1. Introduction
Malignant melanoma (MM) is the most aggressive and life-threatening skin cancer with an increasing incidence worldwide. Over 90% of skin cancer deaths are due to MM, principally due to the high tendency to metastasize [
1,
2]. Many treatments have been tested to try to reduce MM early initiation of metastasis (chemotherapy, immunotherapy, radiotherapy, etc.) with modest or no efficacy. Several studies have shown that a postoperative adjuvant therapy with interferons (IFNs) might improve the recurrence-free survival (RFS) and overall survival (OS) of patients with high risk of suffering tumor recurrence [
3]. In fact, systemic application of IFNs has been the only effective therapeutic option used in the dermato-oncological routine until the advent of immune-checkpoint inhibitors and BRAF/MEK-directed targeted therapy that are still under investigation in non-metastatic stages [
4]. IFNs are a heterogeneous group of glycoproteins classified into type I (IFN-α, IFN-β), type II (IFN-γ) and type III. The IFN Receptor complex consists of two alpha chains (Type I receptor) in complex with Jak1 and Tyk2. These kinases phosphorylate Stat1 and Stat2, respectively. Finally, the Jak-STAT/IRFs pathway triggers the transcriptional modulation of more than 300 genes [
5]. This activation leads to a wide range of biological responses, including antiviral, anti-proliferative and anti-tumoral effects. In addition, the stimulation of the cytotoxic activity of a variety of cells of the immune system by IFNs, increases the expression of tumor-associated surface antigens [
6]. Despite the proven benefits of IFNs type I, questions regarding the optimal treatment regimen (dosage and duration), along with the significant side effects associated with this treatment, remain subjects of controversy and debate.
High risk MM is characterized by an extraordinary propensity for dissemination to distant organs and resistance to chemotherapy, in part due to the existence of MM cancer stem cells (CSCs) subpopulations. In fact, one of the major issues in the fight against cancer is that CSCs can survive treatments by slowly dividing, surpassing cytostatic drugs and probably, the immune system activity contributing to immune-checkpoint inhibitors and BRAF/MEK-directed targeted therapy resistances [
7,
8]. CSCs represent a small fraction of the total cell population in a solid tumor and are defined by their ability of self-renewal and to produce tumor heterogeneity [
9]. Expression of tissue type-specific cell surface markers has been used to isolate and enrich CSCs in vitro from different tumors. MM CSCs overexpress CD20, CD44, CD133 and the membrane transporter ABCB5, have high activity of the enzyme aldehyde dehydrogenase (ALDH) and have been shown, in animal models, to induce primary tumor initiation [
10]. The high metastatic potential of MM is facilitated in part by the interaction between MM-CSCs and the tumor microenvironment (TME). In fact, CSCs secrete soluble factors and extracellular vesicles (EVs), which include exosomes, enclosing specific lipids, proteins and RNAs that contribute to the pre-metastatic niche formation and, consequently, tumor spread [
10,
11]. Thus, targeting CSCs subpopulation within MM would reduce the fatal metastatic potential of the skin cancer cells.
Here, we have analyzed the effect of low and high doses of IFN-α-treatment on melanospheres enriched in CSCs subpopulations from the A375 MM established cell line and the MEL-1 primary cell line. To do so, we firstly measured by flow cytometry the ALDH activity, the side population and the specific stemness surface markers expression. Secondly, we evaluated the clonogenic ability of treated cells by soft-agar assay, the migration capacity of cells by wound-healing assay and the anti-CSCs properties in mice xenotransplant models. Moreover, we studied by miRNAseq and microarrays the effect of IFN-α on the expression of miRNAs related to stemness and the modulation of the expression of tumor related genes. Finally, we studied the effect of IFN-α on exosomes released from MM CSCs. To do so, we characterized and compared the protein content of MM CSCs released exosomes, of control CSCs versus IFN-α treated CSCs, by NanoSight, TEM, SEM Western blot and LC-HRMS-based metabolomic analysis.
Our work suggests the utility of low dose of Interferon together with the analysis of metabolic biomarkers as a potential clinical approach against the aggressiveness of CSCs in melanoma. This novel approach, together with current therapies, could improve MM treatment.
2. Materials and Methods
2.1. Cell Culture, CSC Enrichment and IFN-α Treatments
MM cancer cell lines were obtained from A375 and MEL-1 cell cultures. The line A375 was obtained from American Type Culture Collection (ATCC). Human primary MEL-1 cell line derives from a malignant metastatic melanoma (stage M1a) skin biopsy (BBSPA-Mel#1), and was provided by the Biobank of the Andalusian Public Health System (Spain). This cell line is hipotriploid (complex karyotype with multiple numerical and structural chromosome abnormalities), MelA positive, p53 positive and S100 positive, and with high tumourigenic ability. Melanoma adherent cells lines were maintained in standard culture conditions in a humid incubator at 37 °C and 5% CO2, with DMEM (Dubelcco’s Modified Eagle’s medium, Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% heat inactivated fetal bovine serum (FBS) (Gibco, Grand Island, NY, USA) and 1% Penicillin/Streptomycin (P/S) (Sigma-Aldrich) in 75 cm2 flask culture (Nunc, Roskilde, Denmark) unless otherwise indicated. FBS was inactivated by heating at 56 °C for 45 min. Cells were assayed for mycoplasma contamination.
Enriched MM CSCs subpopulation were obtained after cultivation as primary and secondary spheroids in serum free medium and under anchorage-independent conditions as described by Jiménez et al., 2018 [
10]. Briefly, for primary spheroids culture, cells were plated in serum-free spheres culture medium (DMEM:F12, 1% P/S, B27, 10 µg/mL ITS, 1 µg/mL Hydrocortisone, 4 ng/mL Heparin, 20 ng/mL EGF, 10 ng/mL FGF, 10 ng/mL IL6, 10 ng/mL HGF) in ultra-low adherence 6-well plates (Corning, Corning, NY, USA). For the secondary spheres culture, cells from primary spheroids were collected by centrifugation (1500 rpm for 10min), and, then, the pellet was resuspended in DMEM:F12 sphere medium and mechanically disrupted with a pipette and by syringing three to five times through a sterile 25-gauge needle. After that, cells were plated and incubated for 72 h in spheres culture medium in ultra-low adherence 6-well plates and treated with different IFN-α concentrations: 2000 IU/mL (low dose) and 20,000 IU/mL (high dose). The IFN-α was facilitated by Hospital Pharmacy Service at Hospital Universitario Virgen de las Nieves (Granada, Spain) under the commercial name of INTRON A
® (Interferon ALFA-2b for Injection −10 million I.U. per 1 mL × 1 vial- of MSD laboratories) [
12].
2.2. Sphere-Forming Assay
To determine the self-renewal ability of the MM CSCs population, sphere-forming assay was performed [
13]. For the secondary sphere-forming assay, 2.5 × 10
5 single cells derived from primary spheroids were plated and resuspended in spheres culture medium in ultra-low adherence 6-well plates (Corning) and treated with the same IFN-α concentration detailed above. Secondary spheres > 75 µm diameter were counted after 3 days by light microscopy. Diameters were measured using the ImageJ software.
2.3. Colony-Formation Assay
The clonogenic capability of MM CSCs were determined by colony-formation assay in soft agar as previously described [
13] with minor modifications and treated with IFN-α. Briefly, 10
4 cells coming from secondary spheroids were seeded in 0.4% cell agar base layer, which was on top of 0.8% base agar layer in 6-well culture plates. Then, cells were incubated for a further 27 days at 37 °C and 5% CO
2, adding 100 µL of DMEM (10%FBS, 1% P/S) every 1–2 days. Cell colony formation was then examined under a light microscope after staining with 0.1% Iodonitrotetrazolium Chloride (Sigma-Aldrich) [
13]. The size of colonies were measure using ImageJ™ software.
2.4. Aldefluor Assay and Phenotypic Characterization by Flow Cytometry
The analysis of CD20, CD44 and CD133 surface markers and the ALDH1 activity were performed using a Becton Dickinson FACSCanto II flow cytometer from the CIC Scientific Instrumental Centre (University of Granada) as previously described [
13]. Briefly, aldefluor assays (Stem Cell Technologies, Vancouver, BC, Canada) to detect ALDH1 activity in viable cells were performed according to manufacturer’s instructions. Cell lines were suspended in aldelfuor assay buffer containing ALDH1 substrate (BAAA, 1 μmol/L per 1 × 10
6 cells) and incubated during 45 min at 37 °C in darkness. Dethylaminobenzaldehyde (DEAB) was used as an ALDH1 inhibitor to set ALDH1 gates. The brightly fluorescent ALDH1-expressing cells were detected in the green fluorescent channel (520–540 nm).
Cell surface levels of CD20, CD44 and CD133 were determined with anti-human antibodies CD20-allopycocyanin (APC), CD44-phycoerithrin (PE) and CD133-allopycocyanin (APC) (MiltenyiBiotec, BergischGladbach, Germany), respectively. All samples were analyzed on a FACS CANTO II (BD Biosciences, San Jose, CA, USA) using the FACS DIVA software.
2.5. Side Population Assays
Hoechst 33342 exclusion (Side Population) assays were carried out as previously described to analyze cells overexpressing ABC transporters. Melanospheres were stained with Hoechst 33342 (Sigma-Aldrich) dye. As negative controls, Verapamil (Sigma-Aldrich) was used for maintaining the efflux channel closed inhibiting the capacity to efflux Hoechst 33342 by cells [
14]. The brightly fluorescent cells were measured by flow cytometry in Hoechst blue (440/40) and Hoechst red (695/40) of a FACScan Aria III (BD Biosciences) using FACS DIVA software from the CIC Scientific Instrumental Centre (University of Granada). Cells with the ability to efflux Hoechst 33342 were considered as the side population (SP) [
14].
2.6. Wound-Healing Assay
A375 and MEL-1 MM cells lines were seeded in 6-well low attachment plates and IFN-α treatment was added at 2000 and 20,000 IU/mL. Wounds were created by scraping monolayer cells (after disrupted by syringing and plated in 6-well) with 200 µL pipette tip, and non-adherent cells were washed off with medium. At 0, 24, 48 and 72 h after the creation of wounds, IFN-treated and control non-treated cells were observed and photographed with a 10× objective by light microscopy. Wound distances were measured at each time point and expressed as pixels-area
2 migration of wound closure by comparing the zero time. Image-J software was used to quantify the wound area [
15]. Cell cycle and apoptosis analysis are explained in
Supplementary Materials.
2.7. Microarray Profiling and Analysis
A375 and MEL-1 MM CSCs were treated with IFN-α for 24 h 2000 IU/mL, which will be referred to as low dose, hereafter. Non-treated cells were used as control. Total RNA was extracted using Qiagen extraction kit, according to the manufacturer’s protocol. Transcriptome microarray profiling was carried out using Clariom™ S Assay, Affymetrix Human arrays according to the Affymetrix standard protocol. Data analysis was performed using TAC 4.0 (Transcriptome Analysis Console) Thermofisher software, R and the CRAN package VennDiagram [
16]. In order to establish relationships between selected genes, String data-base was used [
17].
2.8. miRNA NGS Profiling of MM CSCs and Differential Expression Analysis
For each cell line, A375 and MEL-1, three different types of libraries were prepared: (i) untreated MM adhered (non-stem-like) cells; (ii) untreated MM CSCs; and (iii) IFN-α stimulated MM CSCs. Treated MM CSCs were exposed to a low dose of IFN-α for 24 h prior to RNA extraction. Each of the cell-line-condition combinations described were performed and profiled in duplicate (6 conditions, 12 RNA libraries). For sequencing library preparation, 1 μg total RNA was used and libraries were prepared according to the TruSeq Small RNA Sample Prep Kit (Illumina) protocol with automated pooled library size selection using Pippin Prep (Thermo Fisher Scientific). Concentration and size profile of the sequencing libraries were measured using Bioanalyzer (DNA 1000 assay) and KAPA library quantitation kit qPCR determined that the pool concentration was 11.91 nM. The pool of samples was run in one lane on a HiSeq2500 instrument (Illumina) for 50 cycles. Resulting sequencing files were processed with sRNAbench [
18] and using miRBase (release 22) as miRNA annotation [
19]. Quality control was performed using mirnaQC with all samples passing minimum quality criteria and no outliers detected [
20].
Count values were normalized using the Variance Stabilizing Transformation and differential expression analysis was performed using DESeq2 [
21]. miRNAs were considered to be differentially expressed for fold changes above 2 or below 0.5 and False Discovery Rate (FDR) corrected
p-values below 0.05. Consistently under- or overexpressed genes across cell types were systematically detected for each given comparison and included in downstream validation. Quantitative real time-PCR (qRT-PCR) is explained in
Supplementary Materials.
2.9. In Vivo Tumor Xenograft Assays
CSCs from A375 and MEL-1 MM cell lines were used for xenograft assays. After 72 h of treatment with low dose of IFN-α, 500 cells were injected in 0.05 mL matrigel and 0.05 mL of culture medium by subcutaneous injections to 8-week-old NOD scid mice gamma (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, NSG). Tumor growth was assessed twice a week using a digital caliper and the tumor volume was calculated by the formula V = length2 × width × π/6. Animal experimentation was performed according to the protocols reviewed and approved by the Institutional Animal Care and Use Committee of the University of Granada (13/08/2020/095). After 375 and 91 days, in A375 and MEL-1, respectively, animals were sacrificed and the tumors were sectioned and embedded in paraformaldehyde (PFA).
2.10. Immunofluorescence Assay
Tumors of different conditions were immersed in 4% PFA in 0.1 M PBS for 4 h at 4 °C, washed in 0.1M PBS and embedded in paraffin in an automatic tissue processor (TP1020, Leica, Germany). The paraffin blocks were cut into 4 mm sections and subjected to immunofluorescence assay. Antigen retrieval was performed at 121 °C for 15 min in a sodium citrate buffer solution (pH 6.0), and then, sections were deparaffinized with xylene and hydrated with decreasing alcohol concentrations (absolute to 70%).
The tissue sections were then incubated with rabbit anti-p75 antibody (Abcam), rabbit anti-SAMD9 antibody (Abcam), rabbit anti-CD133 antibody (Abcam) and mouse anti-CD44 (Santa Cruz Biotechnology) in phosphate-buffered saline (PBS) containing 1% bovine serum albumin (BSA) overnight at 4 °C. Next day, samples were washed thrice with PBS and incubated with the secondary antibodies (Alexa) for 1 h at RT, after washing thrice with PBS and mounted with DAPI-containing mounting medium. Negative control tissue sections were prepared by omitting the primary antibody. Observation under light microscopy and digital image acquisition were carried out with an inverted microscope (Nikon H550s). Its immunofluorescence intensity was qualified using ImageJ™ software (NIH Image, Bethesda, MD, USA).
2.11. Exosome Isolation and Purification
Exosomes were collected from culture supernatants secondary spheres generated from A375 and MEL-1 MM CSCs and from serum of MM patients by ultracentrifugation as previously described by Costa-Silva et al. with minor modifications [
22]. We set out from ~200 mL of supernatant fractions collected from cell cultures at 72 h with incubation of low dose IFN-α in each purification procedure. Supernatants were first centrifuged at 500×
g for 10 min. Next, the pellet was discarded and the remaining supernatant was ultracentrifuged at 12,000×
g for 20 min in a JS-24-38 rotor (Beckman Coulter Inc., Fullerton, CA, USA). The pellet contained microvesicles (MVs), which were resuspended in 100 µL of Dulbecco’s Phosphate Buffered Saline modified without calcium chloride and magnesium chloride and sterile filtered (modified PBS; Sigma-Aldrich, St. Louis, MO, USA), whereas the supernatant was ultracentrifuged at 100,000×
g for 70 min. The resulting pellet was washed in 35 mL PBS and pelleted again by ultracentrifugation at 100,000×
g for 70 min. Finally, exosomes were obtained in the pellet, which was resuspended into 100 µL of modified PBS and stored frozen at −80 °C for further analyses. Repeated freezing and thawing of the exosome suspensions were avoided. Transmission and Scanning Electron Microscopy Atomic Force Microscopy are explained in
Supplementary Materials.
2.12. Exosome Size Analysis
Size analyses were performed on NanoSightNS500 instruments (Malvern Instruments, UK). The instrument was equipped with a 488 nm laser, a high sensitivity CMOS camera and a syringe pump. Exosomes were diluted 1:1000 in PBS buffer to obtain a concentration range (1–10 × 10
8 particles/mL). The measurements were analyzed using the NTA2.3 software (Malvern) after capturing 3 videos of 60 s. Inmunogold Labeling by Transmission Electron Microscopy are explained in
Supplementary Materials.
2.13. Western Blot Analysis
The final pellets of cell culture supernatants of CSCs were resuspended in 100 μL of PBS and stored at 4 °C for further protein quantification. The protein concentrations were measured using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA) according to manufacturer’s instructions. Proteins extracts (30 μg) were denatured at 95 °C for 5 min in loading buffer (containing Tris—pH 6.8, SDS, glycerol, β-mercaptoethanol and bromophenol blue). Proteins were subjected to 4–20% Mini-PROTEAN TGX (Bio-Rad, Hercules, CA, USA) gel together with Precision Plus ProteinTM Kaleidoscope Prestained Protein Standards (Bio-Rad, USA). The samples were transferred to a nitrocellulose membrane (Trans-Blot, Mini Format, Bio-Rad) using a transfer apparatus according to the manufacturer’s protocols (standard program: 25 V for 30 min) (Bio-Rad).
After incubation with 5% skimmed milk in PBS-Tween 0.1% for 1 h at room temperature, the membranes were incubated overnight with antibodies against CD9 (dilution 1/1500, eBioscience), CD63 (dilution 1/500, Santa Cruz Biotechnology), p75 (dilution 1/500, Abcam), Hsp-70 and Alix (dilution 1/1000, Cell Signaling). Membranes were then incubated with conjugated goat anti-mouse secondary antibody and goat anti-rabbit secondary antibody for 2 h and signals were detected using the ECL-PLUS (Amersham Biosciences). The bands were visualized with medicals photographic films (AGFA) or detected using the Infrared Odyssey Imager (LI-COR Biotechnology, Lincoln, NE, USA).
2.14. LC-HRMS Analysis of Exosomes
The metabolomic analyses of exosomes isolated from cell culture supernatant were performed in Fundación MEDINA (Centro de Excelencia en Investigación de Medicamentos Innovadores en Andalucía) as described by García-Fontana et al., with minor modifications [
23]. Sample preparation for LC-HRMS analysis was performed as follows: Exosome samples were thawed on ice, vortexed and kept at 4 °C during the analytical process. Proteins were withdrawn using methanol (1:3), shaken, sonicated (1 min) and shaken again. Samples were then centrifuged at 13,300 rpm for 15 min at 4 °C. Supernatants were collected and dried under an N2 air stream. Dried samples were reconstituted in 90 μL of mobile phase (50% H
2O and 50% acetonitrile at 0.1% of formic acid) and transferred to the analytical vials. Then, samples were analyzed in triplicate using AB SCIEX TripleTOF 5600 quadrupole-time-of-flight mass spectrometer (Q-TOF-MS) (AB SCIEX, Concord, Canada) coupled to a high-performance liquid chromatography (HPLC) system, in positive electrospray ionization (ESI) mode.
Previous to HRMS analysis, chromatographic separation was carried out by an Agilent Series 1290 LC system (Agilent Technologies), equipped with a reverse phase Atlantis T3 HPLC C18 column (C18: 2.1 mm × 150 mm, 3 mm) (Waters). Samples were injected randomly (5 μL per sample) into the HPLC system. Blank solvent (BS) and quality control (QC) samples were also injected throughout the sequence run. The QC samples were prepared by pooling an equal volume of all exosome samples and injected every five samples in order to assess the stability and performance of the system. The BS samples were also run interspersed in the sequence to detect possible impurities of the solvents or extraction procedure and to check carryover contamination from intense analytes. Generic parameter settings for chromatographic separation and MS detection were used to obtain specific metabolomic fingerprints of the exosome preparations. HRMS analysis was performed using an information-dependent acquisition (IDA) method to collect full scan MS and MS/MS information simultaneously. The method consisted of high-resolution survey spectra from m/z 50 to m/z 1600 and the 8 most intense ions were selected for acquiring MS/MS fragmentation spectra after each scan. An Automated Calibration Delivery System performed an exact mass calibration prior to each analysis.
Data set creation: PeakView software (AB SCIEX) was used in order to evaluate the analytical drift in terms of mass and retention time shift. MarkerView software (version 1.2.1.1, AB SCIEX) was used for processing the LC-HRMS raw data. This software performs peak detection, alignment and data filtering, generating a feature table which defines measured m/z, retention time (RT) and integrated ion intensity. An automated algorithm in the RT range 0.8–19 min and m/z range 50–1600 was used for data mining. The intensity threshold of extraction was established at 50 counts per second. RT and m/z tolerances of 0.1 min and 15 ppm, respectively, were used for peak alignment. Background noise was removed by using a specific tool of MarkerView software. The analytical replicates of each sample were averaged.
Analytical validation: QC distribution on PCA plot was used for analytical validation prior to the following analysis. Variables with unacceptable reproducibility (RSD > 30%) or detected in less than 50% of QC samples were also rejected from the data matrix.
Data treatment: Statistical analyses were carried out using MetaboAnalyst 4.0 Web Server [
24] as previously described [
23]. Briefly, after dataset creation, raw data were normalized (median normalization), transformed (cube root transformation) and scaled (Pareto scaling) in order to achieve a more Gaussian type distribution [
25]. Then, filtering according to significant differences was performed based on statistical analysis including both univariate (UVA) and multivariate (MVA) in order to identify variables (metabolites) that were significantly different between the groups compared. For UVA, a first double filtering procedure with
t-test (
p-value < 0.05) and fold-change (FC > 1.5) was applied to identify differentially expressed mass signals between BS and exosome samples and therefore discard them as background noise, preserving the peaks from true biological samples.
Then, a
t-test based filtering (
p-value < 0.05) was used to detect differential metabolites between the sample groups, providing a quality criterion to evaluate variable relevance for further data analysis. For MVA, principal component analysis (PCA) and partial least squares regression (PLS-DA) were carried out after
t-test filtering. PCA was applied to assess quality of the analytical system performance. PLS-DA allowed discriminating variables that are responsible for variation between the comparison groups. For statistical validation, quality description by goodness of fit (R2) and goodness of prediction (Q2) was used. A powerful model for diagnostics should show high values of R2 and Q2 but also not vary more than 0.2–0.3. For metabolomics data, R2 > 0.7 and Q2 > 0.4 are considered acceptable values [
25]. The models were also validated using 10-fold cross validation.
2.15. Statistical Analysis
All data are presented as the mean ± standard deviation. Differences between groups were analyzed for statistical significance using the two-tailed Student’s t-test. p-value of 0.05 was accepted as the statistical significance level. The different statistical studies have already been reflected in each section of materials and methods.
4. Discussion
CSCs in MM are resistant to chemo- and radiotherapy and are often involved in the recurrence of the disease. There are currently several clinical trials being conducted to test the efficacy of new selective drugs targeting this subpopulation of tumor cells. While IFN-α has been shown to potentially improve recurrence-free survival (RFS) and overall survival (OS) in patients with high-risk melanoma, its effects on OS remain questionable according to some oncologists [
28]. Furthermore, adjuvant therapy with PD-1 or BRAF/MEK inhibitors has shown superior results and is being tested in combination with other options such as immunotherapy, further questioning the efficacy of IFN-α as a standalone treatment [
29,
30]. Despite extensive research, the exact mechanism of IFN-α action in MM remains unclear [
3].
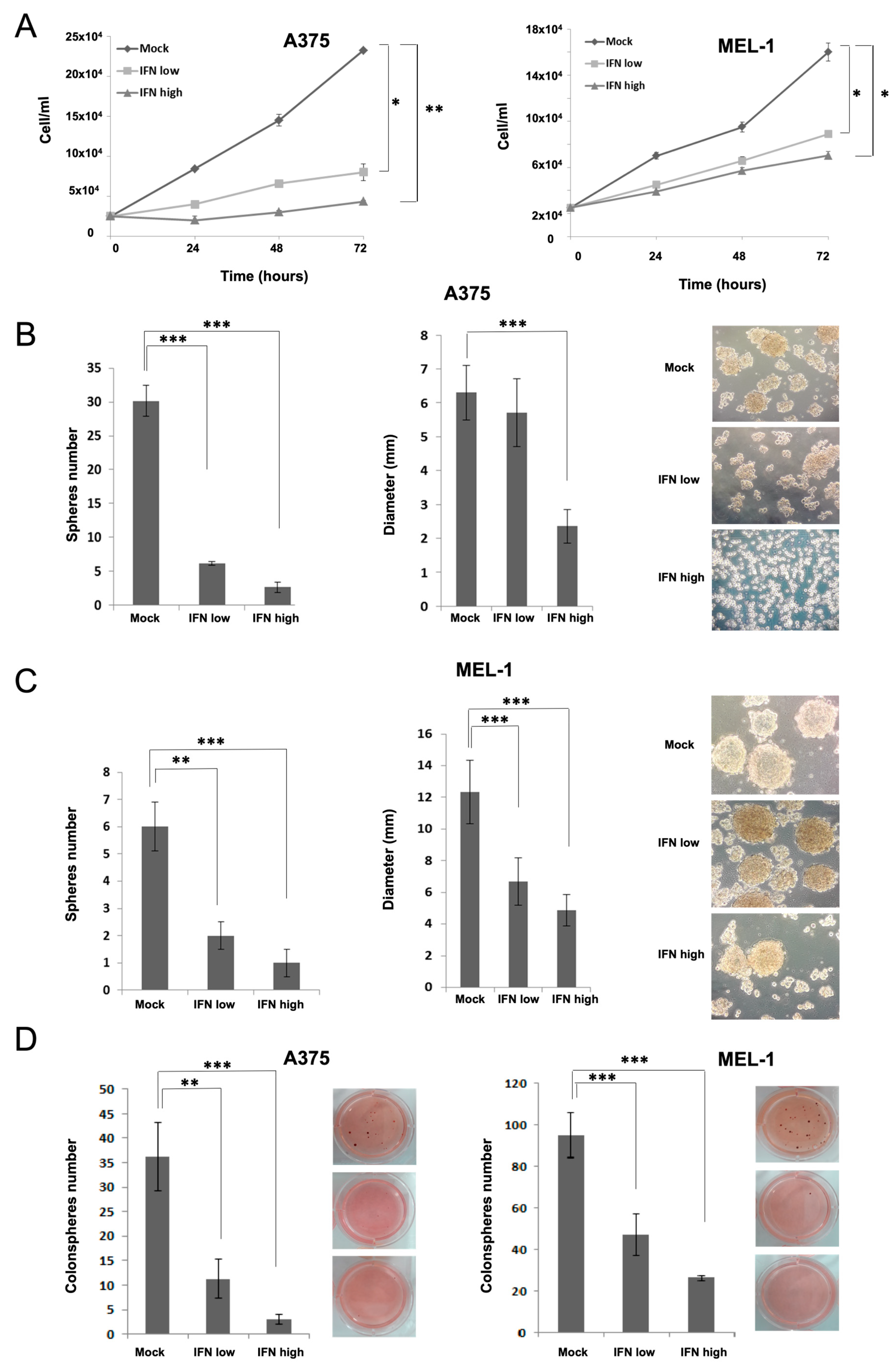
Here, we have analyzed the effects of IFN-α on aggressive CSCs established from primary MM cell lines. Our results showed inhibition in melanosphere formation and proliferation, together with a significant reduction in ALDH-positive population and in the expression of CD20, CD44 and CD133 stemness markers after IFN treatment, even at low doses (
Figure 1). One of the first pieces of evidence of IFN-α effect on CSCs was shown in a rat model of ovarian cancer primary tumors, where the authors demonstrated a significant reduction of the aggressive SP after treatment [
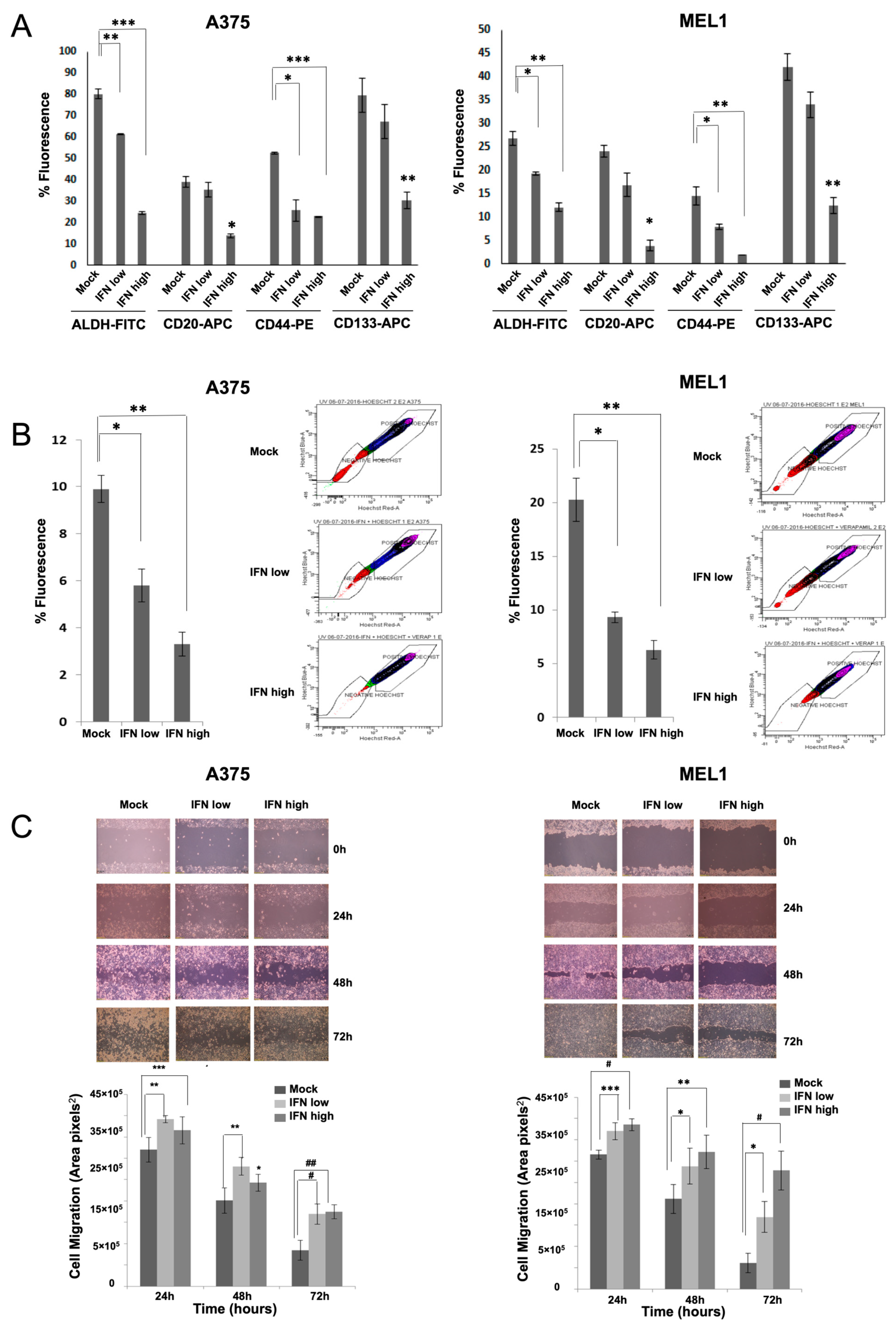
31]. Here, we have also shown such an effect for melanoma, along with the ability to inhibit cell migration, which is another important feature related to stemness (
Figure 2). In agreement with our results, the anti-proliferative effects of IFN-α on triple-negative breast CSCs have also been described, associated with the presence of interferon-regulated signature genes, and improved therapeutic response and overall survival of treated patients [
32]. Other studies have shown the anti-proliferative and anti-angiogenic effects of IFN-α on CSCs in hepatocellular carcinoma, and the benefit of combining IFN with other conventional therapies in different models [
33].
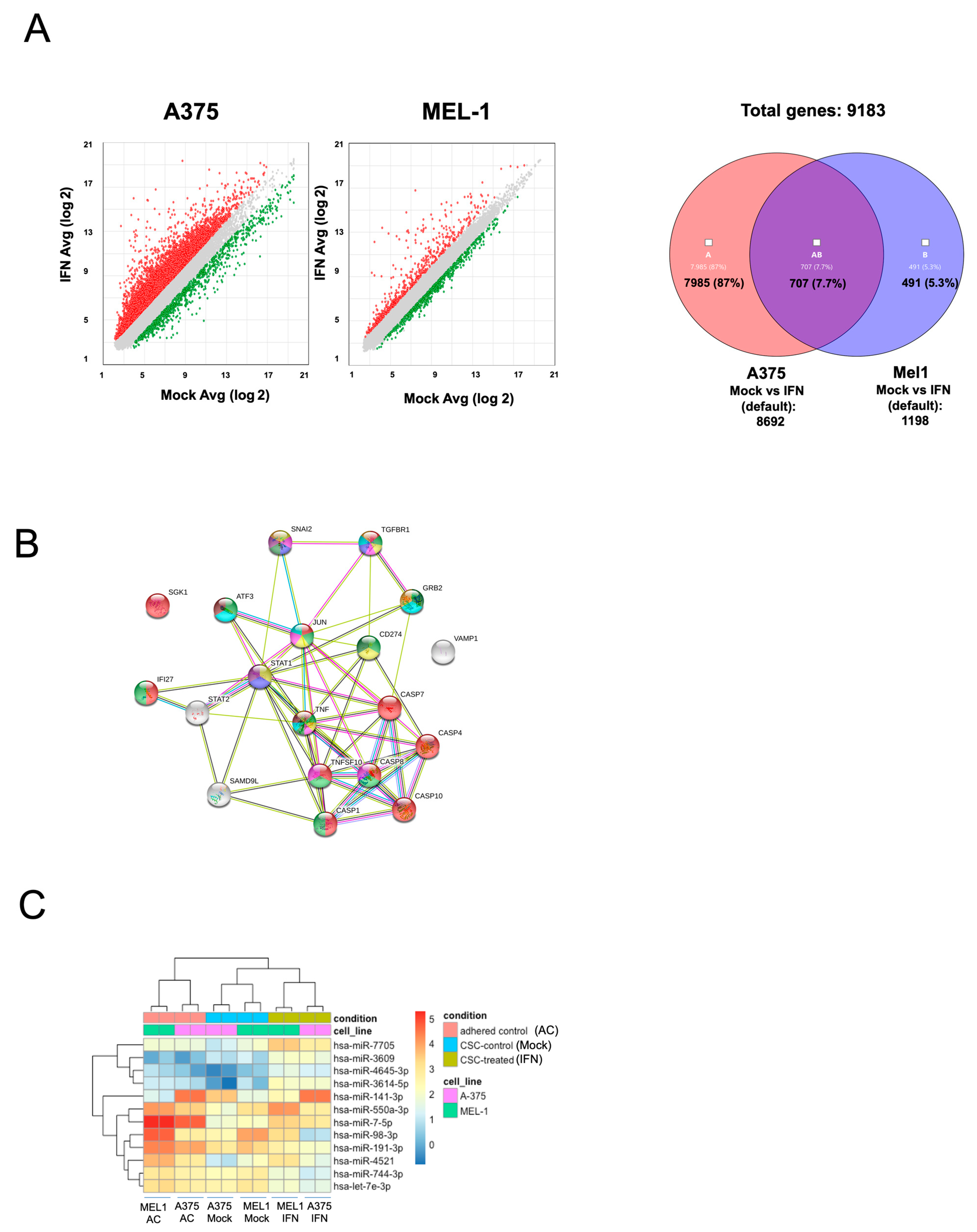
The study conducted gene expression profiling and miRNA sequencing analysis in melanoma CSCs after treatment with low-dose IFN-α, revealing changes in the expression of genes and miRNAs related to stemness as shown in
Figure 3 and
Supplementary Tables S1 and S2. Up-regulated miRNAs included miR7-5p, miR141-3p, miR425, miR550a, miR3614-5p, miR4521 and miR4645, which have been shown to act on several cancer pathways as tumor suppressors. For example, miR7-5p enhances temozolomide sensitivity of drug-resistant glioblastoma cells [
34], miR-425 inhibits melanoma metastasis [
35] miR-3614-5p is a potential biomarker for colorectal cancer [
36] and miR-4521 plays a tumor-repressive role in growth and metastasis of hepatocarcinoma cells [
37]. The miRNAs miR98-5p, miR-191, miR-744-3p and let-7e-3p, known for their involvement in oncogenic mechanisms [
38,
39,
40], were found to be down-regulated after IFN-α treatment.
Microarray analysis revealed that IFN-α treatment led to changes in the expression of genes associated with cancer processes, including migration, apoptosis, vesicle regulation, angiogenesis, and CSC signaling pathways. These findings highlight the potential of validating gene and miRNA signatures after IFN-α treatment to identify new predictive and prognostic biomarkers for MM patients for future studies.
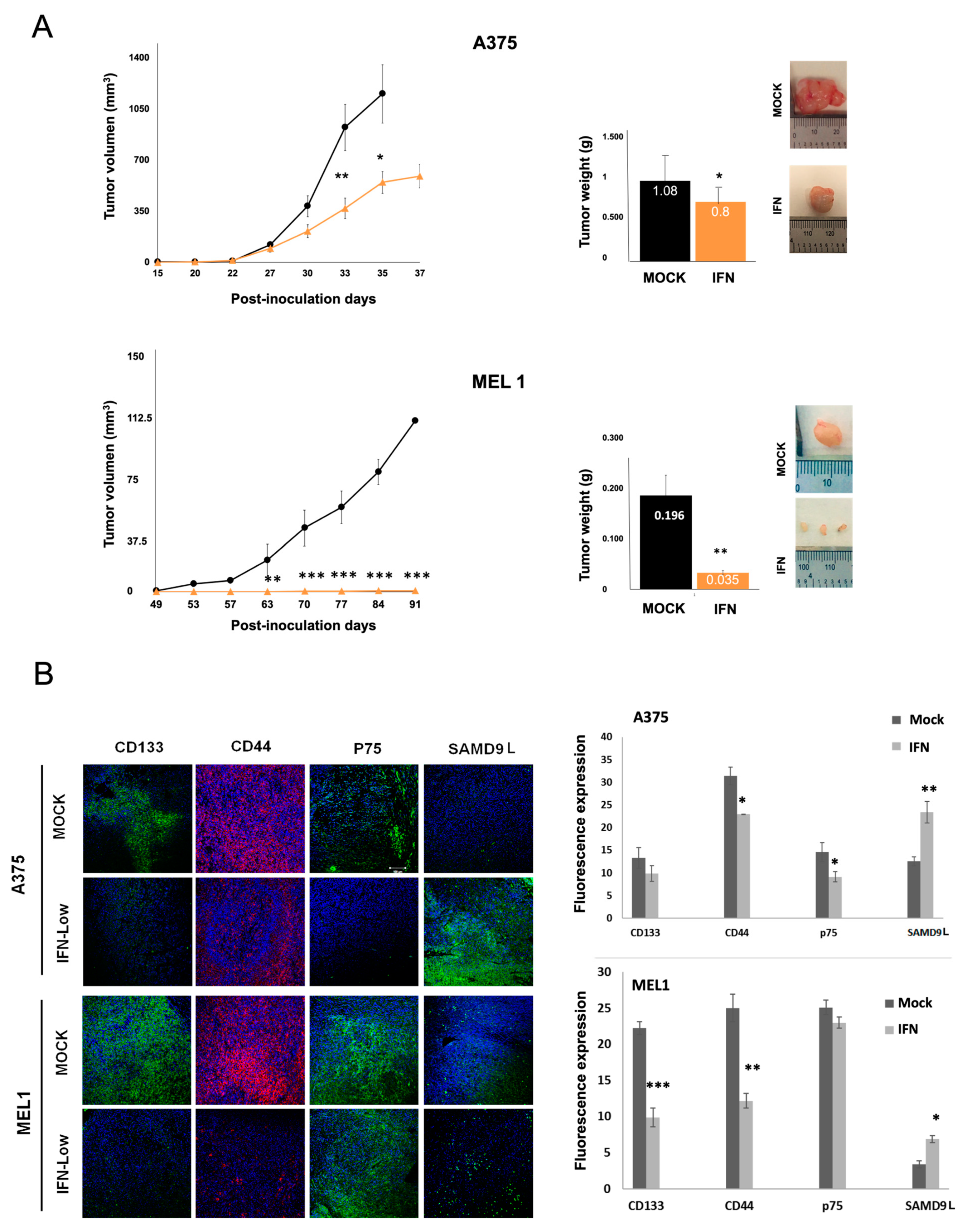
Moreover, we investigated the effect of IFN-α treatment on the in vivo tumourigenic capacity of melanoma CSCs (
Figure 4). The results showed that IFN-α treatment at low doses significantly decreased the ability of CSCs to form tumors. The immunoanalysis of tumors revealed a significant increase in the expression of SAMD9L according to data obtained from previous microarrays analysis. SAMD9L is a protein with antiproliferative function and a tumor suppressor role in various types of cancer, recently identified in genetic signatures of pancreas and melanoma tumors [
41]. While it regulates cell proliferation in hematopoietic tissue by facilitating the degradation of cytokine receptors, its role in stemness processes and cancer needs further analysis. These findings suggest that IFN-α treatment may be a potential therapeutic strategy for melanoma, and further research on the role of SAMD9L in melanoma and other cancers is warranted.
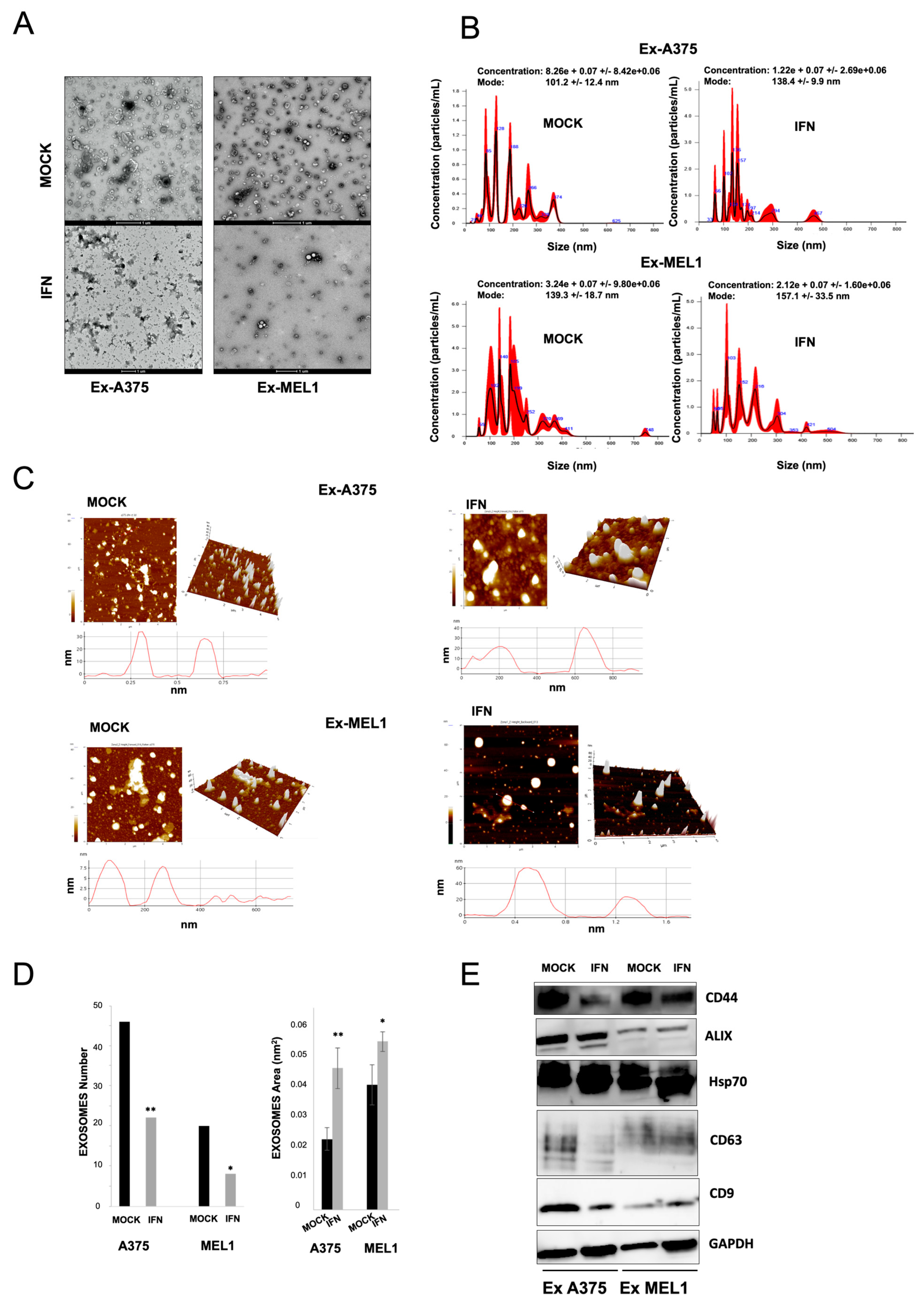
Extracellular vesicles (EVs) have been shown to play a role in cancer progression and the metastatic process by altering the microenvironment of distant sites, promoting angiogenesis and tumor cell migration [
42]. EVs can be isolated by non-invasive liquid biopsy and used as a source of prognostic and predictive biomarkers. In this study, we found that IFN-α treatment decreased EV secretion via exosomes in melanoma CSC subpopulations. Exosomes from melanoma CSCs express the CD44 CSC-related marker, and this expression was reduced after treatment (
Figure 5). The microarrays analysis we conducted also revealed a modulation in mRNA expression of several members of the VAMP family after IFN-α treatment, suggesting their potential involvement in exosome dysregulation since VAMP1 and VAMP8 are essential components of the exocytic machinery and regulate various secretory processes in different systems [
43]. Few recent studies have demonstrated the effect of IFN-gamma on vesicular trafficking from neural CSCs by inducing the generation of altered exosomes [
44]. Further research is necessary to identify the underlying mechanisms responsible for the modulation of CSC-derived exosomes and their potential relationship with the efficacy of IFN-α treatment.
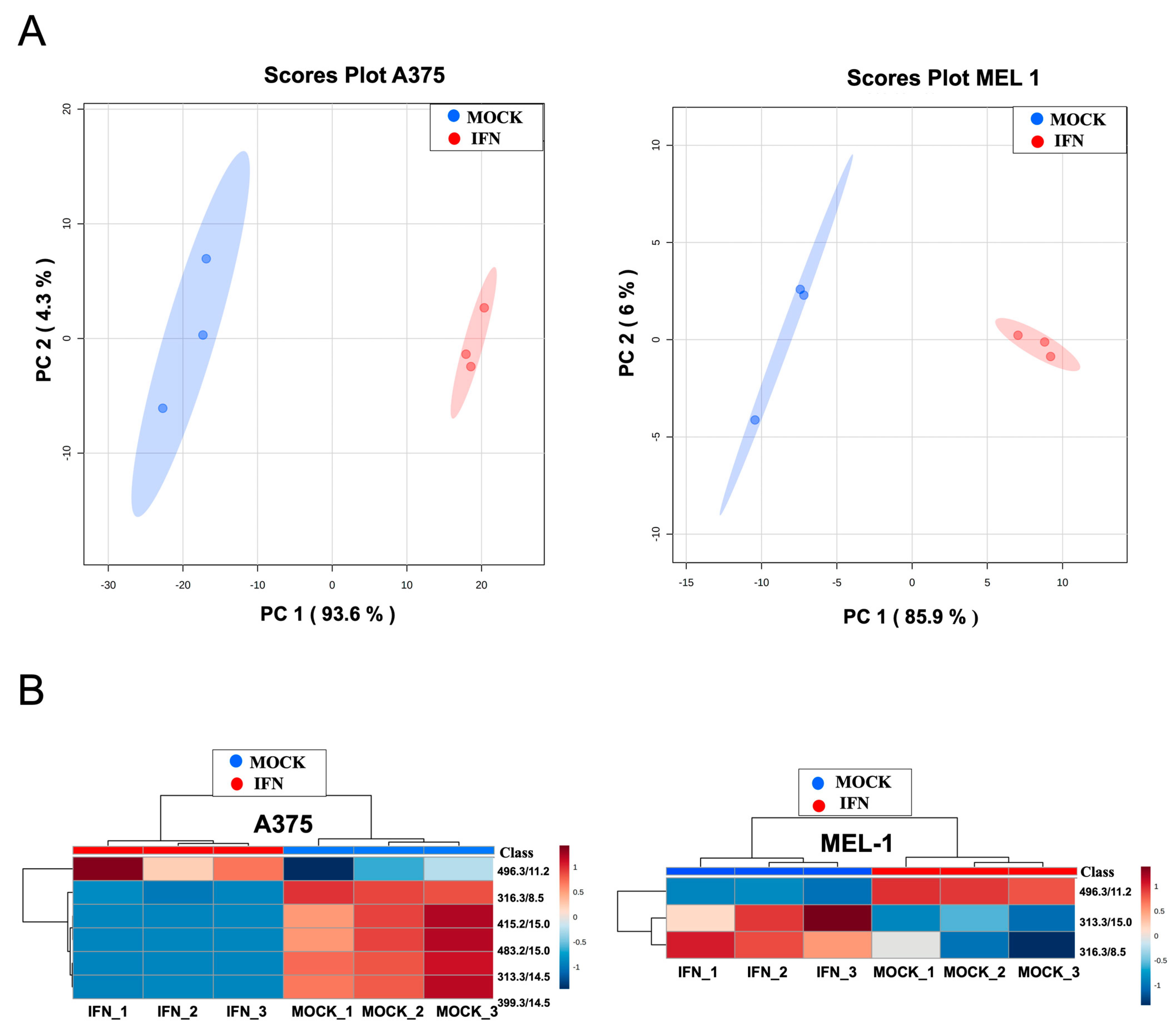
Our previous study, focused on searching potential biomarkers for the diagnosis of melanoma, reported significant metabolomic differences in exosomes derived from MM CSCs compared to those from differentiated tumor cells in A375 and MEL-1 cell line [
27]. We show in this present work the differential abundance of those metabolites in exosomes from IFN-treated CSC-A375 and CSC-MEL1 (
Figure 6). Interestingly, the metabolite with
m/
z 496.3381, which corresponds to 1-hexadecanoyl-sn-glycero-3-phosphocholine (PC 16:0/0:0), was previously found by Palacios-Ferrer and cols. [
27] to be expressed in exosomes derived from both healthy control serum and adherent MEL-1 cells, compared to MM patients’ serum and CSC-enriched subpopulations from MEL-1 cells, respectively. Curiously, PC 16:0/0:0 was higher in IFN-α -treated samples in comparison to controls. The over-expression of this metabolite can be in accordance with previous studies demonstrating its relationship with lower risks of breast, prostate and colorectal cancer [
45]. In fact, it has been suggested that the rapid extracellular hydrolysis of phospholipids like PC 16:0/0:0 by metastatic tumor cells and the subsequent cellular uptake of the resulting free fatty acids (FFA) seems to be a necessary prerequisite for metastatic potential of epithelial tumor cells, probably for generating pro-metastatic lipid second messengers [
27,
46].




 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





