
BRAF Mutations in Melanoma: Biological Aspects, Therapeutic Implications, and Circulating Biomarkers

 ,
,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. BRAF Mutations as Key Players of Genetic Instability in Melanoma
3. Biological Aspects of BRAF Mutations
3.1. BRAF, Melanogenesis, and Phenotype Switching
3.2. BRAF Mutations and Inflammation
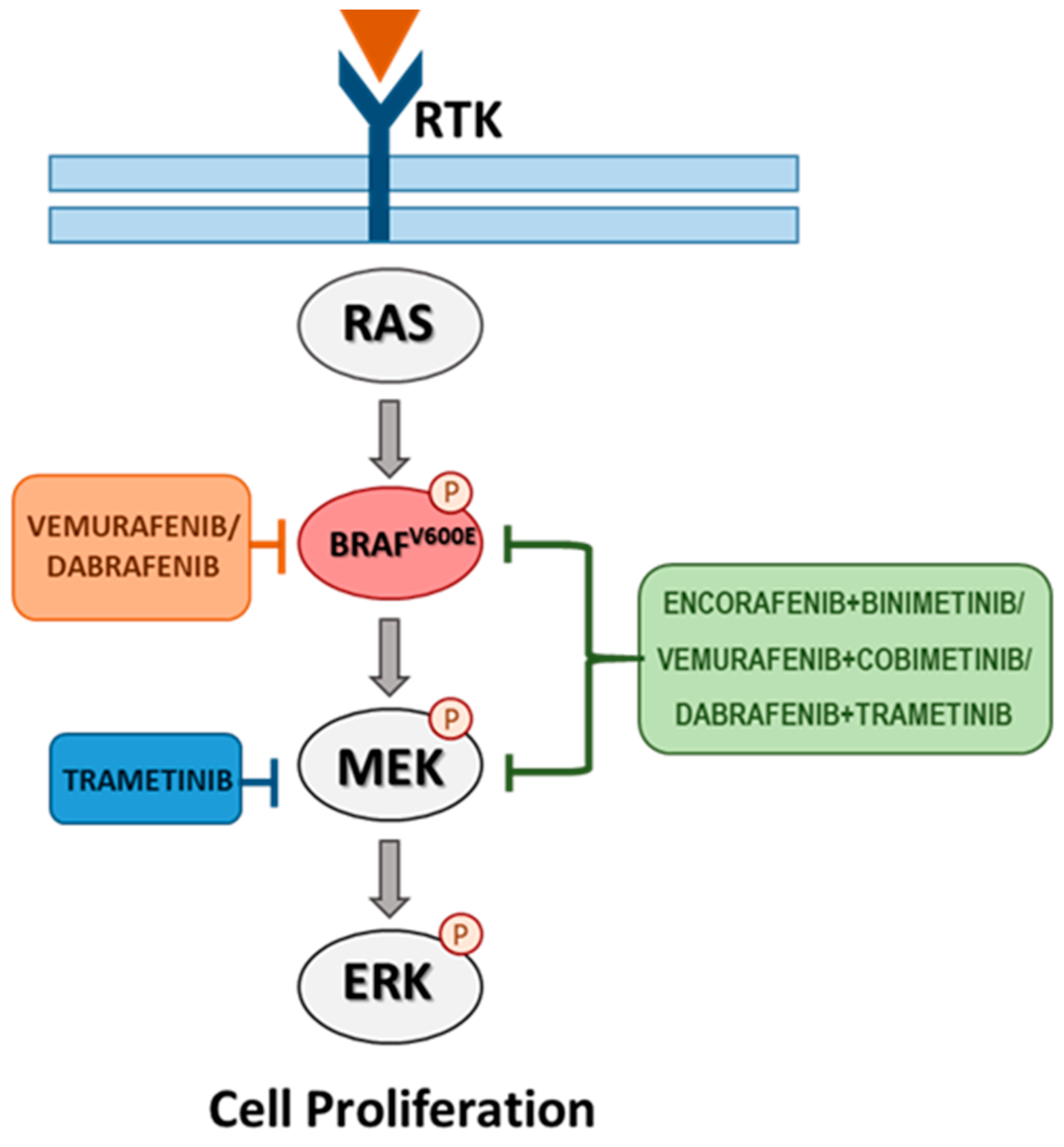
4. Targeted Therapy for BRAF-Mutated Melanoma
5. Resistance Mechanisms to Targeted Therapy in BRAF-Mutated Melanoma
6. Circulating Biomarkers
6.1. Non-Coding RNAs
6.2. Circulating Tumour DNA and Circulating Tumour Cells
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hughes, B.K.; Bishop, C.L. Current Understanding of the Role of Senescent Melanocytes in Skin Ageing. Biomedicines 2022, 10, 3111. [Google Scholar] [CrossRef]
- Nasti, T.H.; Timares, L. MC1R, eumelanin and pheomelanin: Their role in determining the susceptibility to skin cancer. Photochem. Photobiol. 2015, 91, 188–200. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Wakamatsu, K.; Sarna, T. Photodegradation of Eumelanin and Pheomelanin and Its Pathophysiological Implications. Photochem. Photobiol. 2018, 94, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chalouni, C.; Tan, C.; Clark, R.; Venook, R.; Ohri, R.; Raab, H.; Firestein, R.; Mallet, W.; Polakis, P. The melanosomal protein PMEL17 as a target for antibody drug conjugate therapy in melanoma. J. Biol. Chem. 2012, 287, 24082–24091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saginala, K.; Barsouk, A.; Aluru, J.S.; Rawla, P.; Barsouk, A. Epidemiology of Melanoma. Med. Sci. 2021, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- Dantonio, P.M.; Klein, M.O.; Freire, M.R.V.B.; Araujo, C.N.; Chiacetti, A.C.; Correa, R.G. Exploring major signaling cascades in melanomagenesis: A rationale route for targetted skin cancer therapy. Biosci. Rep. 2018, 38, BSR20180511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sample, A.; He, Y.Y. Mechanisms and prevention of UV-induced melanoma. Photodermatol. Photoimmunol. Photomed. 2018, 34, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhang, N.; Yin, C.; Zhu, B.; Li, X. Ultraviolet Radiation and Melanomagenesis: From Mechanism to Immunotherapy. Front. Oncol. 2020, 10, 951. [Google Scholar] [CrossRef]
- Kamiński, K.; Kazimierczak, U.; Kolenda, T. Oxidative stress in melanogenesis and melanoma development. Contemp. Oncol. 2022, 26, 1–7. [Google Scholar] [CrossRef]
- Del Bino, S.; Bernerd, F. Variations in skin colour and the biological consequences of ultraviolet radiation exposure. Br. J. Dermatol. 2013, 169, 33–40. [Google Scholar] [CrossRef]
- García-Guede, Á.; Vera, O.; Ibáñez-de-Caceres, I. When Oxidative Stress Meets Epigenetics: Implications in Cancer Development. Antioxidants 2020, 9, 468. [Google Scholar] [CrossRef]
- Mannavola, F.; D’Oronzo, S.; Cives, M.; Stucci, L.S.; Ranieri, G.; Silvestris, F.; Tucci, M. Extracellular Vesicles and Epigenetic Modifications Are Hallmarks of Melanoma Progression. Int. J. Mol. Sci. 2019, 21, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Yi, X.; Sun, N.; Guo, W.; Li, C. Epigenetics Regulates Antitumor Immunity in Melanoma. Front. Immunol. 2022, 13, 68786. [Google Scholar] [CrossRef]
- Palmieri, G.; Colombino, M.; Casula, M.; Manca, A.; Mandalà, M.; Cossu, A.; Italian Melanoma Intergroup (IMI). Molecular Pathways in Melanomagenesis: What We Learned from Next-Generation Sequencing Approaches. Curr. Oncol. Rep. 2018, 20, 86. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, M.; Giunta, E.F.; Tortora, M.; Curvietto, M.; Attademo, L.; Bosso, D.; Cardalesi, C.; Rosanova, M.; De Placido, P.; Pietroluongo, E.; et al. BRAF Gene and Melanoma: Back to the Future. Int. J. Mol. Sci. 2021, 22, 3474. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Bartnik, E.; Fiedorowicz, M.; Rutkowski, P. Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 2020, 21, 4576. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turski, M.L.; Vidwans, S.J.; Janku, F.; Garrido-Laguna, I.; Munoz, J.; Schwab, R.; Subbiah, V.; Rodon, J.; Kurzrock, R. Genomically Driven Tumors and Actionability across Histologies: BRAF-Mutant Cancers as a Paradigm. Mol. Cancer Ther. 2016, 15, 533–547. [Google Scholar] [CrossRef]
- Owsley, J.; Stein, M.K.; Porter, J.; In, G.K.; Salem, M.; O’Day, S.; Elliott, A.; Poorman, K.; Gibney, G.; VanderWalde, A. Prevalence of class I-III BRAF mutations among 114,662 cancer patients in a large genomic database. Exp. Biol. Med. 2021, 246, 31–39. [Google Scholar] [CrossRef]
- Sarkozy, A.; Carta, C.; Moretti, S.; Zampino, G.; Digilio, M.C.; Pantaleoni, F.; Scioletti, A.P.; Esposito, G.; Cordeddu, V.; Lepri, F.; et al. Germline BRAF mutations in Noonan, LEOPARD, and cardiofaciocutaneous syndromes: Molecular diversity and associated phenotypic spectrum. Hum. Mutat. 2009, 30, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Wagstaff, W.; Mwamba, R.N.; Grullon, K.; Armstrong, M.; Zhao, P.; Hendren-Santiago, B.; Qin, K.H.; Li, A.J.; Hu, D.A.; Youssef, A.; et al. Melanoma: Molecular genetics, metastasis, targeted therapies, immunotherapies, and therapeutic resistance. Genes Dis. 2022, 9, 1608–1623. [Google Scholar] [CrossRef]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Wan, P.T.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; Barford, D.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.B.; Dahlman, K.B. Class Matters: Sensitivity of BRAF-Mutant Melanoma to MAPK Inhibition. Clin. Cancer Res. 2018, 24, 6107–6109. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Villafane, N.; Dogruluk, T.; Grzeskowiak, C.L.; Kong, K.; Tsang, Y.H.; Zagorodna, O.; Pantazi, A.; Yang, L.; Neill, N.J.; et al. Engineering and Functional Characterization of Fusion Genes Identifies Novel Oncogenic Drivers of Cancer. Cancer Res. 2017, 77, 3502–3512. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Swetter, S.M.; Menzies, A.M.; Gershenwald, J.E.; Scolyer, R.A. Cutaneous melanoma. Lancet 2023, 402, 485–502. [Google Scholar] [CrossRef] [PubMed]
- Sakaizawa, K.; Ashida, A.; Uchiyama, A.; Ito, T.; Fujisawa, Y.; Ogata, D.; Matsushita, S.; Fujii, K.; Fukushima, S.; Shibayama, Y.; et al. Clinical characteristics associated with BRAF, NRAS and KIT mutations in Japanese melanoma patients. J. Dermatol. Sci. 2015, 80, 33–37. [Google Scholar] [CrossRef]
- Alicea, G.M.; Rebecca, V.W. Emerging strategies to treat rare and intractable subtypes of melanoma. Pigment Cell Melanoma Res. 2021, 34, 44–58. [Google Scholar] [CrossRef] [PubMed]
- Elefanti, L.; Zamuner, C.; Del Fiore, P.; Stagni, C.; Pellegrini, S.; Dall’Olmo, L.; Fabozzi, A.; Senetta, R.; Ribero, S.; Salmaso, R.; et al. The Molecular Landscape of Primary Acral Melanoma: A Multicenter Study of the Italian Melanoma Intergroup (IMI). Int. J. Mol. Sci. 2021, 22, 3826. [Google Scholar] [CrossRef]
- D’Ecclesiis, O.; Caini, S.; Martinoli, C.; Raimondi, S.; Gaiaschi, C.; Tosti, G.; Queirolo, P.; Veneri, C.; Saieva, C.; Gandini, S.; et al. Gender-Dependent Specificities in Cutaneous Melanoma Predisposition, Risk Factors, Somatic Mutations, Prognostic and Predictive Factors: A Systematic Review. Int. J. Environ. Res. Public Health 2021, 18, 7945. [Google Scholar] [CrossRef]
- Pinto, J.A.; Araujo, J.M.; Gómez, H.L. Sex, immunity, and cancer. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188647. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Artomov, M.; Goggins, W.; Daly, M.; Tsao, H. Gender Disparity and Mutation Burden in Metastatic Melanoma. J. Natl. Cancer Inst. 2015, 107, djv221. [Google Scholar] [CrossRef]
- van der Kooij, M.K.; Dekkers, O.M.; Aarts, M.J.B.; van den Berkmortel, F.W.P.J.; Boers-Sonderen, M.J.; de Groot, J.W.B.; Hospers, G.A.P.; Piersma, D.; van Rijn, R.S.; Suijkerbuijk, K.P.M.; et al. Sex-Based Differences in Treatment with Immune Checkpoint Inhibition and Targeted Therapy for Advanced Melanoma: A Nationwide Cohort Study. Cancers 2021, 13, 4639. [Google Scholar] [CrossRef] [PubMed]
- Lokhandwala, P.M.; Tseng, L.H.; Rodriguez, E.; Zheng, G.; Pallavajjalla, A.; Gocke, C.D.; Eshleman, J.R.; Lin, M.T. Clinical mutational profiling and categorization of BRAF mutations in melanomas using next generation sequencing. BMC Cancer 2019, 19, 665. [Google Scholar] [CrossRef]
- Colombino, M.; Rozzo, C.; Paliogiannis, P.; Casula, M.; Manca, A.; Doneddu, V.; Fedeli, M.A.; Sini, M.C.; Palomba, G.; Pisano, M.; et al. Comparison of BRAF Mutation Screening Strategies in a Large Real-Life Series of Advanced Melanoma Patients. J. Clin. Med. 2020, 9, 2430. [Google Scholar] [CrossRef]
- Yeh, I.; von Deimling, A.; Bastian, B.C. Clonal BRAF mutations in melanocytic nevi and initiating role of BRAF in melanocytic neoplasia. J. Natl. Cancer Inst. 2013, 105, 917–919. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, P.; Friedlander, P.; Zakaria, E.A.; Kandil, E. Impact of BRAF mutation status in the prognosis of cutaneous melanoma: An area of ongoing research. Ann. Transl. Med. 2015, 3, 24. [Google Scholar] [CrossRef] [PubMed]
- Rubió-Casadevall, J.; Carbó-Bagué, A.; Puigdemont, M.; Osca-Gelis, G.; Oliveras, G.; Vilar-Coromina, N.; Ferrer-Fabrega, B.; Urban, A.; Llobet-Roma, M.; Martín-Romero, F.; et al. Population-based analysis of the prevalence of BRAF mutation in patients diagnosed with cutaneous melanoma and its significance as a prognostic factor. Eur. J. Dermatol. 2021, 31, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Hugdahl, E.; Kalvenes, M.B.; Puntervoll, H.E.; Ladstein, R.G.; Akslen, L.A. BRAF-V600E expression in primary nodular melanoma is associated with aggressive tumour features and reduced survival. Br. J. Cancer 2016, 114, 801–808. [Google Scholar] [CrossRef] [Green Version]
- Zerfaoui, M.; Toraih, E.; Ruiz, E.; Errami, Y.; Attia, A.S.; Krzysztof, M.; Abd Elmageed, Z.Y.; Kandil, E. Nuclear Localization of BRAFV600E Is Associated with HMOX-1 Upregulation and Aggressive Behavior of Melanoma Cells. Cancers 2022, 14, 311. [Google Scholar] [CrossRef]
- Menzies, A.M.; Haydu, L.E.; Visintin, L.; Carlino, M.S.; Howle, J.R.; Thompson, J.F.; Kefford, R.F.; Scolyer, R.A.; Long, G.V. Distinguishing clinicopathologic features of patients with V600E and V600K BRAF-mutant metastatic melanoma. Clin. Cancer Res. 2012, 18, 3242–3249. [Google Scholar] [CrossRef] [Green Version]
- Nepote, A.; Avallone, G.; Ribero, S.; Cavallo, F.; Roccuzzo, G.; Mastorino, L.; Conforti, C.; Paruzzo, L.; Poletto, S.; Carnevale Schianca, F.; et al. Current Controversies and Challenges on BRAF V600K-Mutant Cutaneous Melanoma. J. Clin. Med. 2022, 11, 828. [Google Scholar] [CrossRef] [PubMed]
- Liebig, J.K.; Kuphal, S.; Bosserhoff, A.K. HuRdling Senescence: HuR Breaks BRAF-Induced Senescence in Melanocytes and Supports Melanoma Growth. Cancers 2020, 12, 1299. [Google Scholar] [CrossRef]
- Damsky, W.E.; Bosenberg, M. Melanocytic nevi and melanoma: Unraveling a complex relationship. Oncogene 2017, 36, 5771–5792. [Google Scholar] [CrossRef] [Green Version]
- Rotolo, S.; Diotti, R.; Gordon, R.E.; Qiao, R.F.; Yao, Z.; Phelps, R.G.; Dong, J. Effects on proliferation and melanogenesis by inhibition of mutant BRAF and expression of wild-type INK4A in melanoma cells. Int. J. Cancer 2005, 115, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Wellbrock, C.; Rana, S.; Paterson, H.; Pickersgill, H.; Brummelkamp, T.; Marais, R. Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF. PLoS ONE 2008, 3, e2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Jang, G.B.; Yang, X.; Wang, Q.; He, S.; Li, S.; Quach, C.; Zhao, S.; Li, F.; Yuan, Z.; et al. Central role of autophagic UVRAG in melanogenesis and the suntan response. Proc. Natl. Acad. Sci. USA 2018, 115, E7728–E7737. [Google Scholar] [CrossRef] [Green Version]
- Mehdi, A.; Attias, M.; Arakelian, A.; Szyf, M.; Piccirillo, C.A.; Rabbani, S.A. S-adenosylmethionine blocks tumorigenesis and with immune checkpoint inhibitor enhances anti-cancer efficacy against BRAF mutant and wildtype melanomas. Neoplasia 2023, 36, 100874. [Google Scholar] [CrossRef]
- Hossain, S.M.; Eccles, M.R. Phenotype Switching and the Melanoma Microenvironment; Impact on Immunotherapy and Drug Resistance. Int. J. Mol. Sci. 2023, 24, 1601. [Google Scholar] [CrossRef]
- Najem, A.; Soumoy, L.; Sabbah, M.; Krayem, M.; Awada, A.; Journe, F.; Ghanem, G.E. Understanding Molecular Mechanisms of Phenotype Switching and Crosstalk with TME to Reveal New Vulnerabilities of Melanoma. Cells 2022, 11, 1157. [Google Scholar] [CrossRef]
- Estrada, C.; Mirabal-Ortega, L.; Méry, L.; Dingli, F.; Besse, L.; Messaoudi, C.; Loew, D.; Pouponnot, C.; Bertolotto, C.; Eychène, A.; et al. MITF activity is regulated by a direct interaction with RAF proteins in melanoma cells. Commun. Biol. 2022, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Aida, S.; Sonobe, Y.; Tanimura, H.; Oikawa, N.; Yuhki, M.; Sakamoto, H.; Mizuno, T. MITF suppression improves the sensitivity of melanoma cells to a BRAF inhibitor. Cancer Lett. 2017, 409, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, K.M.; Correa, I.; Josephs, D.H.; Karagiannis, P.; Egbuniwe, I.U.; Cafferkey, M.J.; Spicer, J.F.; Harries, M.; Nestle, F.O.; Lacy, K.E.; et al. Effects of BRAF mutations and BRAF inhibition on immune responses to melanoma. Mol. Cancer Ther. 2014, 13, 2769–2783. [Google Scholar] [CrossRef] [Green Version]
- Mandalà, M.; De Logu, F.; Merelli, B.; Nassini, R.; Massi, D. Immunomodulating property of MAPK inhibitors: From translational knowledge to clinical implementation. Lab. Investig. 2017, 97, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Bradley, S.D.; Chen, Z.; Melendez, B.; Talukder, A.; Khalili, J.S.; Rodriguez-Cruz, T.; Liu, S.; Whittington, M.; Deng, W.; Li, F.; et al. BRAFV600E Co-opts a Conserved MHC Class I Internalization Pathway to Diminish Antigen Presentation and CD8+ T-cell Recognition of Melanoma. Cancer Immunol. Res. 2015, 3, 602–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalili, J.S.; Liu, S.; Rodríguez-Cruz, T.G.; Whittington, M.; Wardell, S.; Liu, C.; Zhang, M.; Cooper, Z.A.; Frederick, D.T.; Li, Y.; et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin. Cancer Res. 2012, 18, 5329–5340. [Google Scholar] [CrossRef] [Green Version]
- Sumimoto, H.; Imabayashi, F.; Iwata, T.; Kawakami, Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J. Exp. Med. 2006, 203, 1651–1656. [Google Scholar] [CrossRef]
- Avagliano, A.; Fiume, G.; Pelagalli, A.; Sanità, G.; Ruocco, M.R.; Montagnani, S.; Arcucci, A. Metabolic Plasticity of Melanoma Cells and Their Crosstalk With Tumor Microenvironment. Front. Oncol. 2020, 10, 722. [Google Scholar] [CrossRef] [PubMed]
- Shelton, M.; Anene, C.A.; Nsengimana, J.; Roberts, W.; Newton-Bishop, J.; Boyne, J.R. The role of CAF derived exosomal microRNAs in the tumour microenvironment of melanoma. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188456. [Google Scholar] [CrossRef]
- Gener Lahav, T.; Adler, O.; Zait, Y.; Shani, O.; Amer, M.; Doron, H.; Abramovitz, L.; Yofe, I.; Cohen, N.; Erez, N. Melanoma-derived extracellular vesicles instigate proinflammatory signaling in the metastatic microenvironment. Int. J. Cancer 2019, 145, 2521–2534. [Google Scholar] [CrossRef] [PubMed]
- Anselmi, M.; Fontana, F.; Marzagalli, M.; Gagliano, N.; Sommariva, M.; Limonta, P. Melanoma Stem Cells Educate Neutrophils to Support Cancer Progression. Cancers 2022, 14, 3391. [Google Scholar] [CrossRef]
- Whipple, C.A.; Brinckerhoff, C.E. BRAF(V600E) melanoma cells secrete factors that activate stromal fibroblasts and enhance tumourigenicity. Br. J. Cancer 2014, 111, 1625–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eck, S.M.; Côté, A.L.; Winkelman, W.D.; Brinckerhoff, C.E. CXCR4 and matrix metalloproteinase-1 are elevated in breast carcinoma-associated fibroblasts and in normal mammary fibroblasts exposed to factors secreted by breast cancer cells. Mol. Cancer Res. 2009, 7, 1033–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Garlanda, C.; Allavena, P. Molecular pathways and targets in cancer-related inflammation. Ann. Med. 2010, 42, 161–170. [Google Scholar] [CrossRef]
- Jobe, N.P.; Rösel, D.; Dvořánková, B.; Kodet, O.; Lacina, L.; Mateu, R.; Smetana, K.; Brábek, J. Simultaneous blocking of IL-6 and IL-8 is sufficient to fully inhibit CAF-induced human melanoma cell invasiveness. Histochem. Cell Biol. 2016, 146, 205–217. [Google Scholar] [CrossRef]
- Izar, B.; Joyce, C.E.; Goff, S.; Cho, N.L.; Shah, P.M.; Sharma, G.; Li, J.; Ibrahim, N.; Gold, J.; Hodi, F.S.; et al. Bidirectional cross talk between patient-derived melanoma and cancer-associated fibroblasts promotes invasion and proliferation. Pigment Cell Melanoma Res. 2016, 29, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, P.; Prasad, C.P.; Andersson, T. Combination therapy targeting the elevated interleukin-6 level reduces invasive migration of BRAF inhibitor-resistant melanoma cells. Mol. Oncol. 2019, 13, 480–494. [Google Scholar] [CrossRef] [Green Version]
- Sandri, S.; Faião-Flores, F.; Tiago, M.; Pennacchi, P.C.; Massaro, R.R.; Alves-Fernandes, D.K.; Berardinelli, G.N.; Evangelista, A.F.; de Lima Vazquez, V.; Reis, R.M.; et al. Vemurafenib resistance increases melanoma invasiveness and modulates the tumor microenvironment by MMP-2 upregulation. Pharmacol. Res. 2016, 111, 523–533. [Google Scholar] [CrossRef]
- Smith, M.P.; Sanchez-Laorden, B.; O’Brien, K.; Brunton, H.; Ferguson, J.; Young, H.; Dhomen, N.; Flaherty, K.T.; Frederick, D.T.; Cooper, Z.A.; et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFα. Cancer Discov. 2014, 4, 1214–1229. [Google Scholar] [CrossRef] [Green Version]
- Pandya, P.; Kublo, L.; Stewart-Ornstein, J. p53 Promotes Cytokine Expression in Melanoma to Regulate Drug Resistance and Migration. Cells 2022, 11, 405. [Google Scholar] [CrossRef]
- Hölzel, M.; Bovier, A.; Tüting, T. Plasticity of tumour and immune cells: A source of heterogeneity and a cause for therapy resistance? Nat. Rev. Cancer 2013, 13, 365–376. [Google Scholar] [CrossRef]
- Romano, V.; Belviso, I.; Venuta, A.; Ruocco, M.R.; Masone, S.; Aliotta, F.; Fiume, G.; Montagnani, S.; Avagliano, A.; Arcucci, A. Influence of Tumor Microenvironment and Fibroblast Population Plasticity on Melanoma Growth, Therapy Resistance and Immunoescape. Int. J. Mol. Sci. 2021, 22, 5283. [Google Scholar] [CrossRef] [PubMed]
- Simiczyjew, A.; Dratkiewicz, E.; Mazurkiewicz, J.; Ziętek, M.; Matkowski, R.; Nowak, D. The influence of tumor microenvironment on immune escape of melanoma. Int. J. Mol. Sci. 2020, 21, 8359. [Google Scholar] [CrossRef]
- Mun, J.Y.; Leem, S.H.; Lee, J.H.; Kim, H.S. Dual Relationship Between Stromal Cells and Immune Cells in the Tumor Microenvironment. Front. Immunol. 2022, 13, 864739. [Google Scholar] [CrossRef] [PubMed]
- Montoyo-Pujol, Y.G.; Wang, X.; Bermúdez-Sánchez, S.; Martin, A.; Almazan, F.; López-Nevot, M.Á. Measurement of 45 cytokine, chemokine and growth factors in established cell culture supernatants and autologous serum from advanced melanoma patients. Carcinogenesis 2021, 42, 714–723. [Google Scholar] [CrossRef]
- Bagheri, H.; Pourhanifeh, M.H.; Derakhshan, M.; Mahjoubin-Tehran, M.; Ghasemi, F.; Mousavi, S.; Rafiei, R.; Abbaszadeh-Goudarzi, K.; Mirzaei, H.R.; Mirzaei, H. CXCL-10: A new candidate for melanoma therapy? Cell. Oncol. 2020, 43, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A.; Bhandaru, M.; Zhou, Y.; McElwee, K.J. Immunotherapy of melanoma: Present options and future promises. Cancer Metastasis Rev. 2015, 34, 115–128. [Google Scholar] [CrossRef]
- Domingues, B.; Lopes, J.M.; Soares, P.; Pópulo, H. Melanoma treatment in review. Immunotargets Ther. 2018, 7, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garbe, C.; Eigentler, T.K. Vemurafenib. Recent Results Cancer Res. 2018, 211, 77–89. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [Green Version]
- Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; Testori, A.; Lorigan, P.C.; et al. Vemurafenib in patients with BRAFV600 mutation-positive metastatic melanoma: Final overall survival results of the randomized BRIM-3 study. Ann. Oncol. 2017, 28, 2581–2587. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, S.; Lee, R.; Fusi, A.; Lorigan, P. Dabrafenib and its use in the treatment of metastatic melanoma. Melanoma Manag. 2015, 2, 199–208. [Google Scholar] [CrossRef]
- Rose, A.A.N. Encorafenib and binimetinib for the treatment of BRAF V600E/K-mutated melanoma. Drugs Today 2019, 55, 247–264. [Google Scholar] [CrossRef]
- Solit, D.; Rosen, N. Oncogenic RAF: A brief history of time. Pigment Cell Melanoma Res. 2010, 23, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Eroglu, Z.; Ribas, A. Combination therapy with BRAF and MEK inhibitors for melanoma: Latest evidence and place in therapy. Ther. Adv. Med. Oncol. 2016, 8, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzano, J.L.; Layos, L.; Bugés, C.; de Los Llanos Gil, M.; Vila, L.; Martínez-Balibrea, E.; Martínez-Cardús, A. Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 2016, 4, 237. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Saw, R.P.M.; Lo, S.; Nieweg, O.E.; Shannon, K.F.; Gonzalez, M.; Guminski, A.; Lee, J.H.; Lee, H.; Ferguson, P.M.; et al. Neoadjuvant dabrafenib combined with trametinib for resectable, stage IIIB-C, BRAFV600 mutation-positive melanoma (NeoCombi): A single-arm, open-label, single-centre, phase 2 trial. Lancet Oncol. 2019, 20, 961–971. [Google Scholar] [CrossRef]
- Namikawa, K.; Yamazaki, N. Targeted Therapy and Immunotherapy for Melanoma in Japan. Curr. Treat. Options Oncol. 2019, 20, 7. [Google Scholar] [CrossRef] [Green Version]
- Michielin, O.; Atkins, M.B.; Koon, H.B.; Dummer, R.; Ascierto, P.A. Evolving impact of long-term survival results on metastatic melanoma treatment. J. Immunother. Cancer 2020, 8, e000948. [Google Scholar] [CrossRef]
- Lim, S.Y.; Menzies, A.M.; Rizos, H. Mechanisms and strategies to overcome resistance to molecularly targeted therapy for melanoma. Cancer 2017, 123, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Pappalardo, F.; Russo, G.; Candido, S.; Pennisi, M.; Cavalieri, S.; Motta, S.; McCubrey, J.A.; Nicoletti, F.; Libra, M. Computational Modeling of PI3K/AKT and MAPK Signaling Pathways in Melanoma Cancer. PLoS ONE 2016, 11, 0152104. [Google Scholar] [CrossRef] [PubMed]
- Byeon, S.; Cho, H.J.; Jang, K.T.; Kwon, M.; Lee, J.; Lee, J.; Kim, S.T. Molecular profiling of Asian patients with advanced melanoma receiving check-point inhibitor treatment. ESMO Open 2021, 6, 100002. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, Y.; Ito, T.; Kato, H.; Irie, H.; Kaji, T.; Maekawa, T.; Asai, J.; Yamamoto, Y.; Fujimura, T.; Nakai, Y.; et al. Outcome of combination therapy using BRAF and MEK inhibitors among Asian patients with advanced melanoma: An analysis of 112 cases. Eur. J. Cancer 2021, 145, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Ishitsuka, Y.; Tanaka, R.; Okiyama, N.; Watanabe, R.; Saito, A.; Furuta, J.; Fujisawa, Y. Frequent brain metastases during treatment with BRAF/MEK inhibitors: A retrospective single institutional study. J. Dermatol. 2020, 47, 1191–1194. [Google Scholar] [CrossRef]
- Tawbi, H.A.; Boutros, C.; Kok, D.; Robert, C.; McArthur, G. New Era in the Management of Melanoma Brain Metastases. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 741–750. [Google Scholar] [CrossRef]
- Khaliq, M.; Fallahi-Sichani, M. Epigenetic Mechanisms of Escape from BRAF Oncogene Dependency. Cancers 2019, 11, 1480. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Roberto, M.; Panebianco, M.; Botticelli, A.; Mazzuca, F.; Marchetti, P. Drug resistance of BRAF-mutant melanoma: Review of up-to-date mechanisms of action and promising targeted agents. Eur. J. Pharmacol. 2019, 862, 172621. [Google Scholar] [CrossRef]
- Proietti, I.; Skroza, N.; Bernardini, N.; Tolino, E.; Balduzzi, V.; Marchesiello, A.; Michelini, S.; Volpe, S.; Mambrin, A.; Mangino, G.; et al. Mechanisms of Acquired BRAF Inhibitor Resistance in Melanoma: A Systematic Review. Cancers 2020, 12, 2801. [Google Scholar] [CrossRef]
- Tangella, L.P.; Clark, M.E.; Gray, E.S. Resistance mechanisms to targeted therapy in BRAF-mutant melanoma–A mini review. Biochim. Biophys. Acta Gen. Subj. 2021, 1865, 129736. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [Green Version]
- Nathanson, K.L.; Martin, A.; Wubbenhorst, B.; Greshock, J.; Letrero, R.; D’Andrea, K.; O’Day, S.; Infante, J.R.; Falchook, G.S.; Arkenau, H.T.; et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin. Cancer Res. 2013, 17, 4868–4878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paraiso, K.H.T.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 7, 2750–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 6166, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blateau, P.; Coyaud, E.; Laurent, E.; Béganton, B.; Ducros, V.; Chauchard, G.; Vendrell, J.A.; Solassol, J. TERT Promoter Mutation as an Independent Prognostic Marker for Poor Prognosis MAPK Inhibitors-Treated Melanoma. Cancers 2020, 12, 2224. [Google Scholar] [CrossRef] [PubMed]
- Thielmann, C.M.; Matull, J.; Zaremba, A.; Murali, R.; Chorti, E.; Lodde, G.; Jansen, P.; Herbst, R.; Terheyden, P.; Utikal, J. TERT promoter mutations are associated with longer progression-free and overall survival in patients with BRAF-mutant melanoma receiving BRAF and MEK inhibitor therapy. Eur. J. Cancer 2022, 161, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Liu, R.; Zhu, G.; Umbricht, C.B.; Xing, M. TERT promoter mutation determines apoptotic and therapeutic responses of BRAF-mutant cancers to BRAF and MEK inhibitors: Achilles Heel. Proc. Natl. Acad. Sci. USA 2020, 117, 15846–15851. [Google Scholar] [CrossRef]
- Delyon, J.; Vallet, A.; Bernard-Cacciarella, M.; Kuzniak, I.; Reger de Moura, C.; Louveau, B.; Jouenne, F.; Mourah, S.; Lebbé, C.; Dumaz, N. TERT Expression Induces Resistance to BRAF and MEK Inhibitors in BRAF-Mutated Melanoma In Vitro. Cancers 2023, 15, 2888. [Google Scholar] [CrossRef]
- Delmas, A.; Cherier, J.; Pohorecka, M.; Medale-Giamarchi, C.; Meyer, N.; Casanova, A.; Sordet, O.; Lamant, L.; Savina, A.; Pradines, A.; et al. The c-Jun/RHOB/AKT pathway confers resistance of BRAF-mutant melanoma cells to MAPK inhibitors. Oncotarget 2015, 17, 15250–15264. [Google Scholar] [CrossRef] [Green Version]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012, 7408, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Margue, C.; Philippidou, D.; Kozar, I.; Cesi, G.; Felten, P.; Kulms, D.; Letellier, E.; Haan, C.; Kreis, S. Kinase inhibitor library screening identifies synergistic drug combinations effective in sensitive and resistant melanoma cells. J. Exp. Clin. Cancer Res. 2019, 38, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, X.; Zhang, T.; Wang, Q.; Rathore, M.G.; Reddy, K.; Chen, H.; Shin, S.H.; Ma, W.Y.; Bode, A.M.; Dong, Z. HI-511 overcomes melanoma drug resistance via targeting AURKB and BRAF V600E. Theranostics 2020, 10, 9721–9740. [Google Scholar] [CrossRef] [PubMed]
- Misek, S.A.; Foda, B.M.; Dexheimer, T.S.; Akram, M.; Conrad, S.E.; Schmidt, J.C.; Neubig, R.R.; Gallo, K.A. BRAF Inhibitor Resistance Confers Increased Sensitivity to Mitotic Inhibitors. Front. Oncol. 2022, 12, 766794. [Google Scholar] [CrossRef]
- Zhou, A.Y.; Johnson, D.B. Combinatorial Therapies in Melanoma: MAPK Inhibitors and Beyond. Am. J. Clin. Dermatol. 2018, 2, 181–193. [Google Scholar] [CrossRef]
- Duggan, M.C.; Stiff, A.R.; Bainazar, M.; Regan, K.; Olaverria Salavaggione, G.N.; Maharry, S.; Blachly, J.S.; Krischak, M.; Walker, C.J.; Latchana, N.; et al. Identification of NRAS isoform 2 overexpression as a mechanism facilitating BRAF inhibitor resistance in malignant melanoma. Proc. Natl. Acad. Sci. USA 2017, 36, 9629–9634. [Google Scholar] [CrossRef]
- Germann, U.A.; Furey, B.F.; Markland, W.; Hoover, R.R.; Aronov, A.M.; Roix, J.J.; Hale, M.; Boucher, D.M.; Sorrell, D.A.; Martinez-Botella, G.; et al. Targeting the MAPK Signaling Pathway in Cancer: Promising Preclinical Activity with the Novel Selective ERK1/2 Inhibitor BVD-523 (Ulixertinib). Mol. Cancer Ther. 2017, 11, 2351–2363. [Google Scholar] [CrossRef] [Green Version]
- Fattore, L.; Marra, E.; Pisanu, M.E.; Noto, A.; de Vitis, C.; Belleudi, F.; Aurisicchio, L.; Mancini, R.; Torrisi, M.R.; Ascierto, P.A.; et al. Activation of an early feedback survival loop involving phospho-ErbB3 is a general response of melanoma cells to RAF/MEK inhibition and is abrogated by anti-ErbB3 antibodies. J. Transl. Med. 2013, 11, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Tran, L.; Park, Y.; Chen, I.; Lan, J.; Xie, Y.; Semenza, G.L. Reciprocal Regulation of DUSP9 and DUSP16 Expression by HIF1 Controls ERK and p38 MAP Kinase Activity and Mediates Chemotherapy-Induced Breast Cancer Stem Cell Enrichment. Cancer Res. 2018, 78, 4191–4202. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.S.; Fong, C.W.; Lim, J.; Yusoff, P.; Low, B.C.; Langdon, W.Y.; Guy, G.R. Sprouty2 attenuates epidermal growth factor receptor ubiquitylation and endocytosis, and consequently enhances Ras/ERK signalling. EMBO J. 2002, 21, 4796–4808. [Google Scholar] [CrossRef] [Green Version]
- Lake, D.; Corrêa, S.A.; Müller, J. Negative feedback regulation of the ERK1/2 MAPK pathway. Cell. Mol. Life Sci. 2016, 73, 4397–4413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saei, A.; Eichhorn, P.J.A. Adaptive Responses as Mechanisms of Resistance to BRAF Inhibitors in Melanoma. Cancers 2019, 11, 1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojha, R.; Leli, N.M.; Onorati, A.; Piao, S.; Verginadis, I.I.; Tameire, F.; Rebecca, V.W.; Chude, C.I.; Murugan, S.; Fennelly, C.; et al. ER Translocation of the MAPK Pathway Drives Therapy Resistance in BRAF-Mutant Melanoma. Cancer Discov. 2019, 3, 396–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Song, Y.; Quach, C.; Guo, H.; Jang, G.; Maazi, H.; Zhao, S.; Sands, N.A.; Liu, Q.; In, G.K.; et al. Transcriptional regulation of autophagy-lysosomal function in BRAF-driven melanoma progression and chemoresistance. Nat. Commun. 2019, 1, 1693. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat. Med. 2019, 4, 620–627. [Google Scholar] [CrossRef]
- Sun, C.; Wang, L.; Huang, S.; Heynen, G.J.; Prahallad, A.; Robert, C.; Haanen, J.; Blank, C.; Wesseling, J.; Willems, S.M.; et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 2014, 508, 118–122. [Google Scholar] [CrossRef]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Rambow, F.; Marine, J.C.; Goding, C.R. Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 2019, 33, 1295–1318. [Google Scholar] [CrossRef] [Green Version]
- Berico, P.; Cigrang, M.; Davidson, G.; Braun, C.; Sandoz, J.; Legras, S.; Vokshi, B.H.; Slovic, N.; Peyresaubes, F.; Gene Robles, C.M.; et al. CDK7 and MITF repress a transcription program involved in survival and drug tolerance in melanoma. EMBO Rep. 2021, 22, e51683. [Google Scholar] [CrossRef]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.; Geukes Foppen, M.H.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, S.C.; Krishnan, B.; Bailey, S.T.; Moschos, S.J.; Kuan, P.F.; Shimamura, T.; Osborne, L.D.; Siegel, M.B.; Duncan, L.M.; O’Brien, E.T., 3rd; et al. HIF1α and HIF2α independently activate SRC to promote melanoma metastases. J. Clin. Investig. 2013, 123, 2078–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaverdashvili, K.; Wong, P.; Ma, J.; Zhang, K.; Osman, I.; Bedogni, B. MT1-MMP modulates melanoma cell dissemination and metastasis through activation of MMP2 and RAC1. Pigment Cell Melanoma Res. 2014, 27, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Obenauf, A.; Zou, Y.; Ji, A.L.; Vanharanta, S.; Shu, W.; Shi, H.; Kong, X.; Bosenberg, M.C.; Wiesner, T.; Rosen, N.; et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015, 520, 368–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, E.; Girotti, M.R.; Viros, A.; Hooper, S.; Spencer-Dene, B.; Matsuda, M.; Larkin, J.; Marais, R.; Sahai, E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin Β1/FAK signaling. Cancer Cell 2015, 27, 574–588. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Xiao, M.; Ge, Y.; Krepler, C.; Belser, E.; Lopez-Coral, A.; Xu, X.; Zhang, G.; Azuma, R.; Liu, Q.; et al. BRAF inhibition stimulates melanoma-associated macrophages to drive tumor growth. Clin. Cancer Res. 2015, 21, 1652–1664. [Google Scholar] [CrossRef]
- Tabolacci, C.; Cordella, M.; Mariotti, S.; Rossi, S.; Senatore, C.; Lintas, C.; Levati, L.; D’Arcangelo, D.; Facchiano, A.; D’Atri, S.; et al. Melanoma Cell Resistance to Vemurafenib Modifies Inter-Cellular Communication Signals. Biomedicines 2021, 9, 79. [Google Scholar] [CrossRef]
- Steinberg, S.M.; Zhang, P.; Malik, B.T.; Boni, A.; Shabaneh, T.B.; Byrne, K.T.; Mullins, D.W.; Brinckerhoff, C.E.; Ernstoff, M.S.; Bosenberg, M.W.; et al. BRAF inhibition alleviates immune suppression in murine autochthonous melanoma. Cancer Immunol. Res. 2014, 2, 1044–1050. [Google Scholar] [CrossRef] [Green Version]
- Kuske, M.; Westphal, D.; Wehner, R.; Schmitz, M.; Beissert, S.; Praetorius, C.; Meier, F. Immunomodulatory effects of BRAF and MEK inhibitors: Implications for Melanoma therapy. Pharmacol. Res. 2018, 136, 151–159. [Google Scholar] [CrossRef]
- Kakavand, H.; Wilmott, J.S.; Menzies, A.M.; Vilain, R.; Haydu, L.E.; Yearley, J.H.; Thompson, J.F.; Kefford, R.F.; Hersey, P.; Long, G.V.; et al. PD-L1 Expression and Tumor-Infiltrating Lymphocytes Define Different Subsets of MAPK Inhibitor-Treated Melanoma Patients. Clin. Cancer Res. 2015, 21, 3140–3148. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Zhou, J.; Giobbie-Hurder, A.; Wargo, J.; Hodi, F.S. The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin. Cancer Res. 2013, 19, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Boni, A.; Cogdill, A.P.; Dang, P.; Udayakumar, D.; Njauw, C.N.; Sloss, C.M.; Ferrone, C.R.; Flaherty, K.T.; Lawrence, D.P.; Fisher, D.E.; et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010, 70, 5213–5219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.A. Metabolic rewiring in melanoma. Oncogene 2017, 36, 147–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Kluza, J.; Corazao-Rozas, Y.; Touil, P.; Jendoubi, M.; Maire, C.; Guerreschi, P.; Jonneaux, A.; Ballot, C.; Balayssac, S.; Valable, S.; et al. Inactivation of the HIF-1alpha/PDK3 signaling axis drives melanoma toward mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res. 2012, 72, 5035–5047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmenter, T.J.; Kleinschmidt, M.; Kinross, K.M.; Bond, S.T.; Li, J.; Kaadige, M.R.; Rao, A.; Sheppard, K.E.; Hugo, W.; Pupo, G.M.; et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014, 4, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, G.; Capone, M.; Ascierto, M.L.; Gentilcore, G.; Stroncek, D.F.; Casula, M.; Sini, M.C.; Palla, M.; Mozzillo, N.; Ascierto, P.A. Main roads to melanoma. J. Transl. Med. 2009, 7, 86. [Google Scholar] [CrossRef] [Green Version]
- Denko, N.C. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 2008, 8, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Bristot, I.J.; Kehl Dias, C.; Chapola, H.; Parsons, R.B.; Klamt, F. Metabolic rewiring in melanoma drug-resistant cells. Crit. Rev. Oncol. Hematol. 2020, 153, 102995. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V.; Managò, A.; La Vecchia, S.; Zamporlini, F.; Vitale, N.; Baroni, G.; Cignetto, S.; Serra, S.; Bologna, C.; Stingi, A.; et al. Nicotinamide Phosphoribosyltransferase (NAMPT) as a Therapeutic Target in BRAF-Mutated Metastatic Melanoma. J. Natl. Cancer Inst. 2018, 110, 290–303. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zanca, C.; Rajkumar, U.; Koga, T.; Diao, Y.; Raviram, R.; Liu, F.; Turner, K.; Yang, H.; Brunk, E.; et al. NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nat. Cell Biol. 2019, 569, 570–575. [Google Scholar] [CrossRef]
- Haq, R.; Fisher, D.E. Biology and clinical relevance of the micropthalmia family of transcription factors in human cancer. J. Clin. Oncol. 2011, 29, 3474–3482. [Google Scholar] [CrossRef] [PubMed]
- Goel, V.K.; Lazar, A.J.; Warneke, C.L.; Redston, M.S.; Haluska, F.G. Examination of mutations in BRAF, NRAS, and PTEN in primary cutaneous melanoma. J. Investig. Dermatol. 2006, 126, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Xu, X.; Li, S.; Ma, J.; Shi, Q.; Guo, S.; Liu, L.; Guo, W.; Xu, P.; He, Y.; et al. SOX4 promotes proliferative signals by regulating glycolysis through AKT activation in melanoma cells. J. Investig. Dermatol. 2017, 137, 2407–2416. [Google Scholar] [CrossRef] [Green Version]
- Laurenzana, A.; Chillà, A.; Luciani, C.; Peppicelli, S.; Biagioni, A.; Bianchini, F.; Tenedini, E.; Torre, E.; Mocali, A.; Calorini, L.; et al. uPA/uPAR system activation drives a glycolytic phenotype in melanoma cells. Int. J. Cancer 2017, 141, 1190–1200. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, P.; Trinh, A.; Khamari, R.; Kluza, J. Melanoma metabolism contributes to the cellular responses to MAPK/ERK pathway inhibitors. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 999–1005. [Google Scholar] [CrossRef]
- Peppicelli, S.; Bianchini, F.; Calorini, L. Extracellular acidity, a “reappreciated” trait of tumor environment driving malignancy: Perspectives in diagnosis and therapy. Cancer Metastasis Rev. 2014, 33, 823–832. [Google Scholar] [CrossRef]
- Choi, Y.K.; Park, K.G. Targeting glutamine metabolism for cancer treatment. Biomol. Ther. 2018, 26, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Huang, S.K.; Marzese, D.M.; Hsu, S.C.; Kawas, N.P.; Chong, K.K.; Long, G.V.; Menzies, A.M.; Scolyer, R.A.; Izraely, S.; et al. Epigenetic changes of EGFR have an important role in BRAF inhibitor-resistant cutaneous melanomas. J. Investig. Dermatol. 2015, 135, 532–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soumoy, L.; Schepkens, C.; Krayem, M.; Najem, A.; Tagliatti, V.; Ghanem, G.E.; Saussez, S.; Colet, J.M.; Journe, F. Metabolic Reprogramming in Metastatic Melanoma with Acquired Resistance to Targeted Therapies: Integrative Metabolomic and Proteomic Analysis. Cancers 2020, 12, 1323. [Google Scholar] [CrossRef]
- Haq, R.; Fisher, D.E.; Widlund, H.R. Molecular pathways: BRAF induces bioenergetic adaptation by attenuating oxidative phosphorylation. Clin. Cancer Res. 2014, 20, 2257–2263. [Google Scholar] [CrossRef] [Green Version]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1alpha in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar] [PubMed]
- Audrito, V.; Managò, A.; Gaudino, F.; Deaglio, S. Targeting metabolic reprogramming in metastatic melanoma: The key role of nicotinamide phosphoribosyltransferase (NAMPT). Semin. Cell Dev. Biol. 2020, 98, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Long, G.V.; Eroglu, Z.; Infante, J.; Patel, S.; Daud, A.; Johnson, D.B.; Gonzalez, R.; Kefford, R.; Hamid, O.; Schuchter, L.; et al. Long-term outcomes in patients with BRAF V600-mutant metastatic melanoma who received dabrafenib combined with trametinib. J. Clin. Oncol. 2018, 36, 667–673. [Google Scholar] [CrossRef]
- Pisanu, M.E.; Maugeri-Saccà, M.; Fattore, L.; Bruschini, S.; De Vitis, C.; Tabbì, E.; Bellei, B.; Migliano, E.; Kovacs, D.; Camera, E.; et al. Inhibition of Stearoyl-CoA desaturase 1 reverts BRAF and MEK inhibition-induced selection of cancer stem cells in BRAF-mutated melanoma. J. Exp. Clin. Cancer Res. 2018, 37, 318. [Google Scholar] [CrossRef] [Green Version]
- Stamatakos, S.; Beretta, G.L.; Vergani, E.; Dugo, M.; Corno, C.; Corna, E.; Tinelli, S.; Frigerio, S.; Ciusani, E.; Rodolfo, M.; et al. Deregulated FASN Expression in BRAF Inhibitor-Resistant Melanoma Cells Unveils New Targets for Drug Combinations. Cancers 2021, 13, 2284. [Google Scholar] [CrossRef]
- Vergani, E.; Beretta, G.; Aloisi, M.; Costantino, M.; Corno, C.; Frigerio, S.; Tinelli, S.; Dugo, M.; Accattatis, F.M.; Granata, A.; et al. Targeting of the Lipid Metabolism Impairs Resistance to BRAF Kinase Inhibitor in Melanoma. Front. Cell Dev. Biol. 2022, 10, 927118. [Google Scholar] [CrossRef]
- Zhang, G.; Frederick, D.T.; Wu, L.; Wei, Z.; Krepler, C.; Srinivasan, S.; Chae, Y.C.; Xu, X.; Choi, H.; Dimwamwa, E.; et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J. Clin. Investig. 2016, 126, 1834–1856. [Google Scholar] [CrossRef]
- Ruocco, M.R.; Avagliano, A.; Granato, G.; Vigliar, E.; Masone, S.; Montagnani, S.; Arcucci, A. Metabolic flexibility in melanoma: A potential therapeutic target. Semin. Cancer Biol. 2019, 59, 187–207. [Google Scholar] [CrossRef]
- Tabolacci, C.; Giordano, D.; Rossi, S.; Cordella, M.; D’Arcangelo, D.; Moschella, F.; D’Atri, S.; Biffoni, M.; Facchiano, A.; Facchiano, F. Identification of Dihydrolipoamide Dehydrogenase as Potential Target of Vemurafenib-Resistant Melanoma Cells. Molecules 2022, 27, 7800. [Google Scholar] [CrossRef] [PubMed]
- Falletta, P.; Goding, C.R.; Vivas-García, Y. Connecting Metabolic Rewiring With Phenotype Switching in Melanoma. Front. Cell Dev. Biol. 2022, 10, 930250. [Google Scholar] [CrossRef]
- Talebi, A.; Dehairs, J.; Rambow, F.; Rogiers, A.; Nittner, D.; Derua, R.; Vanderhoydonc, F.; Duarte, J.A.G.; Bosisio, F.; Van den Eynde, K.; et al. Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat. Commun. 2018, 9, 2500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, Y.; Zhao, B.; Zhou, L.; Zhang, Z.; Shen, Y.; Lv, H.; Hameed AlQudsy, L.H.; Shang, P. Ferroptosis, a novel pharmacological mechanism of anti-cancer drugs. Cancer Lett. 2020, 483, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.; Reid, M.A.; Lowman, X.H.; Kulkarni, R.P.; Tran, T.Q.; Liu, X.; Yang, Y.; Hernandez-Davies, J.E.; Rosales, K.K.; Li, H.; et al. Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat. Cell Biol. 2016, 18, 1090–1101. [Google Scholar] [CrossRef]
- Rubanov, A.; Berico, P.; Hernando, E. Epigenetic Mechanisms Underlying Melanoma Resistance to Immune and Targeted Therapies. Cancers 2022, 14, 5858. [Google Scholar] [CrossRef] [PubMed]
- Ohanna, M.; Bonet, C.; Bille, K.; Allegra, M.; Davidson, I.; Bahadoran, P.; Lacour, J.; Ballotti, R.; Bertolotto, C. SIRT1 promotes proliferation and inhibits the senescence-like phenotype in human melanoma cells. Oncotarget 2014, 5, 2085–2095. [Google Scholar] [CrossRef] [Green Version]
- Bajpe, P.K.; Prahallad, A.; Horlings, H.; Nagtegaal, I.; Beijersbergen, R.; Bernards, R. A chromatin modifier genetic screen identifies SIRT2 as a modulator of response to targeted therapies through the regulation of MEK kinase activity. Oncogene 2014, 34, 531–536. [Google Scholar] [CrossRef]
- Strub, T.; Ghiraldini, F.G.; Carcamo, S.; Li, M.; Wroblewska, A.; Singh, R.; Goldberg, M.S.; Hasson, D.; Wang, Z.; Gallagher, S.J.; et al. SIRT6 haploinsufficiency induces BRAFV600E melanoma cell resistance to MAPK inhibitors via IGF signalling. Nat. Commun. 2018, 9, 3440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strub, T.; Ballotti, R.; Bertolotto, C. The “ART” of Epigenetics in Melanoma: From histone “Alterations, to Resistance and Therapies”. Theranostics 2020, 10, 1777–1797. [Google Scholar] [CrossRef] [PubMed]
- Zecena, H.; Tveit, D.; Wang, Z.; Farhat, A.; Panchal, P.; Liu, J.; Singh, S.J.; Sanghera, A.; Bainiwal, A.; Teo, S.Y.; et al. Systems biology analysis of mitogen activated protein kinase inhibitor resistance in malignant melanoma. BMC Syst. Biol. 2018, 12, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Gorain, M.; Kundu, G.; Kundu, G.C. Therapeutic implications of cellular and molecular biology of cancer stem cells in melanoma. Mol. Cancer 2017, 16, 7. [Google Scholar] [CrossRef] [Green Version]
- Fisher, M.L.; Grun, D.; Adhikary, G.; Xu, W.; Eckert, R.L. Inhibition of YAP function overcomes BRAF inhibitor resistance in melanoma cancer stem cells. Oncotarget 2017, 8, 110257–110272. [Google Scholar] [CrossRef] [Green Version]
- Motti, M.L.; Minopoli, M.; Di Carluccio, G.; Ascierto, P.A.; Carriero, M.V. MicroRNAs as Key Players in Melanoma Cell Resistance to MAPK and Immune Checkpoint Inhibitors. Int. J. Mol. Sci. 2020, 21, 4544. [Google Scholar] [CrossRef]
- Varrone, F.; Caputo, E. The miRNAs Role in Melanoma and in Its Resistance to Therapy. Int. J. Mol. Sci. 2020, 21, 878. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Ahn, J.H.; Lee, M. Upregulation of MicroRNA-1246 Is Associated with BRAF Inhibitor Resistance in Melanoma Cells with Mutant BRAF. Cancer Res. Treat. 2017, 49, 947–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattore, L.; Ruggiero, C.F.; Pisanu, M.E.; Liguoro, D.; Cerri, A.; Costantini, S.; Capone, F.; Acunzo, M.; Romano, G.; Nigita, G.; et al. Reprogramming miRNAs global expression orchestrates development of drug resistance in BRAF mutated melanoma. Cell Death Differ. 2019, 26, 1267–1282. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Martínez, M.; Benito-Jardón, L.; Alonso, L.; Koetz-Ploch, L.; Hernando, E.; Teixidó, J. miR-204-5p and miR-211-5p Contribute to BRAF Inhibitor Resistance in Melanoma. Cancer Res. 2018, 78, 1017–1030. [Google Scholar] [CrossRef] [Green Version]
- Lazăr, A.D.; Dinescu, S.; Costache, M. The Non-Coding Landscape of Cutaneous Malignant Melanoma: A Possible Route to Efficient Targeted Therapy. Cancers 2020, 12, 3378. [Google Scholar] [CrossRef] [PubMed]
- Sanlorenzo, M.; Vujic, I.; Esteve-Puig, R.; Lai, K.; Vujic, M.; Lin, K.; Posch, C.; Dimon, M.; Moy, A.; Zekhtser, M.; et al. The lincRNA MIRAT binds to IQGAP1 and modulates the MAPK pathway in NRAS mutant melanoma. Sci. Rep. 2018, 8, 10902. [Google Scholar] [CrossRef] [Green Version]
- Joung, J.; Engreitz, J.M.; Konermann, S.; Abudayyeh, O.O.; Verdine, V.K.; Aguet, F.; Gootenberg, J.S.; Sanjana, N.E.; Wright, J.B.; Fulco, C.P.; et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature 2017, 548, 343–346. [Google Scholar] [CrossRef] [Green Version]
- Leucci, E.; Vendramin, R.; Spinazzi, M.; Laurette, P.; Fiers, M.; Wouters, J.; Radaelli, E.; Eyckerman, S.; Leonelli, C.; Vanderheyden, K.; et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature 2016, 531, 518–522. [Google Scholar] [CrossRef]
- Gambi, G.; Mengus, G.; Davidson, G.; Demesmaeker, E.; Cuomo, A.; Bonaldi, T.; Katopodi, V.; Malouf, G.G.; Leucci, E.; Davidson, I. The LncRNA LENOX Interacts with RAP2C to Regulate Metabolism and Promote Resistance to MAPK Inhibition in Melanoma. Cancer Res. 2022, 82, 4555–4570. [Google Scholar] [CrossRef]
- Palmer, S.R.; Erickson, L.A.; Ichetovkin, I.; Knauer, D.J.; Markovic, S.N. Circulating serologic and molecular biomarkers in malignant melanoma. Mayo Clin. Proc. 2011, 86, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gassenmaier, M.; Lenders, M.M.; Forschner, A.; Leiter, U.; Weide, B.; Garbe, C.; Eigentler, T.K.; Wagner, N.B. Serum S100B and LDH at Baseline and During Therapy Predict the Outcome of Metastatic Melanoma Patients Treated with BRAF Inhibitors. Target Oncol. 2021, 16, 197–205. [Google Scholar] [CrossRef]
- Ene, C.D.; Anghel, A.E.; Neagu, M.; Nicolae, I. 25-OH Vitamin D and Interleukin-8: Emerging Biomarkers in Cutaneous Melanoma Development and Progression. Mediat. Inflamm. 2015, 2015, 904876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichrath, J.; Biersack, F.; Wagenpfeil, S.; Schöpe, J.; Pföhler, C.; Saternus, R.; Vogt, T. Low Vitamin D Status Predicts Poor Clinical Outcome in Advanced Melanoma Treated With Immune Checkpoint or BRAF/MEK Inhibitors: A Prospective Non-Interventional Side-by-Side Analysis. Front. Oncol. 2022, 12, 839816. [Google Scholar] [CrossRef] [PubMed]
- De Falco, V.; Napolitano, S.; Esposito, D.; Guerrera, L.P.; Ciardiello, D.; Formisano, L.; Troiani, T. Comprehensive Review on the Clinical Relevance of Long Non-Coding RNAs in Cutaneous Melanoma. Int. J. Mol. Sci. 2021, 22, 1166. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, T.; Doss C., G. P. Non-coding RNAs in human health and disease: Potential function as biomarkers and therapeutic targets. Funct. Integr. Genom. 2023, 23, 33. [Google Scholar] [CrossRef] [PubMed]
- Kolenda, T.; Rutkowski, P.; Michalak, M.; Kozak, K.; Guglas, K.; Ryś, M.; Galus, Ł.; Woźniak, S.; Ługowska, I.; Gos, A.; et al. Plasma lncRNA expression profile as a prognostic tool in BRAF-mutant metastatic melanoma patients treated with BRAF inhibitor. Oncotarget 2019, 10, 3879–3893. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Tetzlaff, M.T.; Wang, T.; Yang, R.; Xie, L.; Zhang, G.; Krepler, C.; Xiao, M.; Beqiri, M.; Xu, W.; et al. MiR-200c/Bmi1 axis and epithelial–mesenchymal transition contribute to acquired resistance to BRAF inhibitor treatment. Pigment Cell Melanoma Res. 2015, 28, 431–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fattore, L.; Mancini, R.; Acunzo, M.; Romano, G.; Laganà, A.; Pisanu, M.E.; Malpicci, D.; Madonna, G.; Mallardo, D.; Capone, M.; et al. MiR-579-3p controls melanoma progression and resistance to target therapy. Proc. Natl. Acad. Sci. USA 2016, 113, E5005–E5013. [Google Scholar] [CrossRef] [PubMed]
- Koetz-Ploch, L.; Hanniford, D.; Dolgalev, I.; Sokolova, E.; Zhong, J.; Díaz-Martínez, M.; Bernstein, E.; Darvishian, F.; Flaherty, K.T.; Chapman, P.B.; et al. MicroRNA-125a promotes resistance to BRAF inhibitors through suppression of the intrinsic apoptotic pathway. Pigment Cell Melanoma Res. 2017, 30, 328–338. [Google Scholar] [CrossRef] [Green Version]
- Barbato, A.; Iuliano, A.; Volpe, M.; D’Alterio, R.; Brillante, S.; Massa, F.; De Cegli, R.; Carrella, S.; Salati, M.; Russo, A.; et al. Integrated Genomics Identifies miR-181/TFAM Pathway as a Critical Driver of Drug Resistance in Melanoma. Int. J. Mol. Sci. 2021, 22, 1801. [Google Scholar] [CrossRef]
- Svedman, F.C.; Lohcharoenkal, W.; Bottai, M.; Brage, S.E.; Sonkoly, E.; Hansson, J.; Pivarcsi, A.; Eriksson, H. Extracellular microvesicle microRNAs as predictive biomarkers for targeted therapy in metastastic cutaneous malignant melanoma. PLoS ONE 2018, 13, e0206942. [Google Scholar] [CrossRef] [Green Version]
- Levati, L.; Bassi, C.; Mastroeni, S.; Lupini, L.; Antonini Cappellini, G.C.; Bonmassar, L.; Alvino, E.; Caporali, S.; Lacal, P.M.; Narducci, M.G.; et al. Circulating miR-1246 and miR-485-3p as Promising Biomarkers of Clinical Response and Outcome in Melanoma Patients Treated with Targeted Therapy. Cancers 2022, 14, 3706. [Google Scholar] [CrossRef]
- Ruggiero, C.F.; Fattore, L.; Terrenato, I.; Sperati, F.; Salvati, V.; Madonna, G.; Capone, M.; Valenti, F.; Di Martino, S.; Mandoj, C.; et al. Identification of a miRNA-based non-invasive predictive biomarker of response to target therapy in BRAF-mutant melanoma. Theranostics 2022, 12, 7420–7430. [Google Scholar] [CrossRef]
- Calapre, L.; Warburton, L.; Millward, M.; Ziman, M.; Gray, E.S. Circulating tumour DNA (ctDNA) as a liquid biopsy for melanoma. Cancer Lett. 2017, 404, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Minor, D.; Ribas, A.; Lebbe, C.; O’Hagan, A.; Arya, N.; Guckert, M.; Schadendorf, D.; Kefford, R.F.; Grob, J.-J.; et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 3205–3211. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Rizos, H.; Reid, A.L.; Boyd, S.C.; Pereira, M.R.; Lo, J.; Tembe, V.; Freeman, J.; Lee, J.H.J.; Scolyer, R.A.; et al. Circulating tumor DNA to monitor treatment response and detect acquired resistance in patients with metastatic melanoma. Oncotarget 2015, 6, 42008–42018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanmamed, M.F.; Fernández-Landázuri, S.; Rodríguez, C.; Zárate, R.; Lozano, M.D.; Zubiri, L.; Perez-Gracia, J.L.; Martín-Algarra, S.; González, A. Quantitative cell-free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin. Chem. 2015, 61, 297–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santiago-Walker, A.; Gagnon, R.; Mazumdar, J.; Casey, M.; Long, G.V.; Schadendorf, D.; Flaherty, K.; Kefford, R.; Hauschild, A.; Hwu, P.; et al. Correlation of BRAF mutation status in circulating-free DNA and tumor and association with clinical outcome across four BRAFi and MEKi clinical trials. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Schreuer, M.; Meersseman, G.; Van Den Herrewegen, S.; Jansen, Y.; Chevolet, I.; Bott, A.; Wilgenhof, S.; Seremet, T.; Jacobs, B.; Buyl, R.; et al. Quantitative Assessment of BRAF V600 mutant circulating cell-free tumor DNA as a tool for therapeutic monitoring in metastatic melanoma patients treated with BRAF/MEK inhibitors. J. Transl. Med. 2016, 14, 95. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.J.; Gremel, G.; Marshall, A.; Myers, K.A.; Fisher, N.; Dunn, J.A.; Dhomen, N.; Corrie, P.G.; Middleton, M.R.; Lorigan, P.; et al. Circulating tumor DNA predicts survival in patients with resected high-risk stage II/III melanoma. Ann. Oncol. 2018, 29, 490–496. [Google Scholar] [CrossRef] [Green Version]
- Braune, J.; Keller, L.; Schiller, F.; Graf, E.; Rafei-Shamsabadi, D.; Wehrle, J.; Follo, M.; Philipp, U.; Hussung, S.; Pfeifer, D.; et al. Circulating tumor DNA allows early treatment monitoring in BRAF- and NRAS-mutant malignant melanoma. JCO Precis. Oncol. 2020, 4, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Syeda, M.M.; Wiggins, J.M.; Corless, B.C.; Long, G.V.; Flaherty, K.T.; Schadendorf, D.; Nathan, P.D.; Robert, C.; Ribas, A.; Davies, M.A.; et al. Circulating tumour DNA in patients with advanced melanoma treated with dabrafenib or dabrafenib plus trametinib: A clinical validation study. Lancet Oncol. 2021, 22, 370–380. [Google Scholar] [CrossRef]
- Lee, R.; Rothwell, D.G.; Chow, S.; Shaw, H.M.; Turajlic, S.; Smith, N.; Clipson, A.; Clarke, H.; Kelso, N.; Mitchell, J.; et al. CAcTUS: A parallel arm, biomarker driven, phase II feasibility trial to determine the role of circulating tumor DNA in guiding a switch between targeted therapy and immune therapy in patients with advanced cutaneous melanoma. J. Clin. Oncol. 2021, 39, TPS9587. [Google Scholar] [CrossRef]
- García-Silva, S.; Benito-Martín, A.; Sánchez-Redondo, S.; Hernández-Barranco, A.; Ximénez-Embún, P.; Nogués, L.; Mazariegos, M.S.; Brinkmann, K.; Amor López, A.; Meyer, L.; et al. Use of extracellular vesicles from lymphatic drainage as surrogate markers of melanoma progression and BRAF V600E mutation. J. Exp. Med. 2019, 216, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Zocco, D.; Bernardi, S.; Novelli, M.; Astrua, C.; Fava, P.; Zarovni, N.; Carpi, F.M.; Bianciardi, L.; Malavenda, O.; Quaglino, P.; et al. Isolation of extracellular vesicles improves the detection of mutant DNA from plasma of metastatic melanoma patients. Sci. Rep. 2020, 10, 15745. [Google Scholar] [CrossRef] [PubMed]
- Gray, E.S.; Reid, A.L.; Bowyer, S.; Calapre, L.; Siew, K.; Pearce, R.; Cowell, L.; Frank, M.H.; Millward, M.; Ziman, M. Circulating melanoma cell subpopulations: Their heterogeneity and differential responses to treatment. J. Investig. Dermatol. 2015, 135, 2040–2048. [Google Scholar] [CrossRef] [Green Version]
- Girotti, M.R.; Gremel, G.; Lee, R.; Galvani, E.; Rothwell, D.; Viros, A.; Mandal, A.K.; Lim, K.H.J.; Saturno, G.; Furney, S.J.; et al. Application of sequencing, liquid biopsies, and patient-derived xenografts for personalized medicine in melanoma. Cancer Discov. 2016, 6, 286–299. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.Q.; Raleigh, J.M.; Callahan, J.; Vergara, I.A.; Ftouni, S.; Hatzimihalis, A.; Colebatch, A.J.; Li, J.; Semple, T.; Doig, K.; et al. Circulating tumor DNA analysis and functional imaging provide complementary approaches for comprehensive disease monitoring in metastatic melanoma. JCO Precis. Oncol. 2017, 1, 1–14. [Google Scholar] [CrossRef]
- Khoja, L.; Lorigan, P.; Zhou, C.; Lancashire, M.; Booth, J.; Cummings, J.; Califano, R.; Clack, G.; Hughes, A.; Dive, C. Biomarker utility of circulating tumor cells in metastatic cutaneous melanoma. J. Investig. Dermatol. 2013, 133, 1582–1590. [Google Scholar] [CrossRef] [Green Version]
- Klinac, D.; Gray, E.S.; Freeman, J.B.; Reid, A.; Bowyer, S.; Millward, M.; Ziman, M. Monitoring changes in circulating tumour cells as a prognostic indicator of overall survival and treatment response in patients with metastatic melanoma. BMC Cancer 2014, 14, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiniwa, Y.; Nakamura, K.; Mikoshiba, A.; Ashida, A.; Akiyama, J.; Morimoto, A.; Okuyama, R. Usefulness of monitoring circulating tumor cells as a therapeutic biomarker in melanoma with BRAF mutation. BMC Cancer 2021, 21, 287. [Google Scholar] [CrossRef]
- Kamińska, P.; Buszka, K.; Zabel, M.; Nowicki, M.; Alix-Panabières, C.; Budna-Tukan, J. Liquid biopsy in melanoma: Significance in diagnostics, prediction and treatment monitoring. Int. J. Mol. Sci. 2021, 22, 9714. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BRAF Variant | Frequency in Melanoma | Aminoacid Change |
|---|---|---|
| p.V600E, c.1799 T>A | 70–88% | valine to glutamate |
| p.V600K, c.1798_1799delGTinsAA | 10–20% | valine to lysine |
| p.V600R, c.1798_1799delGTinsAG | <5% | valine to arginine |
| p.V600D, c.1799_1800delTGinsAC | <5% | valine to aspartate |
| p.V600E2, c.1799_1800delTCinsAA | <1% | valine to glutamate |
| p.V600M, c.1798G>A | <1% | valine to methionine |
| p.V600G, c.1799T>G | <1% | valine to glycine |
| p.K601E, c.1801A>G | <1% | lysine to glutamate |
| p.D594N, c.1780G>A | <1% | aspartate to asparagine |
| Non-Coding RNAs | Sample Type | References |
|---|---|---|
| AIR Zeb2NAT Zfas1 7SL | Plasma | [203] |
| miR-200c | Tumour tissue | [204] |
| miR-579-3p | Tumour tissue | [205] |
| miR-125a | Tumour tissue | [206] |
| miR-181a miR-181b | Tumour tissue | [207] |
| miR-4443 miR-4488 miR-204-5p miR-199-5p | Tumour tissue Plasma | [190] |
| miR-497-5p let-7g-5p | Plasma | [208] |
| miR-1246 miR-485-3p | Plasma | [209] |
| miR-579-3p miR-4888 | Serum | [210] |
| NCT No. | Title | Status |
|---|---|---|
| NCT04866680 | Personalized Circulating DNA Follow-up in Melanoma (PERCIMEL) | Recruiting |
| NCT04720768 | Encorafenib, Binimetinib, and Palbociclib in BRAFmutant Metastatic Melanoma CELEBRATE | Recruiting |
| NCT03808441 | CAcTUS—Circulating Tumour DNA Guided Switch | Recruiting |
| NCT03754179 | Dabrafenib/Trametinib/Hydroxychloroquine for Advanced Pretreated BRAFV600 Mutant Melanoma | Unknown |
| NCT03416933 | Therapeutic Drug Monitoring of BRAF-mutated Advanced Melanoma | Active, not recruiting |
| NCT03415126 | A Study of ASN007 in Patients With Advanced Solid Tumours | Completed |
| NCT02862743 | Molecular Characterization of Advanced Stage Melanoma by Blood Sampling | Completed |
| NCT02858921 | Neoadjuvant Dabrafenib, Trametinib, and/or Pembrolizumab in BRAF Mutant Resectable Stage III Melanoma | Active, not recruiting |
| NCT02700763 | [18F]Dabrafenib Molecular Imaging in Melanoma Brain Metastasis | Terminated |
| NCT02537600 | Vemurafenib and Cobimetinib Combination in BRAF Mutated Melanoma With Brain Metastasis | Completed |
| NCT02251314 | Use of Exome Sequence Analysis and Circulating Tumour in Assessing Tumour Heterogeneity in BRAF Mutant Melanoma | Completed |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castellani, G.; Buccarelli, M.; Arasi, M.B.; Rossi, S.; Pisanu, M.E.; Bellenghi, M.; Lintas, C.; Tabolacci, C. BRAF Mutations in Melanoma: Biological Aspects, Therapeutic Implications, and Circulating Biomarkers. Cancers 2023, 15, 4026. https://doi.org/10.3390/cancers15164026
Castellani G, Buccarelli M, Arasi MB, Rossi S, Pisanu ME, Bellenghi M, Lintas C, Tabolacci C. BRAF Mutations in Melanoma: Biological Aspects, Therapeutic Implications, and Circulating Biomarkers. Cancers. 2023; 15(16):4026. https://doi.org/10.3390/cancers15164026
Chicago/Turabian StyleCastellani, Giorgia, Mariachiara Buccarelli, Maria Beatrice Arasi, Stefania Rossi, Maria Elena Pisanu, Maria Bellenghi, Carla Lintas, and Claudio Tabolacci. 2023. "BRAF Mutations in Melanoma: Biological Aspects, Therapeutic Implications, and Circulating Biomarkers" Cancers 15, no. 16: 4026. https://doi.org/10.3390/cancers15164026
APA StyleCastellani, G., Buccarelli, M., Arasi, M. B., Rossi, S., Pisanu, M. E., Bellenghi, M., Lintas, C., & Tabolacci, C. (2023). BRAF Mutations in Melanoma: Biological Aspects, Therapeutic Implications, and Circulating Biomarkers. Cancers, 15(16), 4026. https://doi.org/10.3390/cancers15164026