
Recent Advances in Renal Tumors with TSC/mTOR Pathway Abnormalities in Patients with Tuberous Sclerosis Complex and in the Sporadic Setting


Abstract
:Simple Summary
Abstract
1. Introduction
2. Tuberous Sclerosis Complex Syndrome
3. Renal Tumors with TSC/mTOR Pathway Gene Alterations and Their Sporadic Counterparts
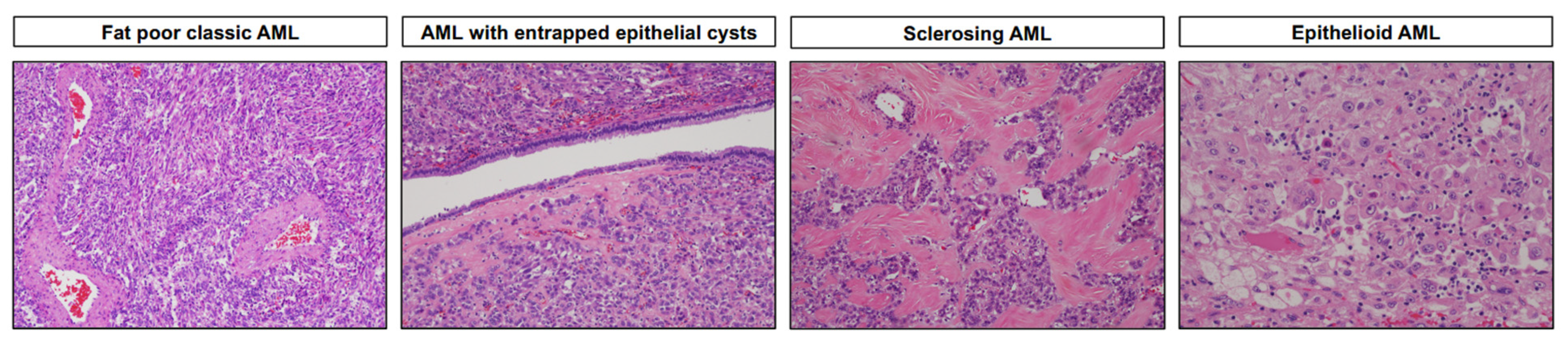
3.1. Angiomyolipoma and Variants
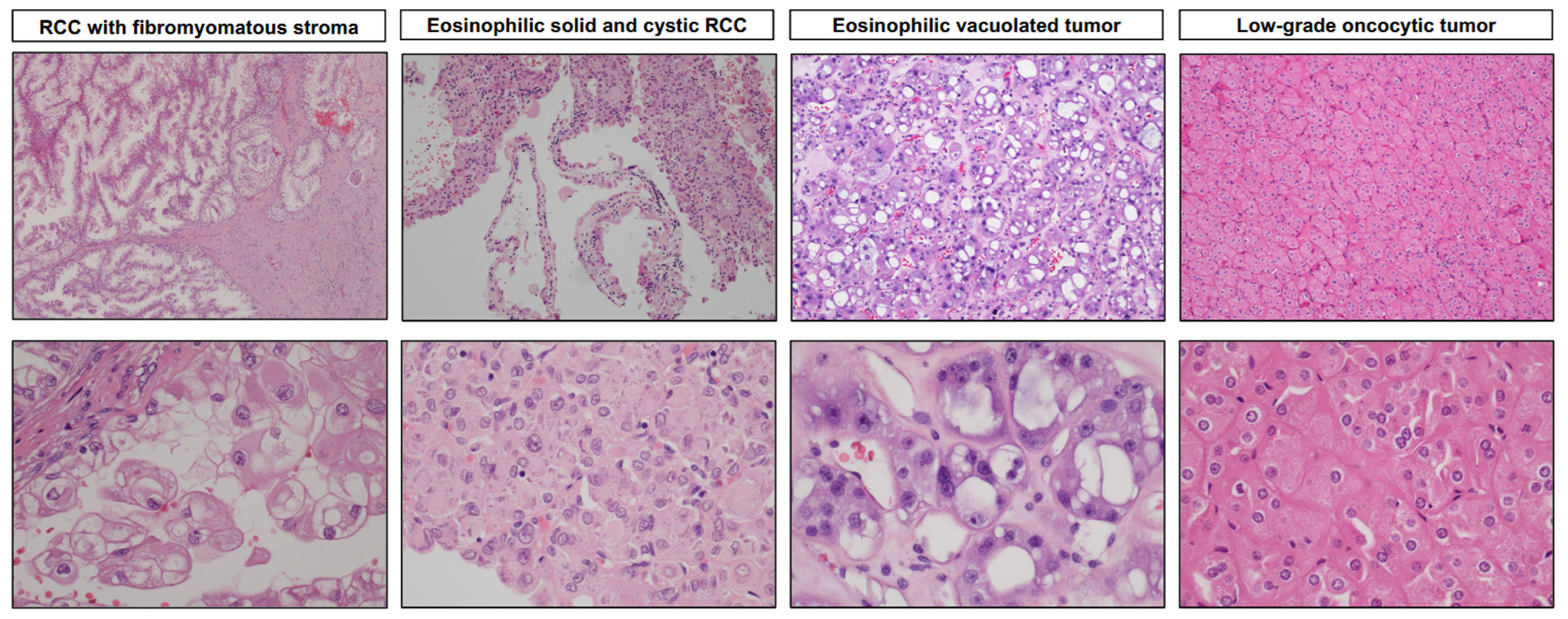
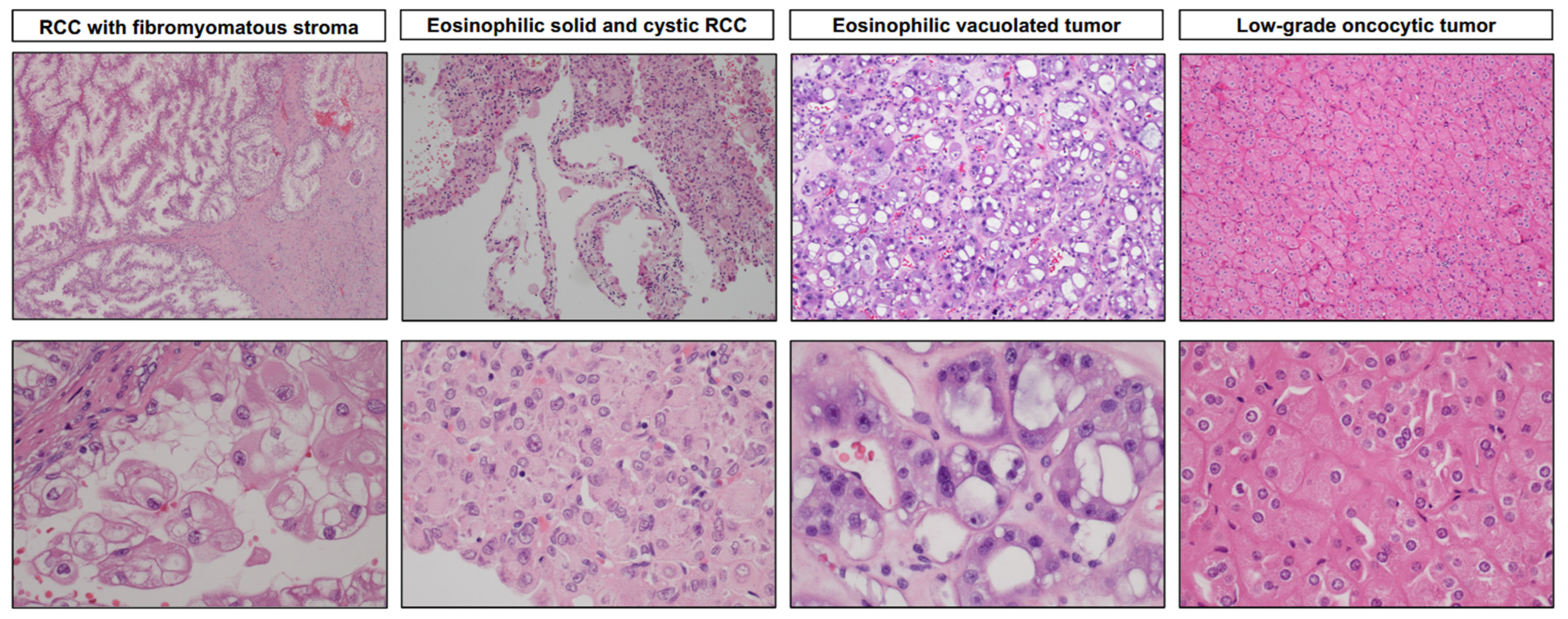
3.2. Renal Cell Carcinoma with Fibromyomatous Stroma (RCC FMS)
3.3. Eosinophilic Solid and Cystic Renal Cell Carcinoma (ESC RCC)
3.4. Eosinophilic Vacuolated Tumor (EVT)
3.5. Low-Grade Oncocytic Tumor (LOT)
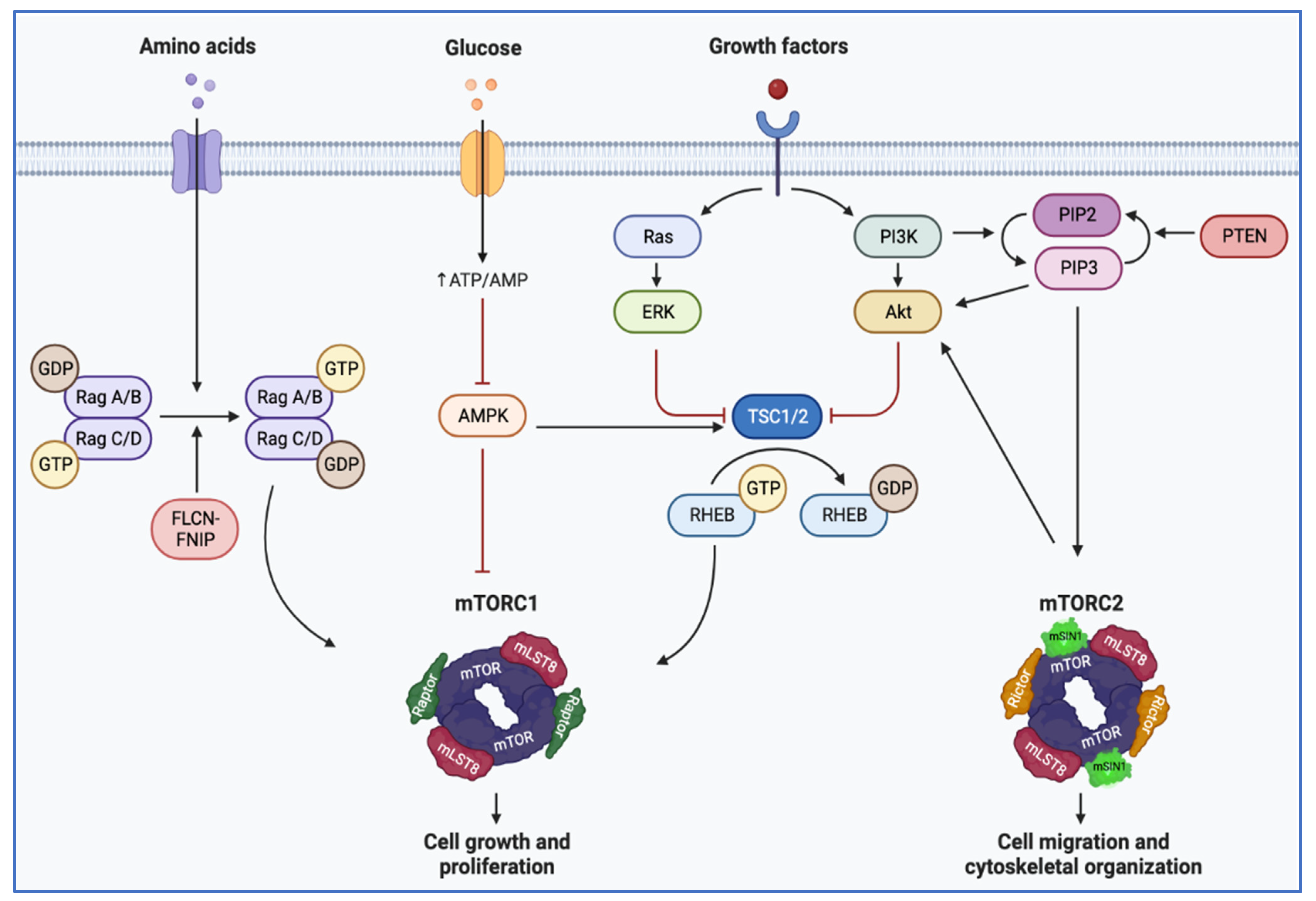
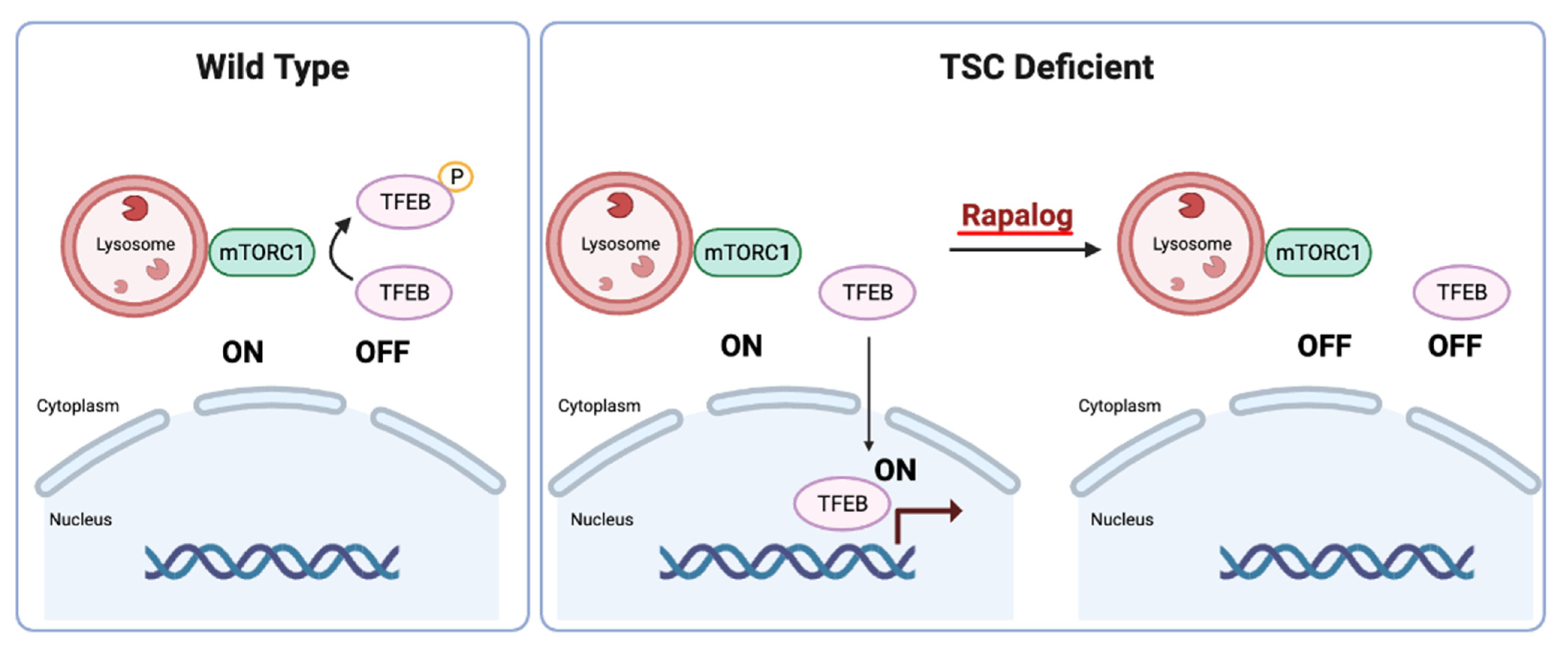
4. Convergence on Mechanistic Target of Rapamycin (mTOR) Complex 1 Pathway
5. Clinical Management and Implications
6. TSC/MTOR Pathway Activated Renal Tumors—Lessons Learned and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Guo, J.; Tretiakova, M.S.; Troxell, M.L.; Osunkoya, A.O.; Fadare, O.; Sangoi, A.R.; Shen, S.S.; Lopez-Beltran, A.; Mehra, R.; Heider, A.; et al. Tuberous Sclerosis-associated Renal Cell Carcinoma: A Clinicopathologic Study of 57 Separate Carcinomas in 18 Patients. Am. J. Surg. Pathol. 2014, 38, 1457–1467. [Google Scholar] [CrossRef]
- Yang, P.; Cornejo, K.M.; Sadow, P.M.; Cheng, L.; Wang, M.; Xiao, Y.; Jiang, Z.; Oliva, E.; Jozwiak, S.; Nussbaum, R.L.; et al. Renal cell carcinoma in tuberous sclerosis complex. Am. J. Surg. Pathol. 2014, 38, 895–909. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Curatolo, P.; Bombardieri, R.; Jozwiak, S. Tuberous sclerosis. Lancet 2008, 372, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Northrup, H.; Krueger, D.A.; International Tuberous Sclerosis Complex Consensus, G. Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr. Neurol. 2013, 49, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northrup, H.; Aronow, M.E.; Bebin, E.M.; Bissler, J.; Darling, T.N.; de Vries, P.J.; Frost, M.D.; Fuchs, Z.; Gosnell, E.S.; Gupta, N.; et al. Updated International Tuberous Sclerosis Complex Diagnostic Criteria and Surveillance and Management Recommendations. Pediatr. Neurol. 2021, 123, 50–66. [Google Scholar] [CrossRef]
- Crino, P.B.; Nathanson, K.L.; Henske, E.P. The tuberous sclerosis complex. N. Engl. J. Med. 2006, 355, 1345–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborne, J.P.; Fryer, A.; Webb, D. Epidemiology of tuberous sclerosis. Ann. N. Y. Acad. Sci. 1991, 615, 125–127. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, F.J.; Noakes, M.J.; Martyn, C.N.; Osborne, J.P. An epidemiological study of renal pathology in tuberous sclerosis complex. BJU Int. 2004, 94, 853–857. [Google Scholar] [CrossRef]
- Cheadle, J.P.; Reeve, M.P.; Sampson, J.R.; Kwiatkowski, D.J. Molecular genetic advances in tuberous sclerosis. Hum. Genet. 2000, 107, 97–114. [Google Scholar] [CrossRef]
- Van Slegtenhorst, M.; de Hoogt, R.; Hermans, C.; Nellist, M.; Janssen, B.; Verhoef, S.; Lindhout, D.; van den Ouweland, A.; Halley, D.; Young, J.; et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997, 277, 805–808. [Google Scholar] [CrossRef]
- Tyburczy, M.E.; Jozwiak, S.; Malinowska, I.A.; Chekaluk, Y.; Pugh, T.J.; Wu, C.L.; Nussbaum, R.L.; Seepo, S.; Dzik, T.; Kotulska, K.; et al. A shower of second hit events as the cause of multifocal renal cell carcinoma in tuberous sclerosis complex. Hum. Mol. Genet. 2015, 24, 1836–1842. [Google Scholar] [CrossRef] [Green Version]
- Sancak, O.; Nellist, M.; Goedbloed, M.; Elfferich, P.; Wouters, C.; Maat-Kievit, A.; Zonnenberg, B.; Verhoef, S.; Halley, D.; van den Ouweland, A. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: Genotype--phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur. J. Hum. Genet. 2005, 13, 731–741. [Google Scholar] [CrossRef] [Green Version]
- Jones, A.C.; Daniells, C.E.; Snell, R.G.; Tachataki, M.; Idziaszczyk, S.A.; Krawczak, M.; Sampson, J.R.; Cheadle, J.P. Molecular genetic and phenotypic analysis reveals differences between TSC1 and TSC2 associated familial and sporadic tuberous sclerosis. Hum. Mol. Genet. 1997, 6, 2155–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabora, S.L.; Jozwiak, S.; Franz, D.N.; Roberts, P.S.; Nieto, A.; Chung, J.; Choy, Y.S.; Reeve, M.P.; Thiele, E.; Egelhoff, J.C.; et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 2001, 68, 64–80. [Google Scholar] [CrossRef] [Green Version]
- Osborne, J.P.; Jones, A.C.; Burley, M.W.; Jeganathan, D.; Young, J.; O’Callaghan, F.J.; Sampson, J.R.; Povey, S. Non-penetrance in tuberous sclerosis. Lancet 2000, 355, 1698. [Google Scholar] [CrossRef]
- Henske, E.P.; Jozwiak, S.; Kingswood, J.C.; Sampson, J.R.; Thiele, E.A. Tuberous sclerosis complex. Nat. Rev. Dis. Primers 2016, 2, 16035. [Google Scholar] [CrossRef]
- Tsai, P.T.; Hull, C.; Chu, Y.; Greene-Colozzi, E.; Sadowski, A.R.; Leech, J.M.; Steinberg, J.; Crawley, J.N.; Regehr, W.G.; Sahin, M. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature 2012, 488, 647–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henske, E.P.; Neumann, H.P.; Scheithauer, B.W.; Herbst, E.W.; Short, M.P.; Kwiatkowski, D.J. Loss of heterozygosity in the tuberous sclerosis (TSC2) region of chromosome band 16p13 occurs in sporadic as well as TSC-associated renal angiomyolipomas. Genes Chromosomes Cancer 1995, 13, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Ewalt, D.H.; Sheffield, E.; Sparagana, S.P.; Delgado, M.R.; Roach, E.S. Renal lesion growth in children with tuberous sclerosis complex. J. Urol. 1998, 160, 141–145. [Google Scholar] [CrossRef]
- Rakowski, S.K.; Winterkorn, E.B.; Paul, E.; Steele, D.J.; Halpern, E.F.; Thiele, E.A. Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors. Kidney Int. 2006, 70, 1777–1782. [Google Scholar] [CrossRef] [Green Version]
- Kingswood, J.C.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; Dahlin, M.; D’Amato, L.; d’Augeres, G.B.; de Vries, P.J.; et al. Renal angiomyolipoma in patients with tuberous sclerosis complex: Findings from the TuberOus SClerosis registry to increase disease Awareness. Nephrol. Dial. Transpl. 2019, 34, 502–508. [Google Scholar] [CrossRef] [Green Version]
- Kingswood, J.C.; Belousova, E.; Benedik, M.P.; Carter, T.; Cottin, V.; Curatolo, P.; Dahlin, M.; D’Amato, L.; Beaure d’Augeres, G.; de Vries, P.J.; et al. Renal Manifestations of Tuberous Sclerosis Complex: Key Findings From the Final Analysis of the TOSCA Study Focussing Mainly on Renal Angiomyolipomas. Front. Neurol. 2020, 11, 972. [Google Scholar] [CrossRef]
- Gupta, S.; Jimenez, R.E.; Herrera-Hernandez, L.; Lohse, C.M.; Thompson, R.H.; Boorjian, S.A.; Leibovich, B.C.; Cheville, J.C. Renal Neoplasia in Tuberous Sclerosis: A Study of 41 Patients. Mayo Clin. Proc. 2021, 96, 1470–1489. [Google Scholar] [CrossRef]
- Brook-Carter, P.T.; Peral, B.; Ward, C.J.; Thompson, P.; Hughes, J.; Maheshwar, M.M.; Nellist, M.; Gamble, V.; Harris, P.C.; Sampson, J.R. Deletion of the TSC2 and PKD1 genes associated with severe infantile polycystic kidney disease--a contiguous gene syndrome. Nat. Genet. 1994, 8, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Stanton, M.L.; Reynolds, J.P.; Whaley, R.D.; Herrera-Hernandez, L.; Jimenez, R.E.; Cheville, J.C. Lessons from histopathologic examination of nephrectomy specimens in patients with tuberous sclerosis complex: Cysts, angiomyolipomas, and renal cell carcinoma. Hum. Pathol. 2022, 129, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Lohse, C.M.; Rowsey, R.; McCarthy, M.R.; Shen, W.; Herrera-Hernandez, L.; Boorjian, S.A.; Houston Thompson, R.; Jimenez, R.E.; Leibovich, B.C.; et al. Renal Neoplasia in Polycystic Kidney Disease: An Assessment of Tuberous Sclerosis Complex-associated Renal Neoplasia and PKD1/TSC2 Contiguous Gene Deletion Syndrome. Eur. Urol. 2022, 81, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Lam, H.C.; Siroky, B.J.; Henske, E.P. Renal disease in tuberous sclerosis complex: Pathogenesis and therapy. Nat. Rev. Nephrol. 2018, 14, 704–716. [Google Scholar] [CrossRef]
- Bjornsson, J.; Short, M.P.; Kwiatkowski, D.J.; Henske, E.P. Tuberous sclerosis-associated renal cell carcinoma. Clinical, pathological, and genetic features. Am. J. Pathol. 1996, 149, 1201–1208. [Google Scholar]
- Duffy, K.; Al-Saleem, T.; Karbowniczek, M.; Ewalt, D.; Prowse, A.H.; Henske, E.P. Mutational analysis of the von hippel lindau gene in clear cell renal carcinomas from tuberous sclerosis complex patients. Mod. Pathol. 2002, 15, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Schreiner, A.; Daneshmand, S.; Bayne, A.; Countryman, G.; Corless, C.L.; Troxell, M.L. Distinctive morphology of renal cell carcinomas in tuberous sclerosis. Int. J. Surg. Pathol. 2010, 18, 409–418. [Google Scholar] [CrossRef]
- Kucejova, B.; Pena-Llopis, S.; Yamasaki, T.; Sivanand, S.; Tran, T.A.; Alexander, S.; Wolff, N.C.; Lotan, Y.; Xie, X.J.; Kabbani, W.; et al. Interplay between pVHL and mTORC1 pathways in clear-cell renal cell carcinoma. Mol. Cancer Res. 2011, 9, 1255–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henske, E.P.; Cornejo, K.M.; Wu, C.L. Renal Cell Carcinoma in Tuberous Sclerosis Complex. Genes 2021, 12, 1585. [Google Scholar] [CrossRef] [PubMed]
- Trpkov, K.; Williamson, S.R.; Gill, A.J.; Adeniran, A.J.; Agaimy, A.; Alaghehbandan, R.; Amin, M.B.; Argani, P.; Chen, Y.B.; Cheng, L.; et al. Novel, emerging and provisional renal entities: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod. Pathol. 2021, 34, 1167–1184. [Google Scholar] [CrossRef] [PubMed]
- Raspolini, M.R.; Amin, M.B.; Moch, H.; Tan, P.H.; Turajlic, S. Renal cell tumours: Introduction. In WHO Classification of Tumours Editon. Urinary and Male Genital Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2022. [Google Scholar]
- Yamakado, K.; Tanaka, N.; Nakagawa, T.; Kobayashi, S.; Yanagawa, M.; Takeda, K. Renal angiomyolipoma: Relationships between tumor size, aneurysm formation, and rupture. Radiology 2002, 225, 78–82. [Google Scholar] [CrossRef]
- Wagner, B.J.; Wong-You-Cheong, J.J.; Davis, C.J., Jr. Adult renal hamartomas. Radiographics 1997, 17, 155–169. [Google Scholar] [CrossRef] [Green Version]
- Giannikou, K.; Malinowska, I.A.; Pugh, T.J.; Yan, R.; Tseng, Y.Y.; Oh, C.; Kim, J.; Tyburczy, M.E.; Chekaluk, Y.; Liu, Y.; et al. Whole Exome Sequencing Identifies TSC1/TSC2 Biallelic Loss as the Primary and Sufficient Driver Event for Renal Angiomyolipoma Development. PLoS Genet. 2016, 12, e1006242. [Google Scholar] [CrossRef] [Green Version]
- Aydin, H.; Magi-Galluzzi, C.; Lane, B.R.; Sercia, L.; Lopez, J.I.; Rini, B.I.; Zhou, M. Renal angiomyolipoma: Clinicopathologic study of 194 cases with emphasis on the epithelioid histology and tuberous sclerosis association. Am. J. Surg. Pathol. 2009, 33, 289–297. [Google Scholar] [CrossRef]
- He, W.; Cheville, J.C.; Sadow, P.M.; Gopalan, A.; Fine, S.W.; Al-Ahmadie, H.A.; Chen, Y.B.; Oliva, E.; Russo, P.; Reuter, V.E.; et al. Epithelioid angiomyolipoma of the kidney: Pathological features and clinical outcome in a series of consecutively resected tumors. Mod. Pathol. 2013, 26, 1355–1364. [Google Scholar] [CrossRef] [Green Version]
- Gournay, M.; Dugay, F.; Belaud-Rotureau, M.A.; Peyronnet, B.; Mathieu, R.; Verhoest, G.; Bensalah, K.; Odent, S.; Denizeau, P.; Vigneau, C.; et al. Renal cell carcinoma with leiomyomatous stroma in tuberous sclerosis complex: A distinct entity. Virchows Arch. 2021, 478, 793–799. [Google Scholar] [CrossRef]
- Nese, N.; Martignoni, G.; Fletcher, C.D.; Gupta, R.; Pan, C.C.; Kim, H.; Ro, J.Y.; Hwang, I.S.; Sato, K.; Bonetti, F.; et al. Pure epithelioid PEComas (so-called epithelioid angiomyolipoma) of the kidney: A clinicopathologic study of 41 cases: Detailed assessment of morphology and risk stratification. Am. J. Surg. Pathol. 2011, 35, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Trpkov, K.; Hes, O. New and emerging renal entities: A perspective post-WHO 2016 classification. Histopathology 2019, 74, 31–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, R.B. Renal Cell Carcinoma with Fibromyomatous Stroma-The Whole Story. Adv. Anat. Pathol. 2022, 29, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Tjota, M.Y.; Sharma, A.; Wanjari, P.; Fitzpatrick, C.; Segal, J.; Antic, T. TSC/MTOR mutated renal cell carcinoma with leiomyomatous stroma is a distinct entity: A comprehensive study of 12 cases. Hum. Pathol. 2023, 134, 124–133. [Google Scholar] [CrossRef]
- Canzonieri, V.; Volpe, R.; Gloghini, A.; Carbone, A.; Merlo, A. Mixed renal tumor with carcinomatous and fibroleiomyomatous components, associated with angiomyolipoma in the same kidney. Pathol. Res. Pr. 1993, 189, 951–956, discussion 957–959. [Google Scholar] [CrossRef]
- Shah, R.B.; Stohr, B.A.; Tu, Z.J.; Gao, Y.; Przybycin, C.G.; Nguyen, J.; Cox, R.M.; Rashid-Kolvear, F.; Weindel, M.D.; Farkas, D.H.; et al. “Renal Cell Carcinoma with Leiomyomatous Stroma” Harbor Somatic Mutations of TSC1, TSC2, MTOR, and/or ELOC (TCEB1): Clinicopathologic and Molecular Characterization of 18 Sporadic Tumors Supports a Distinct Entity. Am. J. Surg. Pathol. 2020, 44, 571–581. [Google Scholar] [CrossRef]
- Lerma, L.A.; Schade, G.R.; Tretiakova, M.S. Co-existence of ESC-RCC, EVT, and LOT as synchronous and metachronous tumors in six patients with multifocal neoplasia but without clinical features of tuberous sclerosis complex. Hum. Pathol. 2021, 116, 1–11. [Google Scholar] [CrossRef]
- Martignoni, G.; Brunelli, M.; Segala, D.; Gobbo, S.; Borze, I.; Atanesyan, L.; Savola, S.; Barzon, L.; Masi, G.; Tardanico, R.; et al. Renal cell carcinoma with smooth muscle stroma lacks chromosome 3p and VHL alterations. Mod. Pathol. 2014, 27, 765–774. [Google Scholar] [CrossRef] [Green Version]
- Parilla, M.; Alikhan, M.; Al-Kawaaz, M.; Patil, S.; Kadri, S.; Ritterhouse, L.L.; Segal, J.; Fitzpatrick, C.; Antic, T. Genetic Underpinnings of Renal Cell Carcinoma with Leiomyomatous Stroma. Am. J. Surg. Pathol. 2019, 43, 1135–1144. [Google Scholar] [CrossRef]
- Williamson, S.R.; Cheng, L.; Eble, J.N.; True, L.D.; Gupta, N.S.; Wang, M.; Zhang, S.; Grignon, D.J. Renal cell carcinoma with angioleiomyoma-like stroma: Clinicopathological, immunohistochemical, and molecular features supporting classification as a distinct entity. Mod. Pathol. 2015, 28, 279–294. [Google Scholar] [CrossRef] [Green Version]
- Hakimi, A.A.; Tickoo, S.K.; Jacobsen, A.; Sarungbam, J.; Sfakianos, J.P.; Sato, Y.; Morikawa, T.; Kume, H.; Fukayama, M.; Homma, Y.; et al. TCEB1-mutated renal cell carcinoma: A distinct genomic and morphological subtype. Mod. Pathol. 2015, 28, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Williamson, S.R.; Hornick, J.L.; Eble, J.N.; Gupta, N.S.; Rogers, C.G.; True, L.; Grignon, D.J.; Cheng, L. Renal Cell Carcinoma with Angioleiomyoma-Like Stroma and Clear Cell Papillary Renal Cell Carcinoma: Exploring SDHB Protein Immunohistochemistry and the Relationship to Tuberous Sclerosis Complex. Hum. Pathol. 2018, 75, 10–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trpkov, K.; Hes, O.; Bonert, M.; Lopez, J.I.; Bonsib, S.M.; Nesi, G.; Comperat, E.; Sibony, M.; Berney, D.M.; Martinek, P.; et al. Eosinophilic, Solid, and Cystic Renal Cell Carcinoma: Clinicopathologic Study of 16 Unique, Sporadic Neoplasms Occurring in Women. Am. J. Surg. Pathol. 2016, 40, 60–71. [Google Scholar] [CrossRef]
- Trpkov, K.; Abou-Ouf, H.; Hes, O.; Lopez, J.I.; Nesi, G.; Comperat, E.; Sibony, M.; Osunkoya, A.O.; Zhou, M.; Gokden, N.; et al. Eosinophilic Solid and Cystic Renal Cell Carcinoma (ESC RCC): Further Morphologic and Molecular Characterization of ESC RCC as a Distinct Entity. Am. J. Surg. Pathol. 2017, 41, 1299–1308. [Google Scholar] [CrossRef]
- Li, Y.; Reuter, V.E.; Matoso, A.; Netto, G.J.; Epstein, J.I.; Argani, P. Re-evaluation of 33 ‘unclassified’ eosinophilic renal cell carcinomas in young patients. Histopathology 2018, 72, 588–600. [Google Scholar] [CrossRef] [PubMed]
- Palsgrove, D.N.; Li, Y.; Pratilas, C.A.; Lin, M.T.; Pallavajjalla, A.; Gocke, C.; De Marzo, A.M.; Matoso, A.; Netto, G.J.; Epstein, J.I.; et al. Eosinophilic Solid and Cystic (ESC) Renal Cell Carcinomas Harbor TSC Mutations: Molecular Analysis Supports an Expanding Clinicopathologic Spectrum. Am. J. Surg. Pathol. 2018, 42, 1166–1181. [Google Scholar] [CrossRef]
- McKenney, J.K.; Przybycin, C.G.; Trpkov, K.; Magi-Galluzzi, C. Eosinophilic solid and cystic renal cell carcinomas have metastatic potential. Histopathology 2018, 72, 1066–1067. [Google Scholar] [CrossRef] [PubMed]
- Tretiakova, M.S. Eosinophilic solid and cystic renal cell carcinoma mimicking epithelioid angiomyolipoma: Series of 4 primary tumors and 2 metastases. Hum. Pathol. 2018, 80, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Mehra, R.; Vats, P.; Cao, X.; Su, F.; Lee, N.D.; Lonigro, R.; Premkumar, K.; Trpkov, K.; McKenney, J.K.; Dhanasekaran, S.M.; et al. Somatic Bi-allelic Loss of TSC Genes in Eosinophilic Solid and Cystic Renal Cell Carcinoma. Eur. Urol. 2018, 74, 483–486. [Google Scholar] [CrossRef]
- Tjota, M.; Chen, H.; Parilla, M.; Wanjari, P.; Segal, J.; Antic, T. Eosinophilic Renal Cell Tumors with a TSC and MTOR Gene Mutations Are Morphologically and Immunohistochemically Heterogenous: Clinicopathologic and Molecular Study. Am. J. Surg. Pathol. 2020, 44, 943–954. [Google Scholar] [CrossRef]
- Parilla, M.; Kadri, S.; Patil, S.A.; Ritterhouse, L.; Segal, J.; Henriksen, K.J.; Antic, T. Are Sporadic Eosinophilic Solid and Cystic Renal Cell Carcinomas Characterized by Somatic Tuberous Sclerosis Gene Mutations? Am. J. Surg. Pathol. 2018, 42, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Munari, E.; Settanni, G.; Calio, A.; Segala, D.; Lonardi, S.; Sandrini, S.; Vacca, P.; Tumino, N.; Marconi, M.; Brunelli, M.; et al. TSC loss is a clonal event in eosinophilic solid and cystic renal cell carcinoma: A multiregional tumor sampling study. Mod. Pathol. 2022, 35, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Aldera, A.P.; Hes, O. Eosinophilic Solid and Cystic Renal Cell Carcinoma with Melanin Pigment-Expanding the Morphological Spectrum. Int. J. Surg. Pathol. 2022, 30, 295–299. [Google Scholar] [CrossRef] [PubMed]
- Lobo, J.; Rechsteiner, M.; Helmchen, B.M.; Rupp, N.J.; Weber, A.; Moch, H. Eosinophilic solid and cystic renal cell carcinoma and renal cell carcinomas with TFEB alterations: A comparative study. Histopathology 2022, 81, 32–43. [Google Scholar] [CrossRef]
- Williamson, S.R.; Cardili, L.; Whiteley, L.J.; Sanchez, J.; Kis, O. Sclerosing TSC1 mutated renal cell carcinoma: An unusual pattern mimicking MITF family translocation renal cell carcinoma. Genes Chromosomes Cancer 2020, 59, 591–594. [Google Scholar] [CrossRef]
- He, H.; Trpkov, K.; Martinek, P.; Isikci, O.T.; Maggi-Galuzzi, C.; Alaghehbandan, R.; Gill, A.J.; Tretiakova, M.; Lopez, J.I.; Williamson, S.R.; et al. “High-grade oncocytic renal tumor”: Morphologic, immunohistochemical, and molecular genetic study of 14 cases. Virchows Arch. 2018, 473, 725–738. [Google Scholar] [CrossRef]
- Chen, Y.B.; Mirsadraei, L.; Jayakumaran, G.; Al-Ahmadie, H.A.; Fine, S.W.; Gopalan, A.; Sirintrapun, S.J.; Tickoo, S.K.; Reuter, V.E. Somatic Mutations of TSC2 or MTOR Characterize a Morphologically Distinct Subset of Sporadic Renal Cell Carcinoma with Eosinophilic and Vacuolated Cytoplasm. Am. J. Surg. Pathol. 2019, 43, 121–131. [Google Scholar] [CrossRef]
- Williamson, S.R.; Gadde, R.; Trpkov, K.; Hirsch, M.S.; Srigley, J.R.; Reuter, V.E.; Cheng, L.; Kunju, L.P.; Barod, R.; Rogers, C.G.; et al. Diagnostic criteria for oncocytic renal neoplasms: A survey of urologic pathologists. Hum. Pathol. 2017, 63, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Hes, O.; Petersson, F.; Kuroda, N.; Hora, M.; Michal, M. Renal hybrid oncocytic/chromophobe tumors—A review. Histol. Histopathol. 2013, 28, 1257–1264. [Google Scholar]
- Trpkov, K.; Hes, O.; Williamson, S.R.; Adeniran, A.J.; Agaimy, A.; Alaghehbandan, R.; Amin, M.B.; Argani, P.; Chen, Y.B.; Cheng, L.; et al. New developments in existing WHO entities and evolving molecular concepts: The Genitourinary Pathology Society (GUPS) update on renal neoplasia. Mod. Pathol. 2021, 34, 1392–1424. [Google Scholar] [CrossRef]
- Trpkov, K.; Bonert, M.; Gao, Y.; Kapoor, A.; He, H.; Yilmaz, A.; Gill, A.J.; Williamson, S.R.; Comperat, E.; Tretiakova, M.; et al. High-grade oncocytic tumour (HOT) of kidney in a patient with tuberous sclerosis complex. Histopathology 2019, 75, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Hes, O.; Trpkov, K. Do we need an updated classification of oncocytic renal tumors?: Emergence of low-grade oncocytic tumor (LOT) and eosinophilic vacuolated tumor (EVT) as novel renal entities. Mod. Pathol. 2022, 35, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Farcas, M.; Gatalica, Z.; Trpkov, K.; Swensen, J.; Zhou, M.; Alaghehbandan, R.; Williamson, S.R.; Magi-Galluzzi, C.; Gill, A.J.; Tretiakova, M.; et al. Eosinophilic vacuolated tumor (EVT) of kidney demonstrates sporadic TSC/MTOR mutations: Next-generation sequencing multi-institutional study of 19 cases. Mod. Pathol. 2022, 35, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Siadat, F.; Trpkov, K. ESC, ALK, HOT and LOT: Three Letter Acronyms of Emerging Renal Entities Knocking on the Door of the WHO Classification. Cancers 2020, 12, 168. [Google Scholar] [CrossRef] [Green Version]
- Kapur, P.; Gao, M.; Zhong, H.; Rakheja, D.; Cai, Q.; Pedrosa, I.; Margulis, V.; Xu, L.; Kinch, L.; Brugarolas, J. Eosinophilic Vacuolated Tumor of the Kidney: A Review of Evolving Concepts in This Novel Subtype with Additional Insights From a Case with MTOR Mutation and Concomitant Chromosome 1 Loss. Adv. Anat. Pathol. 2021, 28, 251–257. [Google Scholar] [CrossRef]
- Trpkov, K.; Williamson, S.R.; Gao, Y.; Martinek, P.; Cheng, L.; Sangoi, A.R.; Yilmaz, A.; Wang, C.; San Miguel Fraile, P.; Perez Montiel, D.M.; et al. Low-grade Oncocytic Tumor of Kidney (CD117 Negative, Cytokeratin 7 Positive): A Distinct Entity? Histopathology 2019, 75, 174–184. [Google Scholar] [CrossRef] [Green Version]
- Kapur, P.; Gao, M.; Zhong, H.; Chintalapati, S.; Mitui, M.; Barnes, S.D.; Zhou, Q.; Miyata, J.; Carrillo, D.; Malladi, V.S.; et al. Germline and sporadic mTOR pathway mutations in low-grade oncocytic tumor of the kidney. Mod. Pathol. 2022, 35, 333–343. [Google Scholar] [CrossRef]
- Kravtsov, O.; Gupta, S.; Cheville, J.C.; Sukov, W.R.; Rowsey, R.; Herrera-Hernandez, L.P.; Lohse, C.M.; Knudson, R.; Leibovich, B.C.; Jimenez, R.E. Low-Grade Oncocytic Tumor of Kidney (CK7-Positive, CD117-Negative): Incidence in a Single Institutional Experience with Clinicopathological and Molecular Characteristics. Hum. Pathol. 2021, 114, 9–18. [Google Scholar] [CrossRef]
- Zhang, H.Z.; Xia, Q.Y.; Wang, S.Y.; Shi, M.J.; Wang, S.Y. Low-grade oncocytic tumor of kidney harboring TSC/MTOR mutation: Clinicopathologic, immunohistochemical and molecular characteristics support a distinct entity. Virchows Arch. 2022, 480, 999–1008. [Google Scholar] [CrossRef]
- Akgul, M.; Al-Obaidy, K.I.; Cheng, L.; Idrees, M.T. Low-grade oncocytic tumour expands the spectrum of renal oncocytic tumours and deserves separate classification: A review of 23 cases from a single tertiary institute. J. Clin. Pathol. 2021, 75, 772–775. [Google Scholar] [CrossRef]
- Guo, Q.; Liu, N.; Wang, F.; Guo, Y.; Yang, B.; Cao, Z.; Wang, Y.; Wang, Y.; Zhang, W.; Huang, Q.; et al. Characterization of a distinct low-grade oncocytic renal tumor (CD117-negative and cytokeratin 7-positive) based on a tertiary oncology center experience: The new evidence from China. Virchows Arch. 2020, 478, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Morini, A.; Drossart, T.; Timsit, M.O.; Sibony, M.; Vasiliu, V.; Gimenez-Roqueplo, A.P.; Favier, J.; Badoual, C.; Mejean, A.; Burnichon, N.; et al. Low-grade oncocytic renal tumor (LOT): Mutations in mTOR pathway genes and low expression of FOXI1. Mod. Pathol. 2022, 35, 352–360. [Google Scholar] [CrossRef]
- Amin, M.B.; McKenney, J.K.; Martignoni, G.; Campbell, S.C.; Pal, S.; Tickoo, S.K. Low grade oncocytic tumors of the kidney: A clinically relevant approach for the workup and accurate diagnosis. Mod. Pathol. 2022, 35, 1306–1316. [Google Scholar] [CrossRef]
- Mohanty, S.K.; Satapathy, A.; Aggarwal, A.; Mishra, S.K.; Sampat, N.Y.; Sharma, S.; Williamson, S.R. Oncocytic renal neoplasms with diffuse keratin 7 immunohistochemistry harbor frequent alterations in the mammalian target of rapamycin pathway. Mod. Pathol. 2022, 35, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.B.; Vazquez, F.; Reddy, A.; Sellers, W.R.; Kaelin, W.G., Jr. TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 2003, 4, 147–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjota, M.Y.; Wanjari, P.; Segal, J.; Antic, T. TSC/MTOR-mutated eosinophilic renal tumors are a distinct entity that is CK7+/CK20-/vimentin-: A validation study. Hum. Pathol. 2021, 115, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.; Hu, Z. FOXI1 expression in chromophobe renal cell carcinoma and renal oncocytoma: A study of The Cancer Genome Atlas transcriptome-based outlier mining and immunohistochemistry. Virchows Arch. 2021, 478, 647–658. [Google Scholar] [CrossRef]
- Skala, S.L.; Wang, X.; Zhang, Y.; Mannan, R.; Wang, L.; Narayanan, S.P.; Vats, P.; Su, F.; Chen, J.; Cao, X.; et al. Next-generation RNA Sequencing-based Biomarker Characterization of Chromophobe Renal Cell Carcinoma and Related Oncocytic Neoplasms. Eur. Urol. 2020, 78, 63–74. [Google Scholar] [CrossRef]
- Fingar, D.C.; Blenis, J. Target of rapamycin (TOR): An integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 2004, 23, 3151–3171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef]
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR kinase structure, mechanism and regulation. Nature 2013, 497, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Sabatini, D.M. mTOR and cancer: Insights into a complex relationship. Nat. Rev. Cancer 2006, 6, 729–734. [Google Scholar] [CrossRef]
- Brugarolas, J.; Kaelin, W.G., Jr. Dysregulation of HIF and VEGF is a unifying feature of the familial hamartoma syndromes. Cancer Cell 2004, 6, 7–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, G.; Di Malta, C.; Ballabio, A. Non-canonical mTORC1 signaling at the lysosome. Trends Cell Biol. 2022, 32, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Vega-Rubin-de-Celis, S.; Pena-Llopis, S.; Konda, M.; Brugarolas, J. Multistep regulation of TFEB by MTORC1. Autophagy 2017, 13, 464–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, G.; Esposito, A.; Choi, H.; Matarese, M.; Benedetti, V.; Di Malta, C.; Monfregola, J.; Medina, D.L.; Lippincott-Schwartz, J.; Ballabio, A. mTOR-dependent phosphorylation controls TFEB nuclear export. Nat. Commun. 2018, 9, 3312. [Google Scholar] [CrossRef] [PubMed]
- Pena-Llopis, S.; Vega-Rubin-de-Celis, S.; Schwartz, J.C.; Wolff, N.C.; Tran, T.A.; Zou, L.; Xie, X.J.; Corey, D.R.; Brugarolas, J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011, 30, 3242–3258. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.; Napolitano, G.; de Araujo, M.E.G.; Esposito, A.; Monfregola, J.; Huber, L.A.; Ballabio, A.; Hurley, J.H. Structure of the lysosomal mTORC1-TFEB-Rag-Ragulator megacomplex. Nature 2023, 614, 572–579. [Google Scholar] [CrossRef]
- Alesi, N.; Akl, E.W.; Khabibullin, D.; Liu, H.J.; Nidhiry, A.S.; Garner, E.R.; Filippakis, H.; Lam, H.C.; Shi, W.; Viswanathan, S.R.; et al. TSC2 regulates lysosome biogenesis via a non-canonical RAGC and TFEB-dependent mechanism. Nat. Commun. 2021, 12, 4245. [Google Scholar] [CrossRef] [PubMed]
- Asrani, K.; Woo, J.; Mendes, A.A.; Schaffer, E.; Vidotto, T.; Villanueva, C.R.; Feng, K.; Oliveira, L.; Murali, S.; Liu, H.B.; et al. An mTORC1-mediated negative feedback loop constrains amino acid-induced FLCN-Rag activation in renal cells with TSC2 loss. Nat. Commun. 2022, 13, 6808. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, G.; Di Malta, C.; Esposito, A.; de Araujo, M.E.G.; Pece, S.; Bertalot, G.; Matarese, M.; Benedetti, V.; Zampelli, A.; Stasyk, T.; et al. A substrate-specific mTORC1 pathway underlies Birt-Hogg-Dube syndrome. Nature 2020, 585, 597–602. [Google Scholar] [CrossRef]
- Agaram, N.P.; Sung, Y.S.; Zhang, L.; Chen, C.L.; Chen, H.W.; Singer, S.; Dickson, M.A.; Berger, M.F.; Antonescu, C.R. Dichotomy of Genetic Abnormalities in PEComas with Therapeutic Implications. Am. J. Surg. Pathol. 2015, 39, 813–825. [Google Scholar] [CrossRef] [Green Version]
- Salles, D.C.; Asrani, K.; Woo, J.; Vidotto, T.; Liu, H.B.; Vidal, I.; Matoso, A.; Netto, G.J.; Argani, P.; Lotan, T.L. GPNMB expression identifies TSC1/2/mTOR-associated and MiT family translocation-driven renal neoplasms. J. Pathol. 2022, 257, 158–171. [Google Scholar] [CrossRef]
- Carlo, M.I.; Hakimi, A.A.; Stewart, G.D.; Bratslavsky, G.; Brugarolas, J.; Chen, Y.B.; Linehan, W.M.; Maher, E.R.; Merino, M.J.; Offit, K.; et al. Familial Kidney Cancer: Implications of New Syndromes and Molecular Insights. Eur. Urol. 2019, 76, 754–764. [Google Scholar] [CrossRef]
- Seyam, R.M.; Bissada, N.K.; Kattan, S.A.; Mokhtar, A.A.; Aslam, M.; Fahmy, W.E.; Mourad, W.A.; Binmahfouz, A.A.; Alzahrani, H.M.; Hanash, K.A. Changing trends in presentation, diagnosis and management of renal angiomyolipoma: Comparison of sporadic and tuberous sclerosis complex-associated forms. Urology 2008, 72, 1077–1082. [Google Scholar] [CrossRef]
- Wagner, A.J.; Malinowska-Kolodziej, I.; Morgan, J.A.; Qin, W.; Fletcher, C.D.; Vena, N.; Ligon, A.H.; Antonescu, C.R.; Ramaiya, N.H.; Demetri, G.D.; et al. Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: Targeting the pathogenic activation of mTORC1 in tumors. J. Clin. Oncol. 2010, 28, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Wolff, N.; Kabbani, W.; Bradley, T.; Raj, G.; Watumull, L.; Brugarolas, J. Sirolimus and temsirolimus for epithelioid angiomyolipoma. J. Clin. Oncol. 2010, 28, e65–e68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trnka, P.; Kennedy, S.E. Renal tumors in tuberous sclerosis complex. Pediatr. Nephrol. 2021, 36, 1427–1438. [Google Scholar] [CrossRef]
- Bissler, J.J.; Kingswood, J.C.; Radzikowska, E.; Zonnenberg, B.A.; Frost, M.; Belousova, E.; Sauter, M.; Nonomura, N.; Brakemeier, S.; de Vries, P.J.; et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2013, 381, 817–824. [Google Scholar] [CrossRef]
- Bissler, J.J.; Franz, D.N.; Frost, M.D.; Belousova, E.; Bebin, E.M.; Sparagana, S.; Berkowitz, N.; Ridolfi, A.; Kingswood, J.C. The effect of everolimus on renal angiomyolipoma in pediatric patients with tuberous sclerosis being treated for subependymal giant cell astrocytoma. Pediatr. Nephrol. 2018, 33, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissler, J.J.; Nonomura, N.; Budde, K.; Zonnenberg, B.A.; Fischereder, M.; Voi, M.; Louveau, A.L.; Herbst, F.; Bebin, E.M.; Curatolo, P.; et al. Angiomyolipoma rebound tumor growth after discontinuation of everolimus in patients with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis. PLoS ONE 2018, 13, e0201005. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.Y.; Wang, X.T.; Zhao, M.; He, H.Y.; Fang, R.; Ye, S.B.; Li, R.; Wang, X.; Zhang, R.S.; Lu, Z.F.; et al. TSC/MTOR -associated Eosinophilic Renal Tumors Exhibit a Heterogeneous Clinicopathologic Spectrum: A Targeted Next-generation Sequencing and Gene Expression Profiling Study. Am. J. Surg. Pathol. 2022, 46, 1562–1576. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Stawiski, E.W.; Pavia-Jimenez, A.; Modrusan, Z.; Kapur, P.; Jaiswal, B.S.; Zhang, N.; Toffessi-Tcheuyap, V.; Nguyen, T.T.; Pahuja, K.B.; et al. Spectrum of diverse genomic alterations define non-clear cell renal carcinoma subtypes. Nat. Genet. 2015, 47, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.F.; Ricketts, C.J.; Wang, M.; Yang, L.; Cherniack, A.D.; Shen, H.; Buhay, C.; Kang, H.; Kim, S.C.; Fahey, C.C.; et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell 2014, 26, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Ricketts, C.J.; De Cubas, A.A.; Fan, H.; Smith, C.C.; Lang, M.; Reznik, E.; Bowlby, R.; Gibb, E.A.; Akbani, R.; Beroukhim, R.; et al. The Cancer Genome Atlas Comprehensive Molecular Characterization of Renal Cell Carcinoma. Cell Rep. 2018, 23, 313–326.e315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Linehan, W.M.; Ricketts, C.J. The Cancer Genome Atlas of renal cell carcinoma: Findings and clinical implications. Nat. Rev. Urol. 2019, 16, 539–552. [Google Scholar] [CrossRef]
- Gu, Y.F.; Cohn, S.; Christie, A.; McKenzie, T.; Wolff, N.; Do, Q.N.; Madhuranthakam, A.J.; Pedrosa, I.; Wang, T.; Dey, A.; et al. Modeling Renal Cell Carcinoma in Mice: Bap1 and Pbrm1 Inactivation Drive Tumor Grade. Cancer Discov. 2017, 7, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, D.J.; Choueiri, T.K.; Fay, A.P.; Rini, B.I.; Thorner, A.R.; de Velasco, G.; Tyburczy, M.E.; Hamieh, L.; Albiges, L.; Agarwal, N.; et al. Mutations in TSC1, TSC2, and MTOR Are Associated with Response to Rapalogs in Patients with Metastatic Renal Cell Carcinoma. Clin. Cancer Res. 2016, 22, 2445–2452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamieh, L.; Choueiri, T.K.; Ogorek, B.; Khabibullin, D.; Rosebrock, D.; Livitz, D.; Fay, A.; Pignon, J.C.; McDermott, D.F.; Agarwal, N.; et al. Mechanisms of acquired resistance to rapalogs in metastatic renal cell carcinoma. PLoS Genet. 2018, 14, e1007679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagle, N.; Grabiner, B.C.; Van Allen, E.M.; Amin-Mansour, A.; Taylor-Weiner, A.; Rosenberg, M.; Gray, N.; Barletta, J.A.; Guo, Y.; Swanson, S.J.; et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N. Engl. J. Med. 2014, 371, 1426–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Butti, R.; Cohn, S.; Tcheuyap, V.T.; Mal, A.; Nguyen, M.; Stevens, C.; Christie, A.; Mishra, A.; Ma, Y.; et al. Unconventional mechanism of action and resistance to rapalogs in renal cancer. Proc. Natl. Acad. Sci. USA, 2023, submitted.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Clinical Features | Morphology | Immunohistochemistry | Molecular Features |
|---|---|---|---|---|
| Renal cell carcinoma with fibromyomatous stroma (RCC FMS) | Mostly sporadic and solitary, indolent | Solid, smaller tumors, tan to brown, may have lobulated appearance, clear cells with voluminous cytoplasm forming nodules, separated, and encircled by fibromuscular stroma | CK7+ CAIX+ (membranous) CD10+ AMACR− | TSC/MTOR mutations |
| Eosinophilic solid and cystic renal cell carcinoma (ESC RCC) | Mostly in females, largely sporadic and solitary, generally indolent | Solid and cystic, voluminous eosinophilic cells, cytoplasmic stippling | CK20+ CK7− CD117− Vimentin+ Cathepsin K+ (focal) | Somatic bi-allelic loss of function mutations in TSC1 and TSC2 |
| Eosinophilic vacuolated tumor (EVT) | Broad age range, sporadic and solitary, rare cases in TSC patients, indolent | Solid, smaller tumors, tan to brown or gray, large vessels often at the periphery, eosinophilic cells with frequent and prominent intracytoplasmic vacuoles, large nucleoli | cathepsin K+ CD117+ CD10+ CK7− (only rare cells +) CK20− Vimentin− | TSC/MTOR pathway mutations (all cases), deletions of chromosome 19 and 1 (in cases with MTOR mutation) |
| Low-grade oncocytic tumor (LOT) | Mostly in older patients, sporadic and solitary, rare cases in TSC patients, indolent | Solid, smaller tumors, tan to mahogany brown, sharp transition to edematous areas with scattered individual cells, round to oval nuclei without irregularities and prominent nucleoli, often perinuclear halos | CK7+ (diffuse) CD117− (rarely weak +) GATA3+ (limited data) FOXI1− CK20− Vimentin− | TSC/MTOR pathway mutations (almost all cases), lack of multiple chromosome losses, deletions of chromosome 19p, 19q, and 1p (in some cases), no CCND1 rearrangements |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapur, P.; Brugarolas, J.; Trpkov, K. Recent Advances in Renal Tumors with TSC/mTOR Pathway Abnormalities in Patients with Tuberous Sclerosis Complex and in the Sporadic Setting. Cancers 2023, 15, 4043. https://doi.org/10.3390/cancers15164043
Kapur P, Brugarolas J, Trpkov K. Recent Advances in Renal Tumors with TSC/mTOR Pathway Abnormalities in Patients with Tuberous Sclerosis Complex and in the Sporadic Setting. Cancers. 2023; 15(16):4043. https://doi.org/10.3390/cancers15164043
Chicago/Turabian StyleKapur, Payal, James Brugarolas, and Kiril Trpkov. 2023. "Recent Advances in Renal Tumors with TSC/mTOR Pathway Abnormalities in Patients with Tuberous Sclerosis Complex and in the Sporadic Setting" Cancers 15, no. 16: 4043. https://doi.org/10.3390/cancers15164043
APA StyleKapur, P., Brugarolas, J., & Trpkov, K. (2023). Recent Advances in Renal Tumors with TSC/mTOR Pathway Abnormalities in Patients with Tuberous Sclerosis Complex and in the Sporadic Setting. Cancers, 15(16), 4043. https://doi.org/10.3390/cancers15164043