, B.P.; Armstrong, J.; Holmes, A.D.; Howard, J.M.; Corbett-Detig, R.B.; Lowe, T.M. Predicting transfer RNA gene activity from sequence and genome context. Genome Res. 2020, 30, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Hah, N.; Danko, C.G.; Core, L.; Waterfall, J.J.; Siepel, A.; Lis, J.T.; Kraus, W.L. A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 2011, 145, 622–634. [Google Scholar] [CrossRef]
- Fang, Z.; Yi, Y.; Shi, G.; Li, S.; Chen, S.; Lin, Y.; Li, Z.; He, Z.; Li, W.; Zhong, S. Role of Brf1 interaction with ERa, and significance of its overexpression in human breast cancer. Mol. Oncol. 2017, 11, 1752–1767. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Loven, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef]
- Lautre, W.; Richard, E.; Feugeas, J.-P.; Dumay-Odelot, H.; Teichmann, M. The POLR3G subunit of human RNA polymerase III regulates tumorigenesis and metastasis in triple-negative breast cancer. Cancers 2022, 14, 5732. [Google Scholar] [CrossRef]
- Scott, P.H.; Cairns, C.A.; Sutcliffe, J.E.; Alzuherri, H.M.; Mclees, A.; Winter, A.G.; White, R.J. Regulation of RNA polymerase III transcription during cell cycle entry. J. Biol. Chem. 2001, 276, 1005–1014. [Google Scholar] [CrossRef]
- Stein, T.; Crighton, D.; Boyle, J.M.; Varley, J.M.; White, R.J. RNA polymerase III transcription can be derepressed by oncogenes or mutations that compromise p53 function in tumours and Li-Fraumeni syndrome. Oncogene 2002, 21, 2961–2970. [Google Scholar] [CrossRef]
- Angelis, E.; Garcia, A.; Chan, S.S.; Schenke-Layland, K.; Ren, S.; Goodfellow, S.J.; Jordan, M.C.; Roos, K.P.; White, R.J.; MacLellan, W.R. A cyclin D2-Rb pathway regulates cardiac myocyte size and RNA polymerase III after biomechanical stress in adult myocardium. Circ. Res. 2008, 102, 1222–1229. [Google Scholar] [CrossRef]
- Woiwode, A.; Johnson, S.A.S.; Zhong, S.; Zhang, C.; Roeder, R.G.; Teichmann, M.; Johnson, D.L. PTEN represses RNA polymerase III-dependent transcription by targeting the TFIIIB complex. Mol. Cell Biol. 2008, 28, 4204–4214. [Google Scholar] [CrossRef] [PubMed]
- Network, T.C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- White, R.J.; Trouche, D.; Martin, K.; Jackson, S.P.; Kouzarides, T. Repression of RNA polymerase III transcription by the retinoblastoma protein. Nature 1996, 382, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Cairns, C.A.; White, R.J. p53 is a general repressor of RNA polymerase III transcription. EMBO J. 1998, 17, 3112–3123. [Google Scholar] [CrossRef]
- Brown, T.R.P.; Scott, P.H.; Stein, T.; Winter, A.G.; White, R.J. RNA polymerase III transcription: Its control by tumor suppressors and its deregulation by transforming agents. Gene Expr. 2000, 9, 15–28. [Google Scholar] [CrossRef] [PubMed]
- White, R.J. RNA polymerase III transcription and cancer. Oncogene 2004, 23, 3208–3216. [Google Scholar] [CrossRef] [PubMed]
- Veras, I.; Rosen, E.M.; Schramm, L. Inhibition of RNA polymerase III transcription by BRCA1. J. Mol. Biol. 2009, 387, 523–531. [Google Scholar] [CrossRef]
- Zhong, Q.; Shi, G.; Zhang, Y.; Lu, L.; Levy, D.; Zhong, S. Alteration of BRCA1 expression affects alcohol-induced transcription of RNA pol III-dependent genes. Gene 2015, 556, 74–79. [Google Scholar] [CrossRef]
- Larminie, C.G.C.; Cairns, C.A.; Mital, R.; Martin, K.; Kouzarides, T.; Jackson, S.P.; White, R.J. Mechanistic analysis of RNA polymerase III regulation by the retinoblastoma protein. EMBO J. 1997, 16, 2061–2071. [Google Scholar] [CrossRef]
- Crighton, D.; Woiwode, A.; Zhang, C.; Mandavia, N.; Morton, J.P.; Warnock, L.J.; Milner, J.; White, R.J.; Johnson, D.L. p53 represses RNA polymerase III transcription by targeting TBP and inhibiting promoter occupancy by TFIIIB. EMBO J. 2003, 22, 2810–2820. [Google Scholar] [CrossRef]
- Ernens, I.; Goodfellow, S.J.; Innes, F.; Kenneth, N.S.; Derblay, L.E.; White, R.J.; Scott, P.H. Hypoxic stress suppresses RNA polymerase III recruitment and tRNA gene transcription in cardiomyocytes. Nucleic Acids Res. 2006, 34, 286–294. [Google Scholar] [CrossRef]
- Steiger, D.; Furrer, M.; Schwinkendorf, D.; Gallant, P. Max-independent functions of Myc in Drosophila melanogaster. Nat. Genet. 2008, 40, 1084–1091. [Google Scholar] [CrossRef]
- Sadeghifar, F.; Bohm, S.; Vintermist, A.; Farrants, A.O. The B-WICH chromatin-remodelling complex regulates RNA polymerase III transcription by promoting Max-dependent c-Myc binding. Nucleic Acids Res. 2015, 43, 4477–4490. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. p53 enters the microRNA world. Cancer Cell 2007, 12, 414–418. [Google Scholar] [CrossRef]
- Finlay-Schultz, J.; Gillen, A.E.; Brechbuhl, H.M.; Ivie, J.J.; Matthews, S.B.; Jacobsen, B.M.; Bentley, D.L.; Kabos, P.; Sartorius, C.A. Breast cancer suppression by progesterone receptors is mediated by their modulation of estrogen receptors and RNA polymerase III. Cancer Res. 2017, 77, 4934–4946. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Smith, D.K.; Ni, H.; Wu, K.Y.; Huang, D.; Pan, S.; Sathe, A.A.; Tang, Y.; Liu, M.-L.; Xing, C.; et al. SOX4-mediated repression of specific tRNAs inhibits proliferation of human glioblastoma cells. Proc. Natl. Acad. Sci. USA 2020, 117, 5782–5790. [Google Scholar] [CrossRef]
- Zhang, J.; Liang, Q.; Lei, Y.; Yao, M.; Li, L.; Gao, X.; Feng, J.; Zhang, Y.; Gao, H.; Liu, D.-X.; et al. SOX4 induces epithelial-mesenchymal transition and contributes to breast cancer progression. Cancer Res. 2012, 72, 4597–4608. [Google Scholar] [CrossRef] [PubMed]
- Harismendy, O.; Gendrel, C.-G.; Soularue, P.; Gidrol, X.; Sentenac, A.; Werner, M.; Lefebvre, O. Genome-wide location of yeast RNA polymerase III transcription machinery. EMBO J. 2003, 22, 4738–4747. [Google Scholar] [CrossRef]
- Roberts, D.N.; Stewart, A.J.; Huff, J.T.; Cairns, B.R. The RNA polymerase III transcriptome revealed by genome-wide localization and activity-occupancy relationships. Proc. Natl. Acad. Sci. USA 2003, 100, 14695–14700. [Google Scholar] [CrossRef]
- Moqtaderi, Z.; Struhl, K. Genome-wide occupancy profile of the RNA polymerase III machinery in Saccharomyces cerevisiae reveals loci with incomplete transcription complexes. Mol. Cell Biol. 2004, 24, 4118–4127. [Google Scholar] [CrossRef]
- Noma, K.; Cam, H.P.; Maraia, R.; Grewal, S.I. A role for TFIIIC transcription factor complex in genome organization. Cell 2006, 125, 859–872. [Google Scholar] [CrossRef]
- Canella, D.; Praz, V.; Reina, J.H.; Cousin, P.; Hernandez, N. Defining the RNA polymerase III transcriptome: Genome-wide localization of the RNA polymerase III transcription machinery in human cells. Genome Res. 2010, 20, 710–721. [Google Scholar] [CrossRef]
- Santos, M.; Fidalgo, A.; Varanda, A.S.; Soares, A.R.; Almeida, G.M.; Martins, D.; Mendes, N.; Oliveira, C.; Santos, M.A.S. Upregulation of tRNA-Ser-AGA-2-1 promotes malignant behavior in normal bronchial cells. Front. Mol. Biosci. 2022, 9, 809985. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butterfield, S.P.; Sizer, R.E.; Rand, E.; White, R.J. Selection of tRNA Genes in Human Breast Tumours Varies Substantially between Individuals. Cancers 2023, 15, 3576. https://doi.org/10.3390/cancers15143576
Butterfield SP, Sizer RE, Rand E, White RJ. Selection of tRNA Genes in Human Breast Tumours Varies Substantially between Individuals. Cancers. 2023; 15(14):3576. https://doi.org/10.3390/cancers15143576
Chicago/Turabian StyleButterfield, Sienna P., Rebecca E. Sizer, Emma Rand, and Robert J. White. 2023. "Selection of tRNA Genes in Human Breast Tumours Varies Substantially between Individuals" Cancers 15, no. 14: 3576. https://doi.org/10.3390/cancers15143576
APA StyleButterfield, S. P., Sizer, R. E., Rand, E., & White, R. J. (2023). Selection of tRNA Genes in Human Breast Tumours Varies Substantially between Individuals. Cancers, 15(14), 3576. https://doi.org/10.3390/cancers15143576