
Development of an Enzyme-Linked Immunosorbent Assay (ELISA) for the Quantification of ARID1A in Tissue Lysates
 ,
,
Abstract
:Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Lysate Preparation
2.3. Determination of Protein Concentration
2.4. Statistical Analysis
2.5. ELISA Protocol
2.6. Standard Curve Analysis and Acceptance Criteria
2.7. Western Blot
3. Results
3.1. Calibration Curve
3.2. Precision and Accuracy: Standards and Incurred Sample Reanalysis
3.3. Specificity
3.4. Spike Recovery
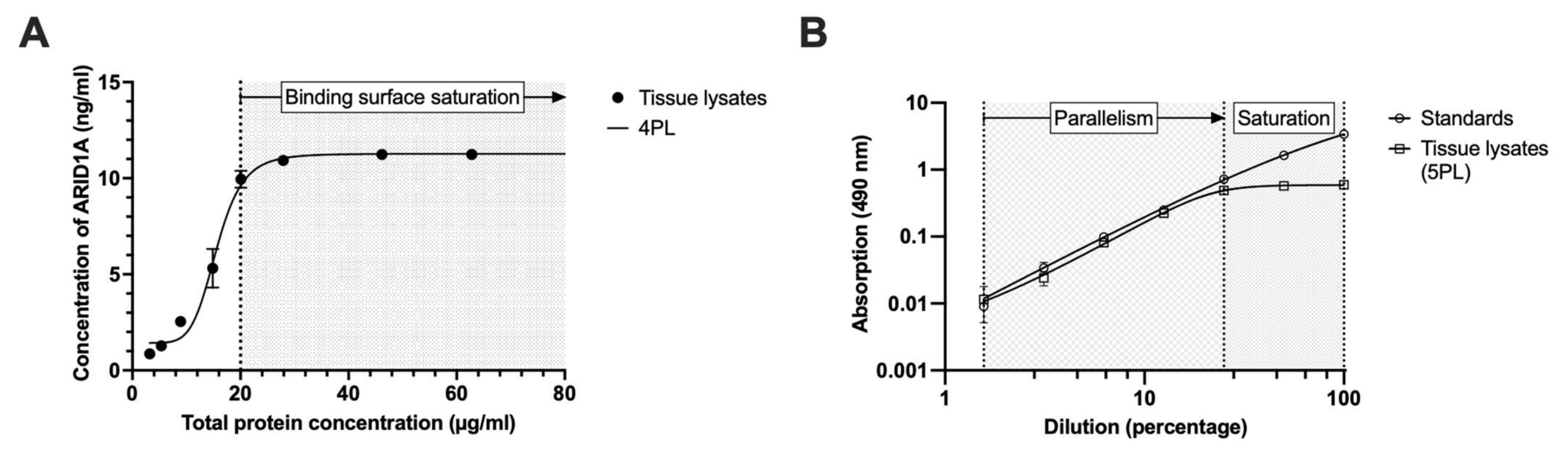
3.5. Minimal Required Dilution (MRD)
3.6. Parallelism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mashtalir, N.; D’Avino, A.; Michel, B.; Luo, J.; Pan, J.; Otto, J.; Zullow, H.; McKenzie, Z.; Kubiak, R.; St Pierre, R.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288.e20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of Action and Regulation of ATP-Dependent Chromatin-Remodelling Complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Giri, M.; Maulik, A.; Singh, M. Signatures of Specific DNA Binding by the AT-Rich Interaction Domain of BAF250a. Biochemistry 2020, 59, 100–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raab, J.R.; Resnick, S.; Magnuson, T. Genome-Wide Transcriptional Regulation Mediated by Biochemically Distinct SWI/SNF Complexes The SWI/SNF Chromatin Remodeling Complex. PLoS ONE 2015, 11, e1005748. [Google Scholar] [CrossRef]
- Shain, A.H.; Pollack, J.R. The Spectrum of SWI/SNF Mutations, Ubiquitous in Human Cancers. PLoS ONE 2013, 8, e55119. [Google Scholar] [CrossRef] [Green Version]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and Bioinformatic Analysis of Mammalian SWI/SNF Complexes Identifies Extensive Roles in Human Malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [Green Version]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A Mutations in Endometriosis-Associated Ovarian Carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.; Wang, T.L.; Shih, I.M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A.; Vogelstein, B.; et al. Frequent Mutations of Chromatin Remodeling Gene ARID1A in Ovarian Clear Cell Carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Roberts, C. ARID1A Mutations in Cancer: Another Epigenetic Tumor Suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Luo, Q.; Wu, X.; Chang, W.; Zhao, P.; Zhu, X.; Chen, H.; Nan, Y.; Luo, A.; Zhou, X.; Su, D.; et al. ARID1A Hypermethylation Disrupts Transcriptional Homeostasis to Promote Squamous Cell Carcinoma Progression. Cancer Res. 2020, 80, 406–417. [Google Scholar] [CrossRef]
- Mandal, J.; Mandal, P.; Wang, T.-L.; Shih, I.-M. Treating ARID1A Mutated Cancers by Harnessing Synthetic Lethality and DNA Damage Response. J. Biomed. Sci. 2022, 29, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Itamochi, H.; Oumi, N.; Oishi, T.; Shoji, T.; Fujiwara, H.; Sugiyama, T.; Suzuki, M.; Kigawa, J.; Harada, T. Loss of ARID1A Expression Is Associated with Poor Prognosis in Patients with Stage I/II Clear Cell Carcinoma of the Ovary. Int. J. Clin. Oncol. 2015, 20, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chhangawala, S.; Cocco, E.; Razavi, P.; Cai, Y.; Otto, J.E.; Ferrando, L.; Selenica, P.; Ladewig, E.; Chan, C.; et al. ARID1A Determines Luminal Identity and Therapeutic Response in Estrogen-Receptor-Positive Breast Cancer. Nat. Genet. 2020, 52, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Nivolumab for the Treatment of Metastatic or Unresectable Solid Tumors With ARID1A Mutation and CXCL13 Expression—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04957615 (accessed on 27 April 2023).
- Phase II Study of Tazemetostat in Solid Tumors Harboring an ARID1A Mutation—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05023655 (accessed on 27 April 2023).
- Walker, R.A. Quantification of Immunohistochemistry—Issues Concerning Methods, Utility and Semiquantitative Assessment I. Histopathology 2006, 49, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.R.; Levenson, R.M. Quantification of Immunohistochemistry—Issues Concerning Methods, Utility and Semiquantitative Assessment II. Histopathology 2006, 49, 411–424. [Google Scholar] [CrossRef]
- McCabe, A.; Dolled-Filhart, M.; Camp, R.L.; Rimm, D.L. Automated Quantitative Analysis (AQUA) of In Situ Protein Expression, Antibody Concentration, and Prognosis. JNCI J. Natl. Cancer Inst. 2005, 97, 1808–1815. [Google Scholar] [CrossRef]
- Camp, R.L.; Chung, G.G.; Rimm, D.L. Automated Subcellular Localization and Quantification of Protein Expression in Tissue Microarrays. Nat. Med. 2002, 8, 1323–1328. [Google Scholar] [CrossRef]
- Azadeh, M.; Gorovits, B.; Kamerud, J.; MacMannis, S.; Safavi, A.; Sailstad, J.; Sondag, P. Calibration Curves in Quantitative Ligand Binding Assays: Recommendations and Best Practices for Preparation, Design, and Editing of Calibration Curves. AAPS J. 2018, 20, 1–16. [Google Scholar] [CrossRef]
- Natarajan, S.; Remick, D.G. The ELISA Standard Save: Calculation of Sample Concentrations in Assays with a Failed Standard Curve. J. Immunol. Methods 2008, 336, 242–245. [Google Scholar] [CrossRef] [Green Version]
- Nowak, J.; Watala, C.; Boncler, M. Antibody Binding, Platelet Adhesion, and Protein Adsorption on Various Polymer Surfaces. Blood Coagul. Fibrinolysis 2014, 25, 52–60. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services. Food and Drug Administration Bioanalytical Method Validation Guidance for Industry; US Department of Health and Human Services Food and Drug Administration: Washington, DC, USA, 2018; pp. 1–41.
- European Medicines Agency. ICH Guideline M10 on Bioanalytical Method Validation. Sci. Med. Health 2019, 44, 57. [Google Scholar]
- Kyte, J.; Doolittle, R.F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as Designed by Its Users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef] [PubMed]
- Barrios-Llerena, M.E.; Chong, P.K.; Gan, C.S.; Snijders, A.P.L.; Reardon, K.F.; Wright, P.C. Shotgun Proteomics of Cyanobacteria—Applications of Experimental and Data-Mining Techniques. Brief. Funct. Genomics 2006, 5, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro. Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Laemmli, U.K. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
- Collins, T.J. ImageJ for Microscopy. Biotechniques 2007, 43, S25–S30. [Google Scholar] [CrossRef]
- Andreasson, U.; Perret-Liaudet, A.; van Waalwijk van Doorn, L.J.C.; Blennow, K.; Chiasserini, D.; Engelborghs, S.; Fladby, T.; Genc, S.; Kruse, N.; Kuiperij, H.B.; et al. A Practical Guide to Immunoassay Method Validation. Front. Neurol. 2015, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kiessig, S.T.; Jahn, S.; Hiepe, F.; Volk, H.D.; Fietze, E.; Zugehör, M.; Porstmann, T.; von Baehr, R. Multireactive Human Monoclonal Antibodies. Allerg. Immunol. 1990, 36, 163–177. [Google Scholar]
- Grebenchtchikov, N.; Brinkman, A.; Van Broekhoven, S.P.J.; De Jong, D.; Geurts-Moespot, A.; Span, P.N.; Peters, H.A.; Portengen, H.; Foekens, J.A.; Sweep, C.G.J.; et al. Development of an ELISA for Measurement of BCAR1 Protein in Human Breast Cancer Tissue. Clin. Chem. 2004, 50, 1356–1363. [Google Scholar] [CrossRef] [Green Version]
- Rodgers, K.J.; Dean, R.T. Assessment of Proteasome Activity in Cell Lysates and Tissue Homogenates Using Peptide Substrates. Int. J. Biochem. Cell Biol. 2003, 35, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Gao, M.; Wu, C.-H.; Wang, T.-L.; Shih, I.-M. Functional Analysis of In-Frame Indel ARID1A Mutations Reveals New Regulatory Mechanisms of Its Tumor Suppressor Functions. Neoplasia 2012, 14, 986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, S.; Grassian, A.R.; Tsuda, M.; Knutson, S.K.; Warholic, N.M.; Kuznetsov, G.; Xu, S.; Xiao, Y.; Pollock, R.M.; Smith, J.S.; et al. Preclinical Evidence of Anti-Tumor Activity Induced by EZH2 Inhibition in Human Models of Synovial Sarcoma. PLoS ONE 2016, 11, e0158888. [Google Scholar] [CrossRef]
- Wang, X.; Nagl, N.G.; Wilsker, D.; Van Scoy, M.; Pacchione, S.; Yaciuk, P.; Dallas, P.B.; Moran, E. Two Related ARID Family Proteins Are Alternative Subunits of Human SWI/SNF Complexes. Biochem. J. 2004, 383, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B Is a Specific Vulnerability in ARID1A-Mutant Cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and Sequence Analysis Tools Services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, gkac240. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A Multiple Sequence Alignment Editor and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Agivetova, R.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bursteinas, B.; et al. UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemicals | Detailed Description |
|---|---|
| Coating solution | Carbonate (NaHCO3/Na2CO3; ratio: 2:1) buffer (50 mM, pH 9.6) |
| Washing solution | Phosphate-buffered saline pH 7.4 (PBS) added with 0.1% Tween-20 (=PBS-T 0.1%) |
| Blocking solution | PBS-T (0.05%) supplemented with 0.2% Casein (I-Block, T2015, Thermo Fisher Scientific, Waltham, MA, USA) |
| Primary antibody | Primary antibody (ab182560, Abcam, UK) diluted 1:1000 in blocking solution |
| Secondary antibody | Secondary antibody (anti-rabbit, K4003, Dako, Glostrup, Denmark) diluted 1:2 in blocking solution |
| Substrate solution | 5 mg OPD (o-phenylenediamine dihydrochloride, 34006, Thermo Fisher Scientific, USA) diluted in 9 mL distilled water and 1 mL Stable Peroxide Substrate Buffer (34062, Thermo Fisher Scientific, USA) |
| Stopping solution | 1403 µL H2SO4 (95–99% purity) diluted in 9 mL distilled water (=2.5 M H2SO4) |
| Steps | Detailed Description | Incubation |
|---|---|---|
| Coating | Prepare tumor lysates by diluting the cellular fractions in coating solution 1:26 (total protein concentration should be at least 20 µg/mL). Prepare minimally 6 standards that are serially diluted in the range between 0.78–50 ng/mL. Apply 100 µL of each standard, lysate fraction, and blanks (e.g., carbonate buffer only) at least in duplicate to corresponding wells. | Seal the plate with parafilm. Then, incubate (for 14 to 18 h) overnight at 4 °C. |
| Washing | Apply 200 µL washing solution. Repeat 2 times. | Incubate at RT for 5 min on a plate shaker. |
| Blocking | Apply 200 µL blocking solution. | Incubate at RT for 2 h on a plate shaker. |
| Washing | Apply 200 µL washing solution. Repeat 2 times. | Incubate at RT for 5 min on a plate shaker. |
| Primary antibody | Apply 100 µL primary antibody diluted 1:1000. | Incubate at RT for 2 h on a plate shaker. |
| Washing | Apply 200 µL washing solution. Repeat 2 times. | Incubate at RT for 5 min on a plate shaker. |
| Secondary antibody | Apply 100 µL secondary antibody diluted 1:2. | Incubate at RT for 2 h on a plate shaker. |
| Washing | Apply 200 µL washing solution. Repeat 2 times. | Incubate at RT for 5 min on a plate shaker. |
| Application of OPD | Apply 100 µL substrate solution. | Incubate at RT for 30 min in the dark. |
| Application of H2SO4 | Apply 100 µL stopping solution. | Measure optical density at 490 nm. |
| Deviation in ARID1A Levels | Standards (AE Model 1) | Standards (EP Model 2) | Same Lysate (n = 18) | Different Lysate (n = 6) | All Repeated Measurements (n = 24) |
|---|---|---|---|---|---|
| Cytosol (%CV) | NA | NA | 10.05% | 12.23% | 10.31% |
| Free nucleus (%CV) | NA | NA | 10.76% | 26.28% | 12.57% |
| Chromatin (%CV) | NA | NA | 11.06% | 19.2% | 12.46% |
| Inter-assay accuracy | 90.26% | 93.16% | NA | NA | NA |
| Inter-assay precision (%CV) | 4.53% | 4.45% | 10.61% | 21.31% | 11.78% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hinsberger, M.; Becker-Kettern, J.; Jürgens-Wemheuer, W.M.; Oertel, J.; Schulz-Schaeffer, W.J. Development of an Enzyme-Linked Immunosorbent Assay (ELISA) for the Quantification of ARID1A in Tissue Lysates. Cancers 2023, 15, 4096. https://doi.org/10.3390/cancers15164096
Hinsberger M, Becker-Kettern J, Jürgens-Wemheuer WM, Oertel J, Schulz-Schaeffer WJ. Development of an Enzyme-Linked Immunosorbent Assay (ELISA) for the Quantification of ARID1A in Tissue Lysates. Cancers. 2023; 15(16):4096. https://doi.org/10.3390/cancers15164096
Chicago/Turabian StyleHinsberger, Manuel, Julia Becker-Kettern, Wiebke M. Jürgens-Wemheuer, Joachim Oertel, and Walter J. Schulz-Schaeffer. 2023. "Development of an Enzyme-Linked Immunosorbent Assay (ELISA) for the Quantification of ARID1A in Tissue Lysates" Cancers 15, no. 16: 4096. https://doi.org/10.3390/cancers15164096