
Cancer Associated Macrophage-like Cells Are Prognostic for Highly Aggressive Prostate Cancer in Both the Non-Metastatic and Metastatic Settings

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. CAML Isolation and Enumeration
2.3. Statistical Analyses
3. Results
3.1. Patient Demographics
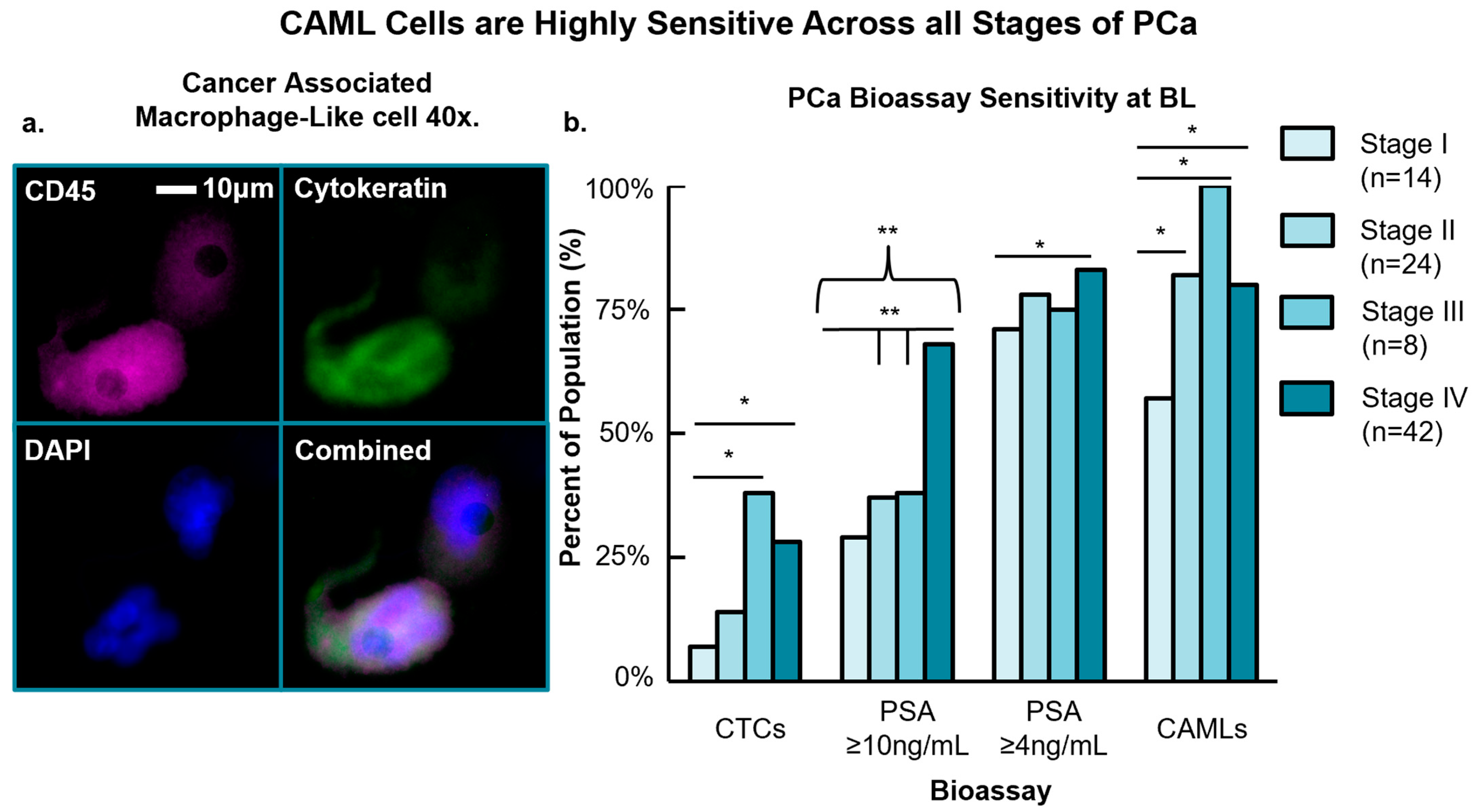
3.2. CAML Cell Presence versus Conventional PCa Bioassays
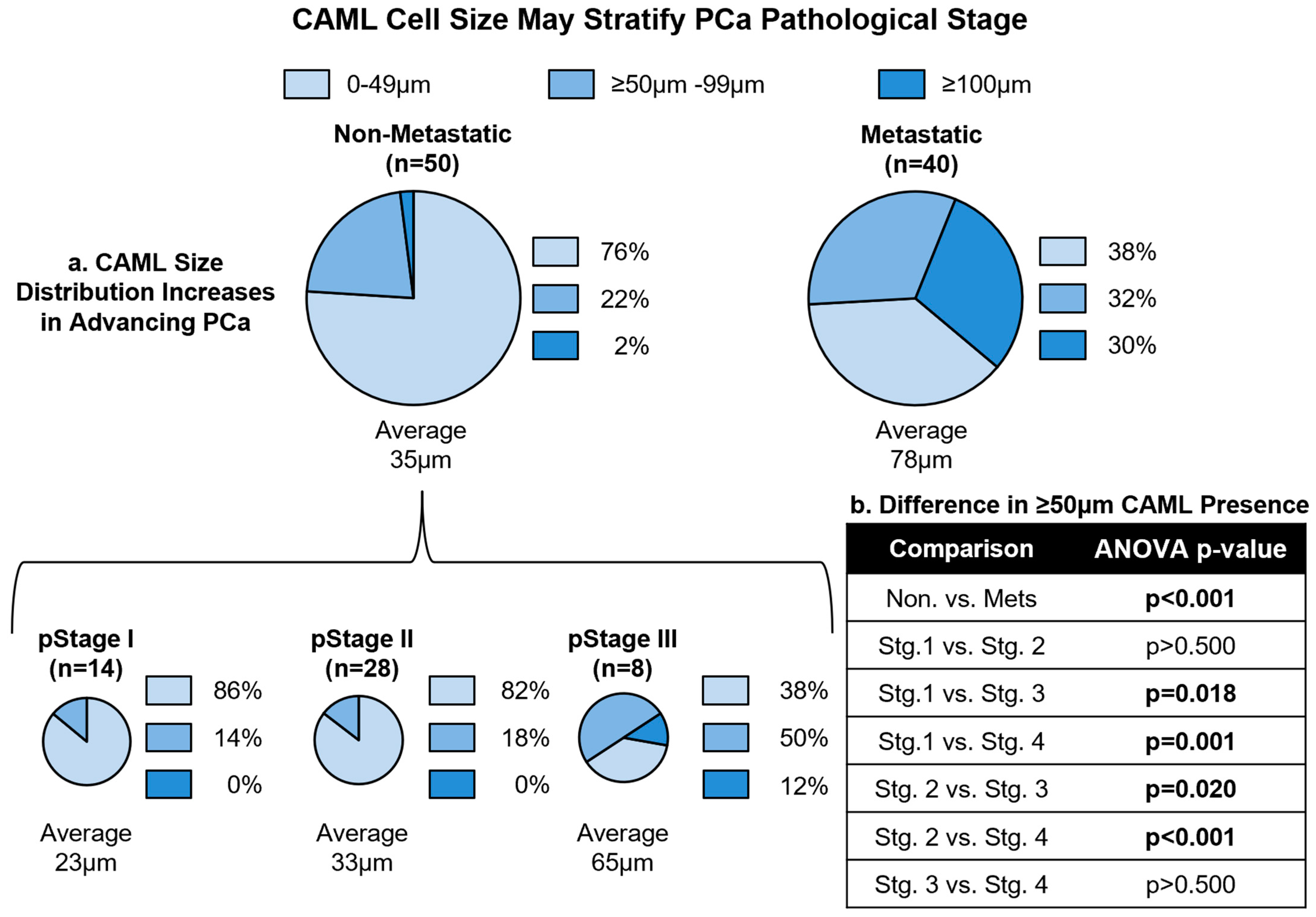
3.3. CAML Size Differentiates Local and Advanced Disease
3.4. Engorged CAMLs Found Prior to Treatment Predict for Early Mortality
3.5. CAML Size Tracking throughout Treatment
3.6. Multivariate Analysis
3.7. Analysis of PSA for Predicting PFS and OS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2016. Natl. Cancer Inst. 2019. Available online: https://seer.cancer.gov/csr/1975_2016/ (accessed on 10 September 2021).
- Pollock, P.A.; Ludgate, A.; Wassersug, R.J. In 2124, half of all men can count on developing prostate cancer. Curr. Oncol. 2015, 22, 10–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culig, Z. Distinguishing indolent from aggressive prostate cancer. In Prostate Cancer Prevention. Recent Results in Cancer Research; Springer: Berlin/Heidelberg, Germany, 2014; Volume 202, pp. 141–147. [Google Scholar] [CrossRef]
- Oto, J.; Fernández-Pardo, Á.; Royo, M.; Hervás, D.; Martos, L.; Vera-Donoso, C.D.; Martínez, M.; Heeb, M.J.; España, F.; Medina, P.; et al. A predictive model for prostate cancer incorporating PSA molecular forms and age. Sci. Rep. 2020, 10, 2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahn, J.L.; Giovannucci, E.L.; Stampfer, M.J. The high prevalence of undiagnosed prostate cancer at autopsy: Implications for epidemiology and treatment of prostate cancer in the Prostate-specific Antigen-era. Int. J. Cancer 2015, 137, 2795–2802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, E.D.; Abrahamsson, P.A. PSA-based screening for prostate cancer: How does it compare with other cancer screening tests? Eur. Urol. 2008, 54, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Barry, M.J. Screening for prostate cancer—The controversy that refuses to die. N. Engl. J. Med. 2009, 360, 1351–1354. [Google Scholar] [CrossRef]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [Green Version]
- Finne, P.; Finne, R.; Auvinen, A.; Juusela, H.; Aro, J.; Määttänen, L.; Hakama, M.; Rannikko, S.; Tammela, T.L.; Stenman, U. Predicting the outcome of prostate biopsy in screen-positive men by a multilayer perceptron network. Urology 2000, 56, 418–422. [Google Scholar] [CrossRef]
- Ford, M.E.; Havstad, S.L.; Demers, R.; Johnson, C.C. Effects of false-positive prostate cancer screening results on subsequent prostate cancer screening behavior. Cancer Epidemiol. Biomark. Prev. 2005, 14, 190–194. [Google Scholar] [CrossRef]
- White, J.; Shenoy, B.V.; Tutrone, R.F.; Karsh, L.I.; Saltzstein, D.R.; Harmon, W.J.; Broyles, D.L.; Roddy, T.E.; Lofaro, L.R.; Paoli, C.J.; et al. Clinical utility of the Prostate Health Index (phi) for biopsy decision management in a large group urology practice setting. Prostate Cancer Prostatic Dis. 2018, 21, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.A.; Andrews, K.S.; Brooks, D.; Fedewa, S.A.; Manassaram-Baptiste, D.; Saslow, D.; Brawley, O.W.; Wender, R.C. Cancer screening in the United States, 2018: A review of current American Cancer Society guidelines and current issues. CA Cancer J. Clin. 2018, 68, 297–316. [Google Scholar] [CrossRef]
- Tutrone, R.; Donovan, M.J.; Torkler, P.; Tadigotla, V.; McLain, T.; Noerholm, M.; Skog, J.; McKiernan, J. Clinical utility of the exosome based ExoDx Prostate (IntelliScore) EPI test in men presenting for initial Biopsy with a PSA 2–10 ng/mL. Prostate Cancer Prostatic Dis. 2020, 23, 607–614. [Google Scholar] [CrossRef]
- Roobol, M.J.; Carlsson, S.V. Risk stratification in prostate cancer screening. Nat. Rev. Urol. 2013, 10, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Schedimann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steyerberg, E.W.; Roobol, M.J.; Kattan, M.W.; van der Kwast, T.H.; de Koning, H.J.; Shröder, F.H. Prediction of indolent prostate cancer: Validation and updating of a prognostic nomogram. J. Urol. 2007, 177, 107–112. [Google Scholar] [CrossRef]
- Schaeffer, E.; Srinivas, S.; Antonarakis, E.S.; Armstrong, A.J.; Bekelman, J.E.; Cheng, H.; D’Amico, A.V.; Davis, B.J.; Desai, N.; Dorff, T.; et al. NCCN Guidelines Insights: Prostate Cancer, Version 1.2021. J. Natl. Compr. Cancer Netw. 2021, 19, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zhu, Y.; Dai, B.; Ye, D.W. National Comprehensive Cancer Network (NCCN) risk classification in predicting biochemical recurrence after radical prostatectomy: A retrospective cohort study in Chinese prostate cancer patients. Asian J. Androl. 2018, 20, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Gejerman, G.; Ciccone, P.; Goldstein, M.; Lanteri, V.; Schlecker, B.; Sanzone, J.; Esposito, M.; Rome, S.; Ciccone, M.; Margolis, E.; et al. US Preventive Services Task Force prostate-specific antigen screening guidelines result in higher Gleason score diagnoses. Investig. Clin. Urol. 2017, 58, 423–428. [Google Scholar] [CrossRef]
- Bratulic, S.; Gatto, F.; Nielsen, J. The Translational Status of Cancer Liquid Biopsies. Regen. Eng. Transl. Med. 2021, 7, 312–352. [Google Scholar] [CrossRef] [Green Version]
- Panebianco, V.; Pecoraro, M.; Fiscon, G.; Paci, P.; Farina, L.; Catalano, C. Prostate cancer screening research can benefit from network medicine: An emerging awareness. npj Syst. Biol. Appl. 2020, 6, 13. [Google Scholar] [CrossRef]
- Scher, H.I.; Jia, X.; de Bono, J.S.; Fleisher, M.; Pienta, K.J.; Raghavan, D.; Heller, G. Circulating tumour cells as prognostic markers in progressive, castration-resistant prostate cancer: A reanalysis of IMMC38 trial data. Lancet Oncol. 2009, 10, 233–239. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, J.C.; Fink, L.M.; Goodman, O.B., Jr.; Symanowski, J.T.; Vogelzang, N.J.; Ward, D.C. Comparison of circulating MicroRNA 141 to circulating tumor cells, lactate dehydrogenase, and prostate-specific antigen for determining treatment response in patients with metastatic prostate cancer. Clin. Genitourin. Cancer 2011, 9, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Zviran, A.; Schulman, R.C.; Shah, M.; Hill, S.T.K.; Deochand, S.; Khamnei, C.C.; Maloney, D.; Patel, K.; Liao, W.; Widman, A.J.; et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nat. Med. 2020, 26, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.L.; Cristofanilli, M. Chapter 5: Detecting and Monitoring Circulating Stromal Cells from Solid Tumors Using Blood-Based Biopsies in the Twenty-First Century: Have Circulating Stromal Cells Come of Age. In Liquid Biopsies in Solid Tumors; Humana Press: Totowa, NJ, USA, 2017; pp. 81–104. ISBN 331950956X/9783319509563. [Google Scholar]
- Danila, D.C.; Heller, G.; Gignac, G.A.; Gonzalez-Espinoza, R.; Anand, A.; Tanaka, E.; Lilja, H.; Schwartz, L.; Larson, S.; Fleisher, M.; et al. Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin. Cancer Res. 2007, 13, 7053–7058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broncy, L.; Paterlini-Bréchot, P. Clinical impact of circulating tumor cells in patients with localized prostate cancer. Cells 2019, 8, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Heliebi, A.; Hille, C.; Laxman, N.; Svedlund, J.; Haudum, C.; Ercan, E.; Kroneis, T.; Chen, S.; Smolle, M.; Rossmann, C.; et al. In Situ Detection and Quantification of AR-V7, AR-FL, PSA, and KRAS Point Mutations in Circulating Tumor Cells. Clin. Chem. 2018, 64, 536–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.J.; Luo, J.; Nanus, D.M.; Giannakakou, P.; Szmulewitz, R.Z.; Danila, D.C.; Healy, P.; Anand, M.; Berry, W.R.; Zhang, T.; et al. Prospective Multicenter Study of Circulating Tumor Cell AR-V7 and Taxane versus Hormonal Treatment Outcomes in Metastatic Castration-Resistant Prostate Cancer. JCO Precis. Oncol. 2020, 4, 1120–1129. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [Green Version]
- Bastos, D.A.; Antonarakis, E.S. CTC-derived AR-V7 detection as a prognostic and predictive biomarker in advanced prostate cancer. Expert Rev. Mol. Diagn. 2018, 18, 155–163. [Google Scholar] [CrossRef]
- Li, H.; Wang, Z.; Xiao, W.; Yan, L.; Guan, W.; Hu, Z.; Wu, L.; Huang, Q.; Wang, J.; Xu, H.; et al. Androgen-receptor splice variant-7-positive prostate cancer: A novel molecular subtype with markedly worse androgen-deprivation therapy outcomes in newly diagnosed patients. Mod. Pathol. 2018, 31, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.L.; Adams, D.K.; Alpaugh, R.K.; Cristofanilli, M.; Martin, S.S.; Chumsri, S.; Tang, C.M.; Marks, J.R. Circulating Cancer-Associated Macrophage-like Cells Differentiate Malignant Breast Cancer and Benign Breast Conditions. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1037–1042. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.L.; Adams, D.K.; He, J.; Kalhor, N.; Zhang, M.; Xu, T.; Gao, H.; Reuben, J.M.; Qiao, Y.; Komaki, R.; et al. Sequential Tracking of PD-L1 Expression and RAD50 Induction in Circulating Tumor and Stromal Cells of Lung Cancer Patients Undergoing Radiotherapy. Clin. Cancer Res. 2017, 23, 5948–5958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, D.L.; Martin, S.S.; Alpaugh, K.R.; Charpentier, M.; Tsai, S.; Bergan, R.C.; Ogden, I.M.; Catalona, W.; Chumsri, S.; Tang, C.M.; et al. Circulating giant macrophages as a potential biomarker of solid tumors. Proc. Natl. Acad. Sci. USA 2014, 111, 3514–3519. [Google Scholar] [CrossRef] [PubMed]
- Gardner, K.P.; Aldakkak, M.; Tang, C.M.; Tsai, S.; Adams, D.L. Circulating stromal cells in resectable pancreatic cancer correlates to pathological stage and predicts for poor clinical outcomes. npj Precis. Oncol. 2021, 5, 25. [Google Scholar] [CrossRef]
- Gironda, D.J.; Adams, D.L.; He, J.; Xu, T.; Gao, H.; Qiao, Y.; Komaki, R.; Reuben, J.M.; Liao, Z.; Blum-Murphy, M.; et al. Cancer associated macrophage-like cells and prognosis of esophageal cancer after chemoradiation therapy. J. Transl. Med. 2020, 18, 413. [Google Scholar] [CrossRef]
- Gardner, K.P.; Tsai, S.; Aldakkak, M.; Gironda, S.; Adams, D.L. CXCR4 expression in tumor associated cells in blood is prognostic for progression and survival in pancreatic cancer. PLoS ONE 2022, 17, e0264763. [Google Scholar] [CrossRef]
- Raghavakaimal, A.; Cristofanilli, M.; Tang, C.M.; Alpaugh, R.K.; Gardner, K.P.; Chumsri, S.; Adams, D.L. CCR5 activation and endocytosis in circulating tumor-derived cells isolated from the blood of breast cancer patients provide information about clinical outcome. Breast Cancer Res. 2022, 24, 35. [Google Scholar] [CrossRef]
- Tang, C.M.; Adams, D.L. Clinical Applications of Cancer-Associated Cells Present in the Blood of Cancer Patients. Biomedicines 2022, 10, 587. [Google Scholar] [CrossRef]
- Augustyn, A.; Adams, D.L.; He, J.; Qiao, Y.; Verma, V.; Liao, Z.; Tang, C.M.; Heymach, J.V.; Tsao, A.S.; Lin, S.H. Giant Circulating Cancer-Associated Macrophage-like Cells Are Associated with Disease Recurrence and Survival in Non-Small-Cell Lung Cancer Treated with Chemoradiation and Atezolizumab. Clin. Lung Cancer 2020, 22, e451–e465. [Google Scholar] [CrossRef]
- Tang, C.M.; Zhu, P.; Li, S.; Makarova, O.V.; Amstutz, P.T.; Adams, D.L. Blood-based biopsies-clinical utility beyond circulating tumor cells. Cytom. Part A 2018, 93, 1246–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manjunath, Y.; Mitchem, J.B.; Suvilesh, K.N.; Avella, D.M.; Kimchi, E.T.; Steveley-O’Carroll, K.F.; Deroche, C.B.; Pantel, K.; Li, G.; Kaifi, J.T. Circulating giant tumor-macrophage fusion cells are independent prognosticators in patients with NSCLC. J. Thorac. Oncol. 2020, 15, 1460–1471. [Google Scholar] [CrossRef]
- Carreta Ruano, A.P.; Guimarães, A.P.G.; Braun, A.C.; Flores, B.C.T.C.P.; Tariki, M.S.; Abdallah, E.A.; Torres, J.A.; Nunes, D.N.; Tirapelli, B.; de Lima, V.C.C.; et al. Fusion cell markers in circulating tumor cells from patients with high-grade ovarian serous carcinoma. Int. J. Mol. Sci. 2022, 23, 14687. [Google Scholar] [CrossRef]
- Sulaiman, R.; De, P.; Aske, J.C.; Lin, X.; Dale, A.; Vaselaar, E.; Ageton, C.; Gaster, K.; Espaillat, L.R.; Starks, D.; et al. Identification and morphological characterization of features of circulating cancer-associated macrophage-like cells (CAMLs) in endometrial cancers. Cancers 2022, 14, 4577. [Google Scholar] [CrossRef] [PubMed]
- Lopresti, A.; Acquaviva, C.; Boudin, L.; Finetti, P.; Garnier, S.; Aulas, A.; Liberatoscioli, M.L.; Cabaud, O.; Guille, A.; de Nonneville, A.; et al. Identification of atypical circulating tumor cells with prognostic value in metastatic breast cancer patients. Cancers 2022, 14, 932. [Google Scholar] [CrossRef] [PubMed]
- Cooperberg, M.R.; Brooks, J.D.; Faino, A.V.; Newcomb, L.F.; Kearns, J.T.; Carroll, P.R.; Dash, A.; Etzioni, R.; Fabrizio, M.D.; Gleave, M.E.; et al. Refined analysis of prostate-specific antigen kinetics to predict prostate cancer active surveillance outcomes. Eur. Urol. 2018, 74, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Gironda, D.J.; Bergan, R.C.; Lin, S.H.; Alpaugh, R.K.; Cristofanilli, M.; Chumsri, S.; Lapidus, R.G.; Williams, W.; Lacher, M.; Danila, D.C.; et al. Hyper engorged cancer associated macrophage-like cells in circulation predict for multi-organ metastatic disease in solid tumors. J. Clin. Oncol. 2021, 39, 3039. [Google Scholar] [CrossRef]
- Adams, D.L.; Stefansson, S.; Haudenschild, C.; Martin, S.S.; Charpentier, M.; Chumsri, S.; Cristofanilli, M.; Tang, C.M.; Alpaugh, R.K. Cytometric characterization of circulating tumor cells captured by microfiltration and their correlation to the CellSearch((R)) CTC test. Cytom. Part A 2015, 87, 137–144. [Google Scholar] [CrossRef] [Green Version]
- Adams, D.L.; Alpaugh, R.K.; Tsai, S.; Tang, C.M.; Stefansson, S. Multi-Phenotypic subtyping of circulating tumor cells using sequential fluorescent quenching and restaining. Sci. Rep. 2016, 6, 33488. [Google Scholar] [CrossRef] [Green Version]
- Santok, G.D.; Raheem, A.A.; Kim, L.H.C.; Chang, K.; Lum, T.G.H.; Chung, B.H.; Choi, Y.D.; Rha, K.H. Prostate-specific antigen 10-20 ng/mL: A predictor of degree of upgrading to >/=8 among patients with biopsy Gleason score 6. Investig. Clin. Urol. 2017, 58, 90–97. [Google Scholar] [CrossRef]
- Wiebe, E.; Rodrigues, G.; Lock, M.; D’Souza, D.; Stitt, L. Outcome analysis of prostate cancer patients with pre-treatment PSA greater than 50 ng/mL. Can. J. Urol. 2008, 15, 4078–4083. [Google Scholar]
- Collette, L.; Burzykowski, T.; Carroll, K.J.; Newling, D.; Morris, T.; Shröder, F.H.; European Organisation for Research and Treatment of Cancer; Limburgs Universitair Centrum; AstraZeneca Pharmaceuticals. Is prostate-specific antigen a valid surrogate end point for survival in hormonally treated patients with metastatic prostate cancer? Joint research of the European Organisation for Research and Treatment of Cancer, the Limburgs Universitair Centrum, and AstraZeneca Pharmaceuticals. J. Clin. Oncol. 2005, 23, 6139–6148. [Google Scholar] [CrossRef]
- Wilbur, J. Prostate cancer screening: The continuing controversy. Am. Fam. Physician 2008, 78, 1377–1384. [Google Scholar] [PubMed]
- Djulbegovic, M.; Beyth, R.J.; Neuberger, M.M.; Stoffs, T.L.; Vieweg, J.; Djulbegovic, B.; Dahm, P. Screening for prostate cancer: Systematic review and meta-analysis of randomised controlled trials. BMJ 2010, 341, c4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moyer, V.A.; U.S. Preventative Services Task Force. Screening for prostate cancer: U.S. Preventive Services Task Force recommendation statement. Ann. Intern. Med. 2012, 157, 120–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zapatero, A.; Gómez-Caamaño, A.; Rodriguez, M.A.C.; Muinelo-Romay, L.; de Vidales, C.M.; Abalo, A.; Crespo, P.C.; Mateos, L.L.; Olivier, C.; Piris, L.V.V. Detection and dynamics of circulating tumor cells in patients with high-risk prostate cancer treated with radiotherapy and hormones: A prospective phase II study. Radiat. Oncol. 2020, 15, 137. [Google Scholar] [CrossRef]
- Adams, D.L.; Lin, S.H.; Raghavakaimal, A.; Weiss, G.; Ford, A.; Brown, C.; Yeh, C.H. Combining circulating stromal cells with cell free DNA for increased sensitivity in profiling oncogenic mutations and indicates highly aggressive non small cell lung cancer. Cancer Res. 2019, 79 (Suppl. S13), 437. [Google Scholar] [CrossRef]
- Kim, M.-Y.; Oskarsson, T.; Acharyya, S.; Nguyen, D.X.; Zhang, X.H.F.; Norton, L.; Massagué, J. Tumor self-seeding by circulating cancer cells. Cell 2009, 139, 1315–1326. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Demographic | Non-Metastatic (n = 50) | Metastatic (n = 42) | Combined (n = 92) |
|---|---|---|---|
| Age (Years): Median (Range) | 66 [50–81] | 73 [48–89] | 69 [48–89] |
| Race | |||
| Caucasian | 43 (86%) | 37 (89%) | 80 (87%) |
| African American | 1 (2%) | 2 (5%) | 3 (3%) |
| Hispanic | 1 (2%) | 1 (2%) | 2 (2%) |
| Asian | 1 (2%) | 1 (2%) | 2 (2%) |
| Unknown | 4 (8%) | 1 (2%) | 5 (6%) |
| Pathological Stage | |||
| I | 14 (28%) | 0 (0%) | 14 (15%) |
| II | 28 (56%) | 0 (0%) | 28 (30%) |
| III | 8 (16%) | 0 (0%) | 8 (9%) |
| IV | 0 (0%) | 42 (100%) | 42 (46%) |
| Histology | |||
| Adenocarcinoma | 46 (92%) | 39 (93%) | 85 (92%) |
| Neuroendocrine | 0 (0%) | 1 (2%) | 1 (1%) |
| Unknown | 4 (8%) | 2 (5%) | 6 (7%) |
| Gleason Score | |||
| 6 | 2 (4%) | 2 (5%) | 4 (4%) |
| 7 | 22 (44%) | 14 (33%) | 36 (39%) |
| 8 | 14 (28%) | 7 (17%) | 21 (23%) |
| 9 | 12 (24%) | 15 (36%) | 27 (30%) |
| 10 | 0 (0%) | 1 (2%) | 1 (1%) |
| Unknown | 0 (0%) | 3 (7%) | 3 (3%) |
| Received Prior Therapy | |||
| Androgen Deprivation Therapy | 14 (28%) | 39 (93%) | 53 (58%) |
| Chemotherapy | 7 (14%) | 30 (71%) | 37 (41%) |
| Average BL PSA (ng/mL) (Median) | 20.5 (7.1) | 186.5 (30.3) | 95.1 (9.4) |
| CAMLs Present (BL) | |||
| Average/7.5 mL blood (Median) | 3 (2) | 6 (2) | 5 (2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gironda, D.J.; Bergan, R.C.; Alpaugh, R.K.; Danila, D.C.; Chuang, T.L.; Hurtado, B.Y.; Ho, T.; Adams, D.L. Cancer Associated Macrophage-like Cells Are Prognostic for Highly Aggressive Prostate Cancer in Both the Non-Metastatic and Metastatic Settings. Cancers 2023, 15, 3725. https://doi.org/10.3390/cancers15143725
Gironda DJ, Bergan RC, Alpaugh RK, Danila DC, Chuang TL, Hurtado BY, Ho T, Adams DL. Cancer Associated Macrophage-like Cells Are Prognostic for Highly Aggressive Prostate Cancer in Both the Non-Metastatic and Metastatic Settings. Cancers. 2023; 15(14):3725. https://doi.org/10.3390/cancers15143725
Chicago/Turabian StyleGironda, Daniel J., Raymond C. Bergan, R. Katherine Alpaugh, Daniel C. Danila, Tuan L. Chuang, Brenda Y. Hurtado, Thai Ho, and Daniel L. Adams. 2023. "Cancer Associated Macrophage-like Cells Are Prognostic for Highly Aggressive Prostate Cancer in Both the Non-Metastatic and Metastatic Settings" Cancers 15, no. 14: 3725. https://doi.org/10.3390/cancers15143725
APA StyleGironda, D. J., Bergan, R. C., Alpaugh, R. K., Danila, D. C., Chuang, T. L., Hurtado, B. Y., Ho, T., & Adams, D. L. (2023). Cancer Associated Macrophage-like Cells Are Prognostic for Highly Aggressive Prostate Cancer in Both the Non-Metastatic and Metastatic Settings. Cancers, 15(14), 3725. https://doi.org/10.3390/cancers15143725