

Relevance of ATM Status in Driving Sensitivity to DNA Damage Response Inhibitors in Patient-Derived Xenograft Models

 , , , ,
, , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. In Vivo Studies
2.2. Tumour Inoculation
2.3. Experimental Design
2.4. In Vivo Data Analysis
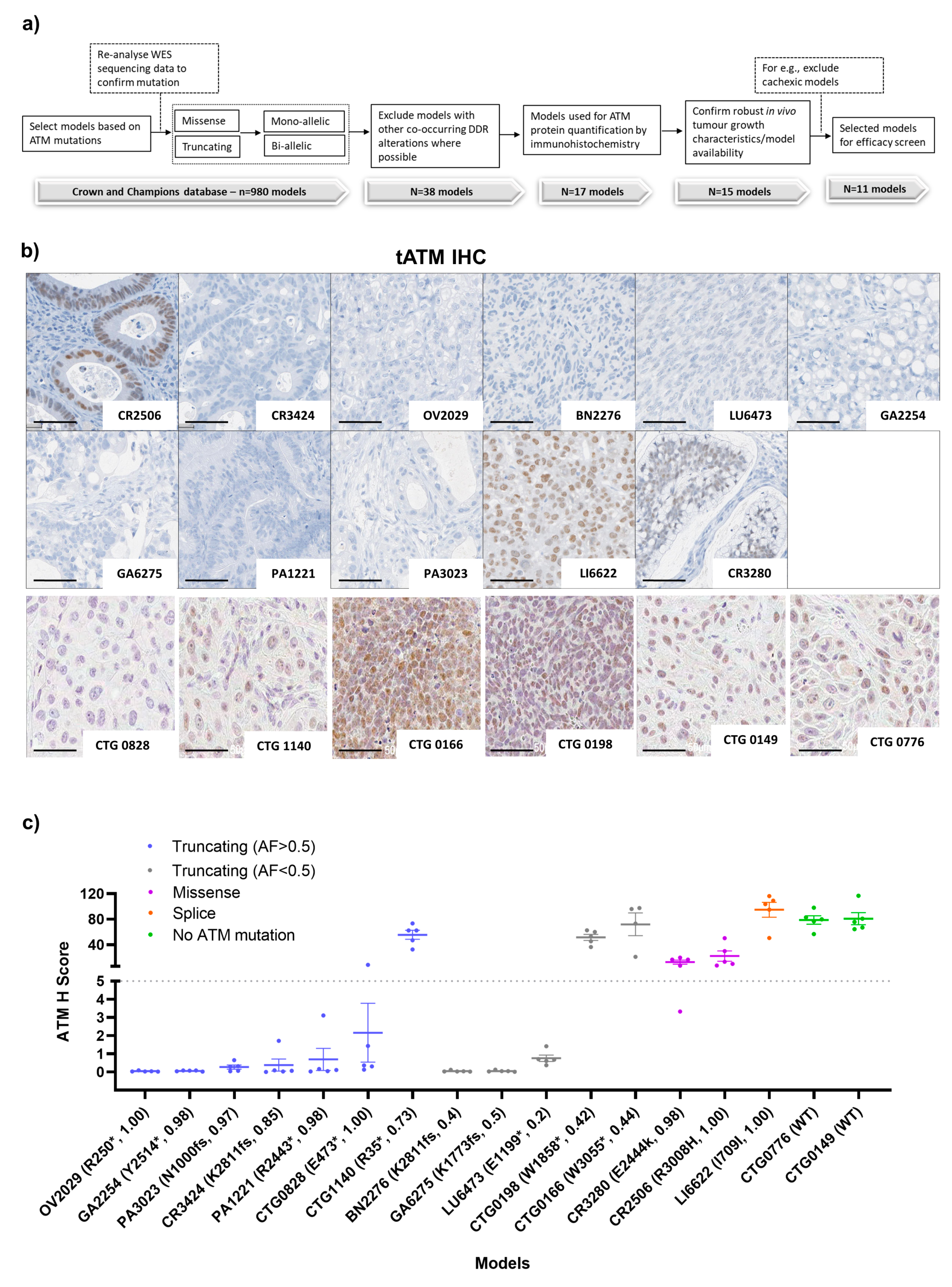
2.5. ATM Assessment
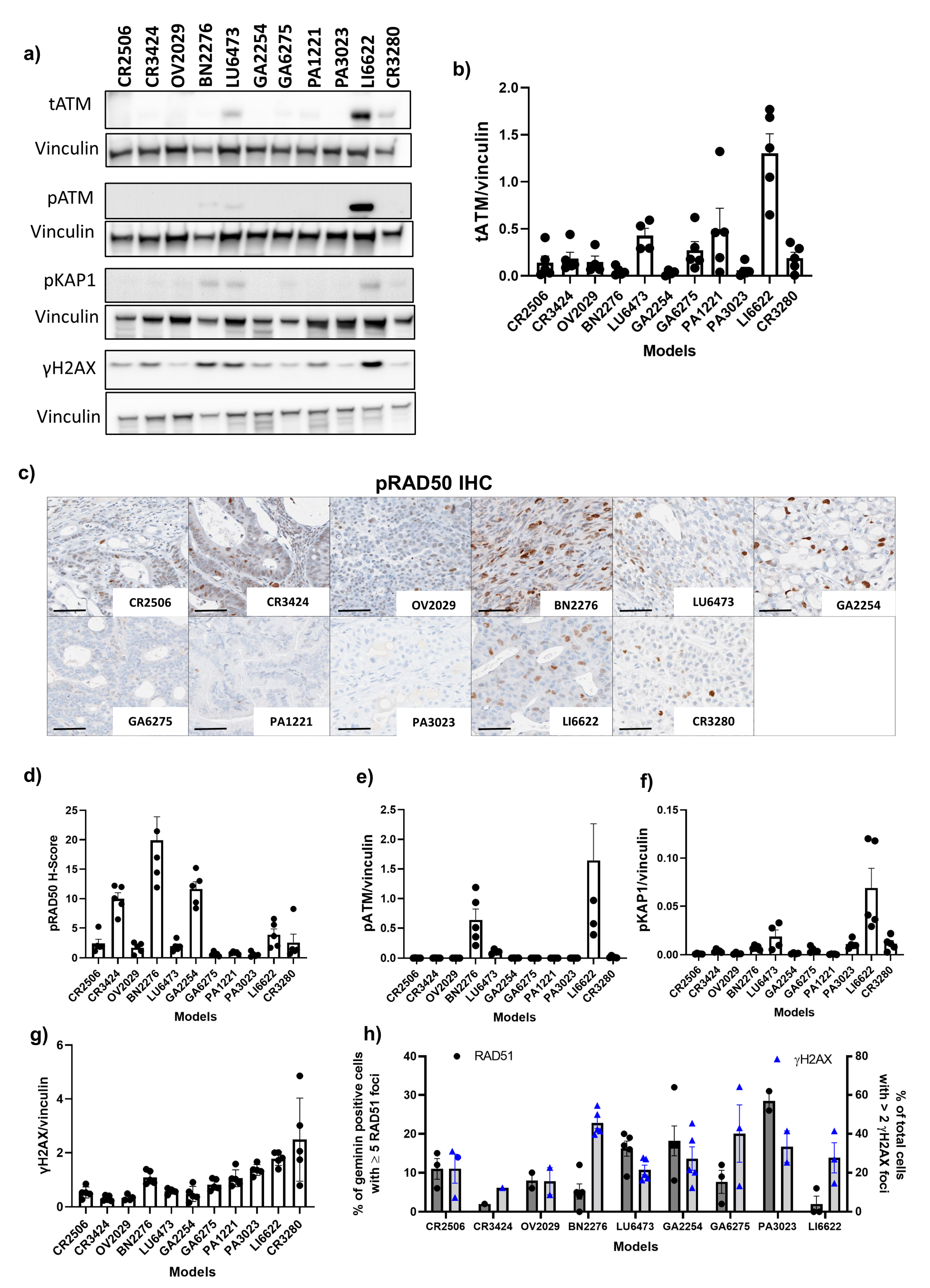
2.6. Histology and IHC
2.7. Tumour Protein Isolation and Immunoblotting
2.8. RAD51 Foci Staining
2.9. Whole-Exome Sequencing (WES)
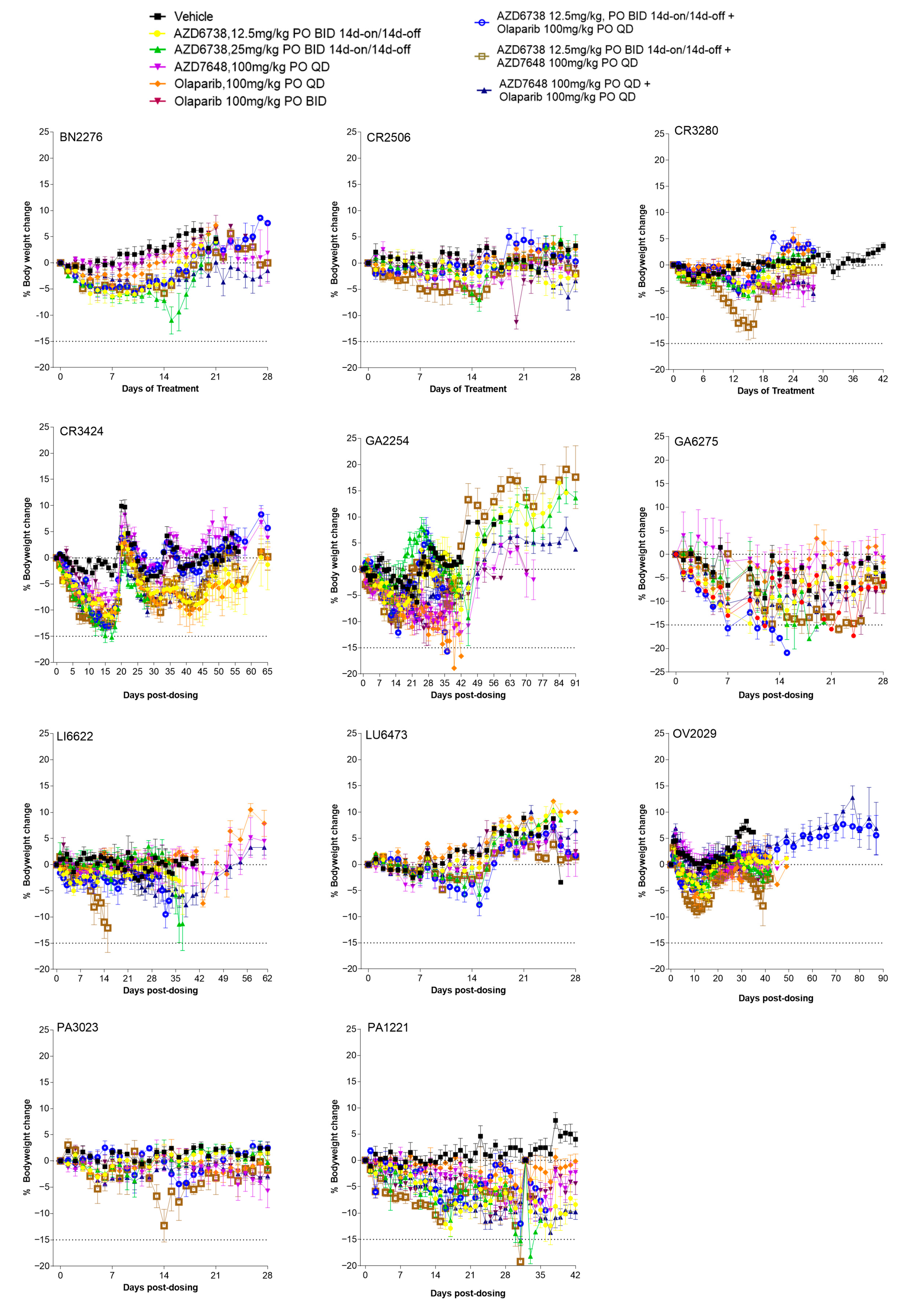
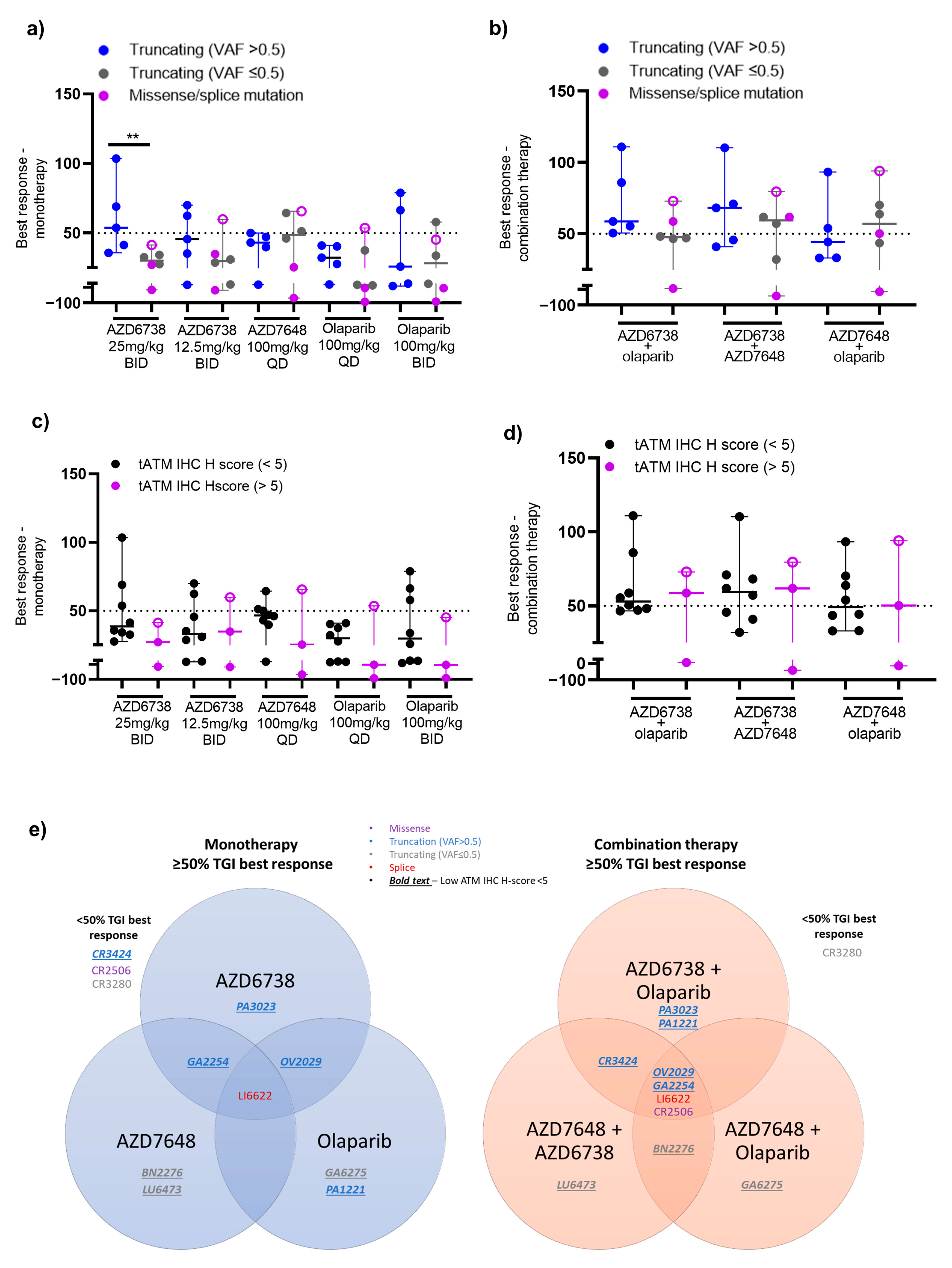
3. Results
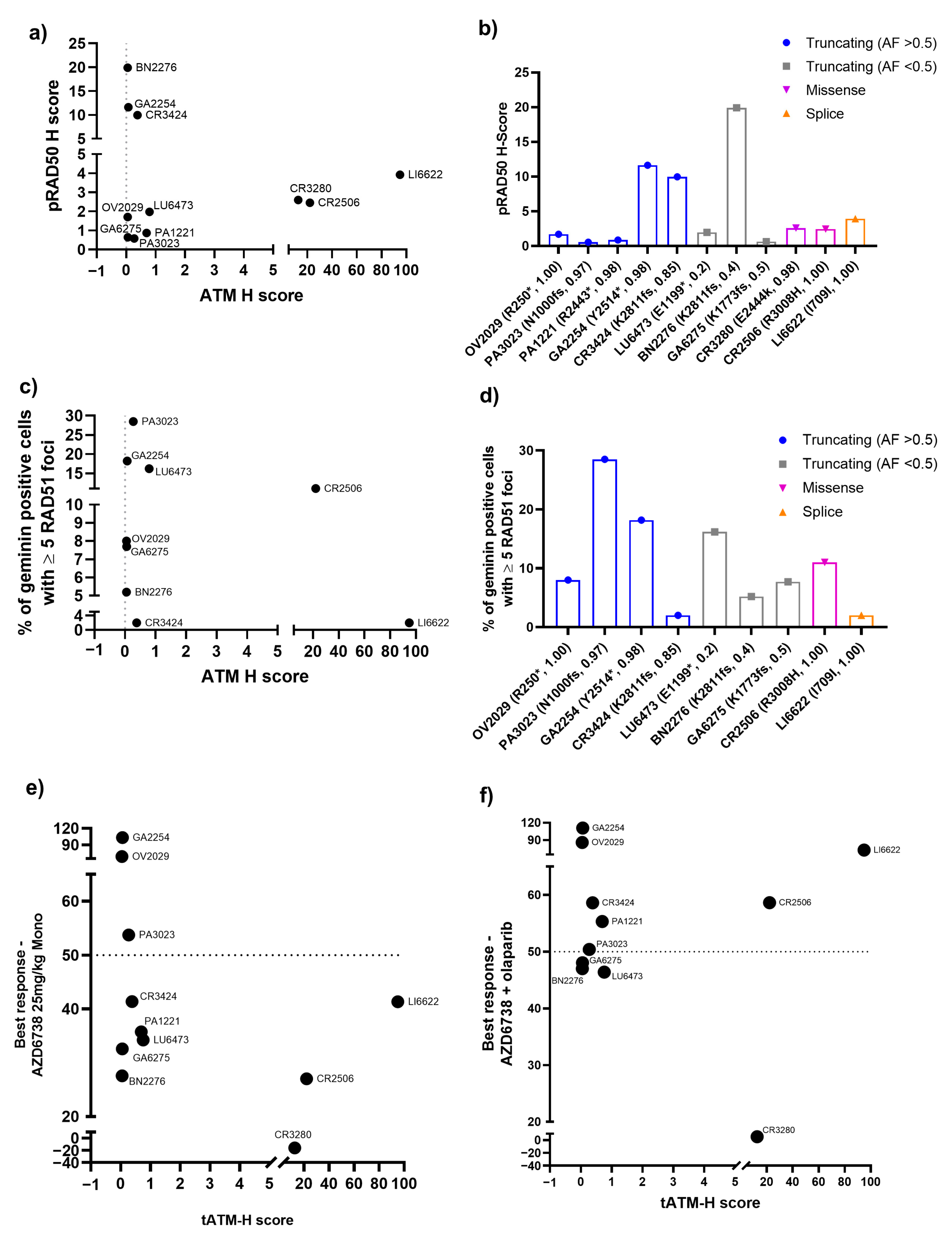
3.1. PDX Model Selection and Baseline Characterisation
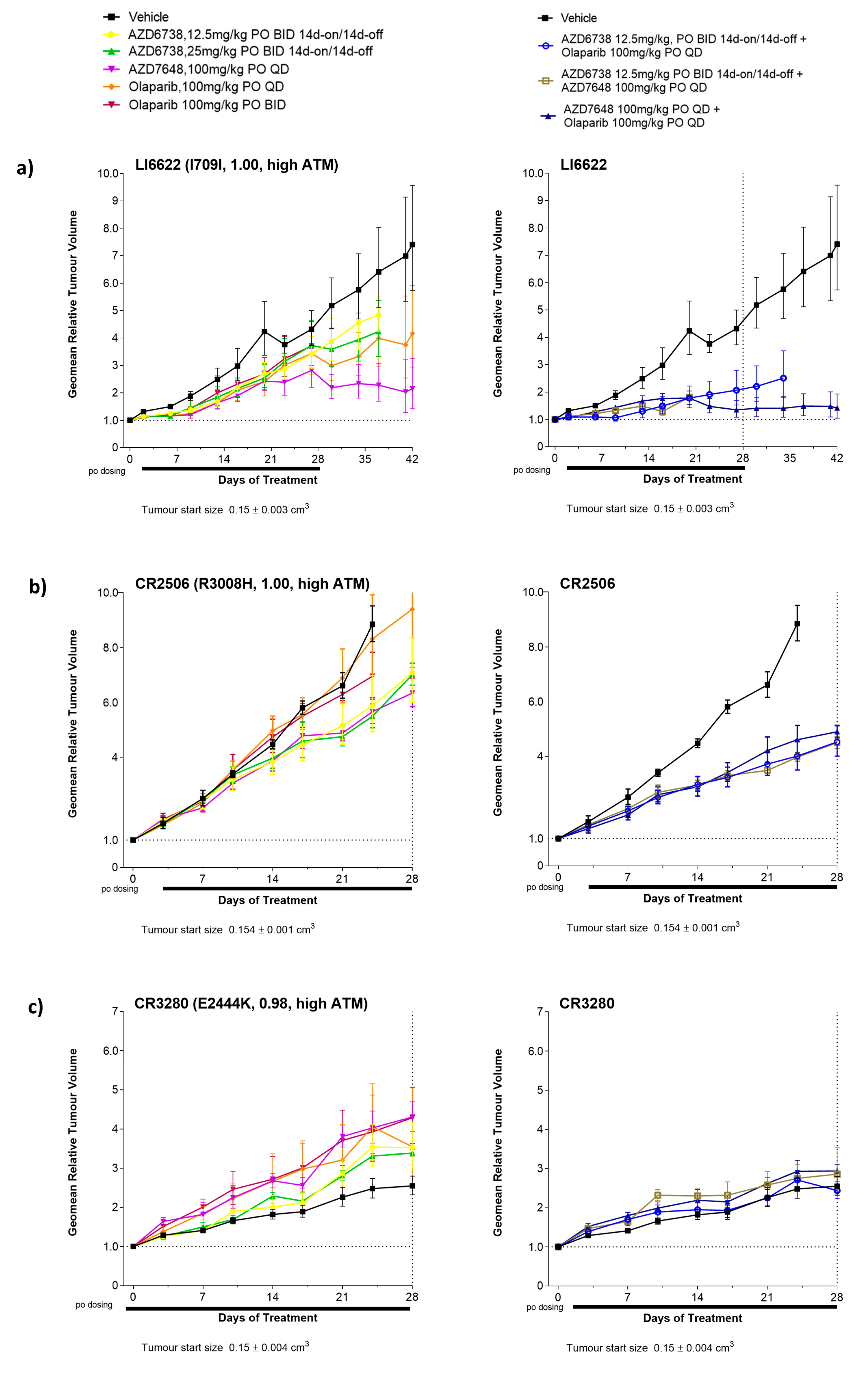
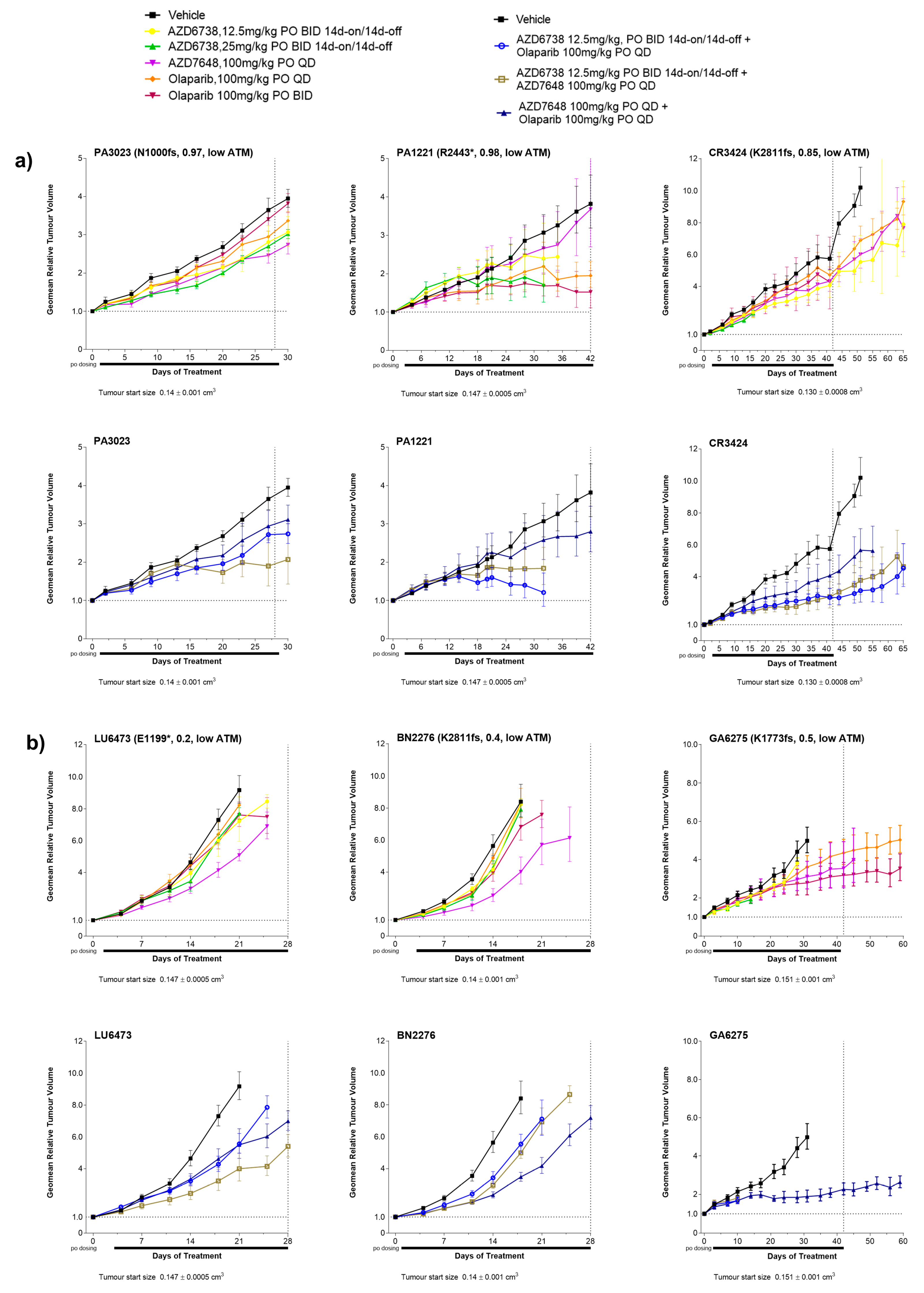
3.2. Antitumour Response to DDRi Agents in PDX Models with ATM Mutations
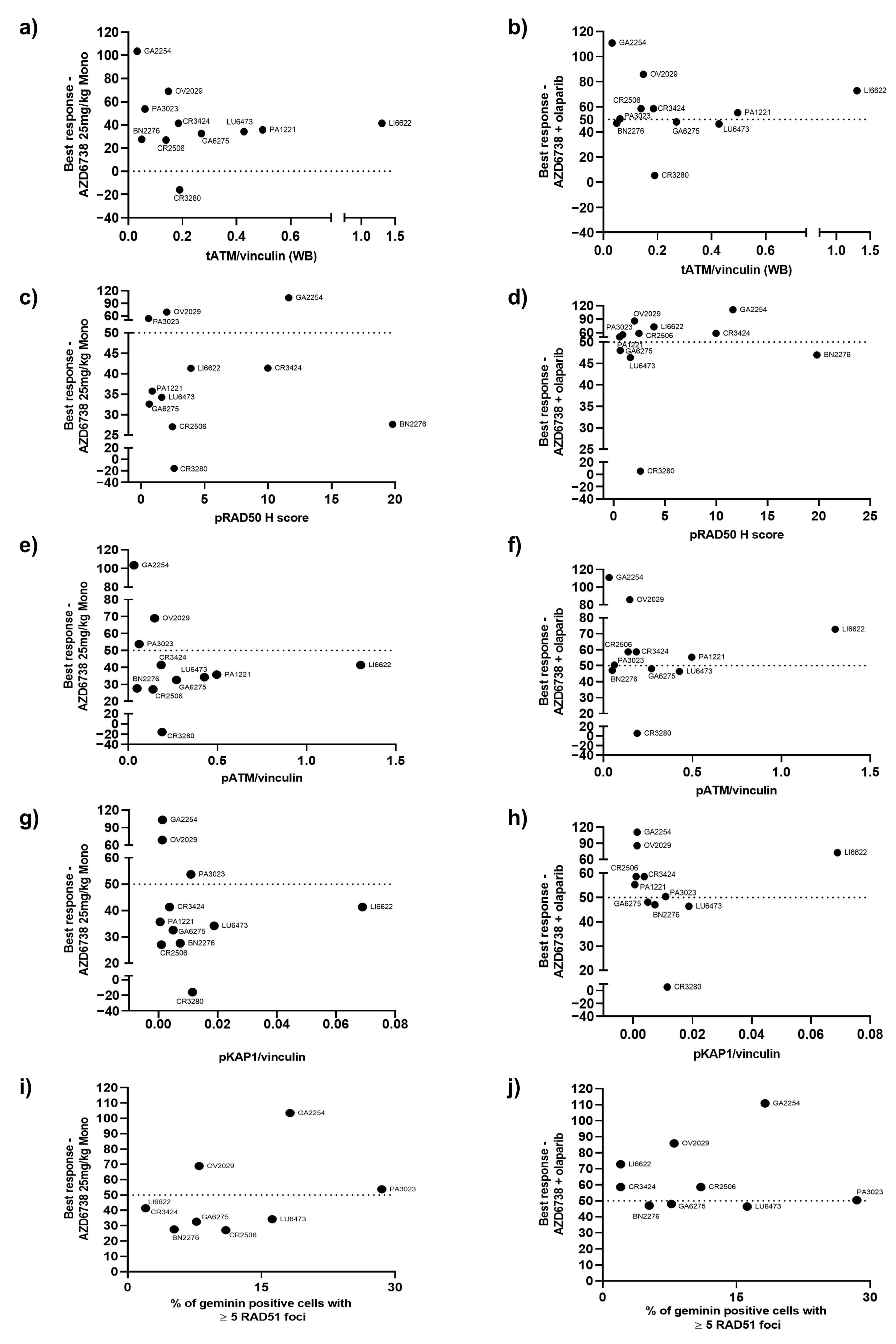
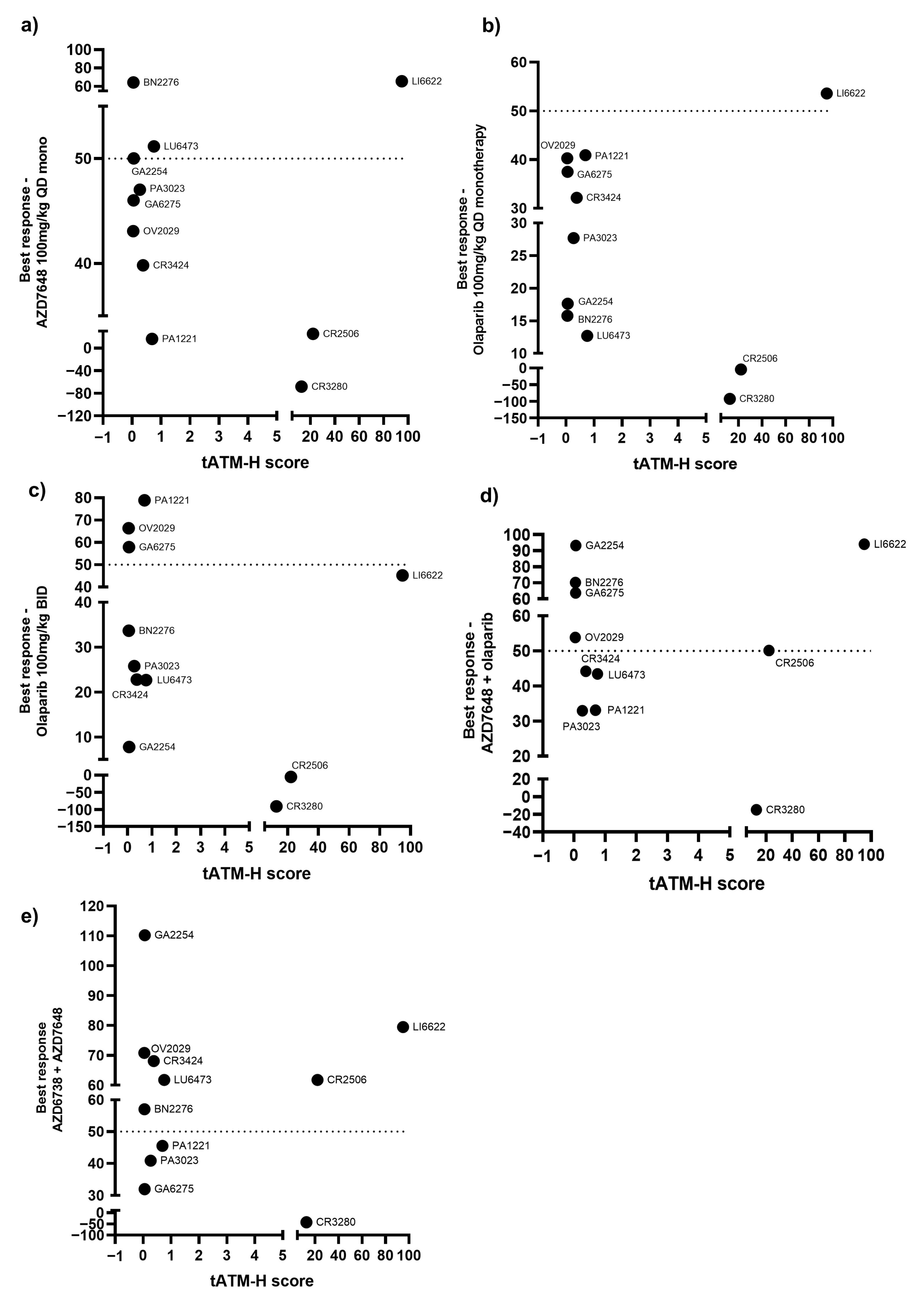
3.3. Correlation of the Best Response in PDX Models with ATM Protein Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model ID | Cancer Type | Subtype | Biopsy Site | Stage | Treatment History | Age | Gender | Ethnicity |
|---|---|---|---|---|---|---|---|---|
| CR2506 | Colorectal Cancer | ADC | Colorectum | NA | NA | 74 | Female | Asian |
| CR3424 | Colorectal Cancer | ADC, mucinous | Primary | NA | NA | NA | Female | Asian |
| OV2029 | Ovarian Cancer | Serous ADC | NA | NA | NA | NA | Female | Caucasian |
| BN2276 | Brain Cancer | Glioblastoma | Brain | NA | NA | 65 | Female | Asian |
| LU6473 | Lung Cancer | LCNEC | NA | NA | NA | 84 | Female | Caucasian |
| GA2254 | Gastric Cancer | ADC | Primary | cT4aN3M1 | NA | 67 | Female | Asian |
| GA6275 | Gastric Cancer | ADC | Primary | NA | NA | 75 | Male | Asian |
| PA1221 | Pancreatic Cancer | Ductal ADC | Pancreas | NA | NA | 67 | Female | Asian |
| PA3023 | Pancreatic Cancer | Ductal ADC | Pancreas | NA | NA | 82 | Female | Asian |
| LI6622 | Liver Cancer | HCC | Liver, right lobe | NA | NA | 34 | Male | Asian |
| CR3280 | Colorectal Cancer | ADC | Lymph node | NA | NA | 63 | Female | Asian |
| CTG 0828 | Lung | NSCLC | Lymph node | II | Naive | 81 | Female | Caucasian |
| CTG 1140 | Head and neck | SCC | Salivary gland | II | NA | 70 | Male | Caucasian |
| CTG 0166 | Lung | NSCLC | Lung | I | Pre-treated | 63 | Female | Caucasian |
| CTG 0198 | Lung | SCLC | Lung | III | Pre-treated | 67 | Male | NA |
| CTG 0149 | Head and neck | SCC | Skin | III | Naive | 77 | Male | Caucasian |
| CTG 0776 | Head and neck | SCC | Tongue | II | Naive | 72 | Female | Caucasian |
| Median Best Response as % TGI (95% CI) | * p-Value | |||||||
|---|---|---|---|---|---|---|---|---|
| Treatment Group | Dose (mg/kg) and Schedule | ATM Truncating VAF > 0.5 (n = 5) A | ATM Truncating VAF ≤ 0.5 (n = 3) B | ATM Missense (n = 3) C | ATM Truncating VAF ≤ 0.5 + Missense (n = 6) B + C | A vs. B | A vs. C | A vs. B + C |
| AZD6738 | 25 BID 14ON 14OFF | 54 (27, 94) | 33 (23, 40) | 27 (−20, 60) | 30 (3, 46) | 0.03 | 0.03 | 0.009 |
| AZD6738 | 12.5 BID 14ON 14OFF | 46 (19, 73) | 29 (9, 43) | 35 (−74, 125) | 30 (−1, 53) | 0.3 | 0.4 | 0.2 |
| AZD7648 | 100 QD continuous | 43 (23, 56) | 51 (30, 77) | 25 (−163, 178) | 49 (−22, 84) | 0.1 | 0.8 | 0.5 |
| Olaparib | 100 QD continuous | 32 (20, 44) | 16 (−12, 56) | −4 (−197, 168) | 14 (−50, 58) | 0.3 | 0.6 | 0.3 |
| Olaparib | 100 BID continuous | 26 (2, 78) | 34 (−7, 83) | −5 (−188, 154) | 28 (−46, 68) | >0.99 | 0.3 | 0.4 |
| AZD6738 + olaparib | 12.5 BID 14ON 14OFF + 100 QD continuous | 59 (40, 104) | 47 (45, 49) | 59 (−43, 134) | 48 (23, 70) | 0.03 | 0.8 | 0.1 |
| AZD6738 + AZD7648 | 12.5 BID 14ON 14OFF + 100 QD continuous | 68 (33, 101) | 57 (11, 90) | 62 (−131, 197) | 59 (−5, 88) | 0.4 | 0.8 | 0.4 |
| AZD7648 + olaparib | 100 QD + 100 QD continuous | 44 (21, 82) | 64 (25, 94) | 50 (−93, 179) | 57 (12, 90) | 0.6 | >0.99 | 0.7 |
| Median Best Response as % TGI (95% CI) | ||||
|---|---|---|---|---|
| Treatment Group | Dose (mg/kg) and Schedule | ATM IHC H Score < 5 (n = 8) | ATM IHC H Score > 5 (n = 3) | * p-Value |
| Monotherapy | ||||
| AZD6738 | 25 BID 14ON 14OFF | 39 (28, 104) | 27 (−16, 41) | 0.1 |
| AZD6738 | 12.5 BID 14ON 14OFF | 33 (16, 70) | 35 (−18, 60) | 0.8 |
| AZD7648 | 100 QD continuous | 47 (16, 64) | 25 (−68, 66) | 0.6 |
| Olaparib | 100 QD continuous | 30 (13, 41) | −4 (−92, 54) | 0.5 |
| Olaparib | 100 BID continuous | 30 (8, 79) | −5 (−91, 45) | 0.2 |
| Combinations | ||||
| AZD6738 + olaparib | 12.5 BID (14ON 14OFF) + 100 QD continuous | 53 (46, 111) | 58 (5, 73) | >0.99 |
| AZD6738 + AZD7648 | 12.5 BID (14ON 14OFF) + 100 QD continuous | 59 (32, 110) | 62 (−43, 80) | >0.99 |
| AZD7648 + olaparib | 100 QD + 100 QD continuous | 49 (33, 93) | 50 (−15, 94) | >0.99 |
| Treatment Group | Dose (mg/kg) and Schedule | Mean % TGI ± SEM | * p-Value Combo vs. Mono | ||
|---|---|---|---|---|---|
| ATM IHC H Score < 5 (n = 8) | ATM IHC H Score > 5 (n = 3) | ATM IHC H Score < 5 | ATM IHC H Score > 5 | ||
| AZD6738 | 25 BID 14ON 14OFF | 50 ± 9 | 18 ± 41 | NA | NA |
| AZD6738 | 12.5 BID 14ON 14OFF | 38 ± 7 | 25 ± 23 | NA | NA |
| AZD7648 | 100 QD continuous | 45 ± 5 | 8 ± 40 | NA | NA |
| Olaparib | 100 QD continuous | 28 ± 4 | −14 ± 42 | NA | NA |
| Olaparib | 100 BID continuous | 40 ± 9 | −17 ± 40 | NA | NA |
| AZD6738 + olaparib | 12.5 BID 14ON 14OFF + 100 QD continuous | 63 ± 8.3 | 46 ± 21 | vs. AZD6738: 0.001 vs. olaparib: 0.007 | vs. AZD6738: 0.03 vs. olaparib: 0.1 |
| AZD6738 + AZD7648 | 12.5 BID 14ON 14OFF + 100 QD continuous | 61 ± 8.6 | 33 ± 38 | vs. AZD6738: 0.001 vs. AZD7648: 0.1 | vs. AZD6738: 0.7 vs. AZD7648: 0.06 |
| AZD7648 + olaparib | 100 QD + 100 QD continuous | 54 ± 7.3 | 43 ± 32 | vs. AZD7648: 0.2 vs. olaparib: 0.03 | vs. AZD7648: 0.06 vs. olaparib: 0.2 |
References
- O’Connor, M.J. Targeting the DNA damage response in cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Capoluongo, E.; Ellison, G.; Lopez-Guerrero, J.A.; Penault-Llorca, F.; Ligtenberg, M.J.L.; Banerjee, S.; Singer, C.; Friedman, E.; Markiefka, B.; Schirmacher, P.; et al. Guidance statement on BRCA1/2 tumor testing in ovarian cancer patients. Semin. Oncol. 2017, 44, 187–197. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- US. Food and Drug Administration. FDA Approves Olaparib for gBRCAm Metastatic Pancreatic Adenocarcinoma; U.S. Food and Drug Administration: Silver Spring, MA, USA, 2019. [Google Scholar]
- Lord, C.J.; McDonald, S.; Swift, S.; Turner, N.C.; Ashworth, A. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair 2008, 7, 2010–2019. [Google Scholar] [CrossRef]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef]
- Turner, N.C.; Lord, C.J.; Iorns, E.; Brough, R.; Swift, S.; Elliott, R.; Rayter, S.; Tutt, A.N.; Ashworth, A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008, 27, 1368–1377. [Google Scholar] [CrossRef]
- Jette, N.R.; Kumar, M.; Radhamani, S.; Arthur, G.; Goutam, S.; Yip, S.; Kolinsky, M.; Williams, G.J.; Bose, P.; Lees-Miller, S.P. ATM-Deficient Cancers Provide New Opportunities for Precision Oncology. Cancers 2020, 12, 687. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Putti, S.; Giovinazzo, A.; Merolle, M.; Falchetti, M.L.; Pellegrini, M. ATM Kinase Dead: From Ataxia Telangiectasia Syndrome to Cancer. Cancers 2021, 13, 5498. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Aguilar-Quesada, R.; Munoz-Gamez, J.A.; Martin-Oliva, D.; Peralta, A.; Valenzuela, M.T.; Matinez-Romero, R.; Quiles-Perez, R.; Menissier-de Murcia, J.; de Murcia, G.; Ruiz de Almodovar, M.; et al. Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol. Biol. 2007, 8, 29. [Google Scholar] [CrossRef]
- Wang, C.; Jette, N.; Moussienko, D.; Bebb, D.G.; Lees-Miller, S.P. ATM-deficient colorectal cancer cells are sensitive to the PARP inhibitor olaparib. Transl. Oncol. 2017, 10, 190–196. [Google Scholar] [CrossRef]
- Weston, V.J.; Oldreive, C.E.; Skowronska, A.; Oscier, D.G.; Pratt, G.; Dyer, M.J.; Smith, G.; Powell, J.E.; Rudzki, Z.; Kearns, P.; et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 2010, 116, 4578–4587. [Google Scholar] [CrossRef]
- Williamson, C.T.; Kubota, E.; Hamill, J.D.; Klimowicz, A.; Ye, R.; Muzik, H.; Dean, M.; Tu, L.; Gilley, D.; Magliocco, A.M.; et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol. Med. 2012, 4, 515–527. [Google Scholar] [CrossRef]
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627. [Google Scholar] [CrossRef]
- Forment, J.V.; O’Connor, M.J. Targeting the replication stress response in cancer. Pharmacol. Ther. 2018, 188, 155–167. [Google Scholar] [CrossRef]
- Wilson, Z.; Odedra, R.; Wallez, Y.; Wijnhoven, P.W.G.; Hughes, A.M.; Gerrard, J.; Jones, G.N.; Bargh-Dawson, H.; Brown, E.; Young, L.A.; et al. ATR inhibitor AZD6738 (ceralasertib) exerts antitumor activity as a monotherapy and in combination with chemotherapy and the PARP inhibitor olaparib. Cancer Res. 2022, 82, 1140–1152. [Google Scholar] [CrossRef]
- Jette, N.R.; Radhamani, S.; Ye, R.; Yu, Y.; Arthur, G.; Goutam, S.; Bismar, T.A.; Kumar, M.; Bose, P.; Yip, S.; et al. ATM-deficient lung, prostate and pancreatic cancer cells are acutely sensitive to the combination of olaparib and the ATR inhibitor AZD6738. Genome Instab. Dis. 2020, 1, 197–205. [Google Scholar] [CrossRef]
- Schmitt, A.; Knittel, G.; Welcker, D.; Yang, T.P.; George, J.; Nowak, M.; Leeser, U.; Buttner, R.; Perner, S.; Peifer, M.; et al. ATM deficiency is associated with sensitivity to PARP1- and ATR inhibitors in lung adenocarcinoma. Cancer Res. 2017, 77, 3040–3056. [Google Scholar] [CrossRef]
- Vendetti, F.P.; Lau, A.; Schamus, S.; Conrads, T.P.; O’Connor, M.J.; Bakkenist, C.J. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget 2015, 6, 44289–44305. [Google Scholar] [CrossRef] [PubMed]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef]
- Kwok, M.; Davies, N.; Agathanggelou, A.; Smith, E.; Oldreive, C.; Petermann, E.; Stewart, G.; Brown, J.; Lau, A.; Pratt, G.; et al. ATR inhibition induces synthetic lethality and overcomes chemoresistance in TP53- or ATM-defective chronic lymphocytic leukemia cells. Blood 2016, 127, 582–595. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.L.; Wijnhoven, P.W.G.; Ramos-Montoya, A.; Wilson, Z.; Illuzzi, G.; Falenta, K.; Jones, G.N.; James, N.; Chabbert, C.D.; Stott, J.; et al. Combined PARP and ATR inhibition potentiates genome instability and cell death in ATM-deficient cancer cells. Oncogene 2020, 39, 4869–4883. [Google Scholar] [CrossRef]
- Min, A.; Im, S.A.; Jang, H.; Kim, S.; Lee, M.; Kim, D.K.; Yang, Y.; Kim, H.J.; Lee, K.H.; Kim, J.W.; et al. AZD6738, a novel oral inhibitor of ATR, induces synthetic lethality with ATM deficiency in gastric cancer cells. Mol. Cancer Ther. 2017, 16, 566–577. [Google Scholar] [CrossRef]
- Neeb, A.; Herranz, N.; Arce-Gallego, S.; Miranda, S.; Buroni, L.; Yuan, W.; Athie, A.; Casals, T.; Carmichael, J.; Rodrigues, D.N.; et al. Advanced prostate cancer with ATM loss: PARP and ATR inhibitors. Eur. Urol. 2021, 79, 200–211. [Google Scholar] [CrossRef]
- Rafiei, S.; Fitzpatrick, K.; Liu, D.; Cai, M.Y.; Elmarakeby, H.A.; Park, J.; Ricker, C.; Kochupurakkal, B.S.; Choudhury, A.D.; Hahn, W.C.; et al. ATM loss confers greater sensitivity to ATR inhibition than PARP inhibition in prostate cancer. Cancer Res. 2020, 80, 2094–2100. [Google Scholar] [CrossRef] [PubMed]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutiérrez-Enríquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzmán, M.; et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef]
- National Library of Medicine. ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/RCV000211973 (accessed on 13 September 2022).
- Takagi, M.; Tsuchida, R.; Oguchi, K.; Shigeta, T.; Nakada, S.; Shimizu, K.; Ohki, M.; Delia, D.; Chessa, L.; Taya, Y.; et al. Identification and characterization of polymorphic variations of the ataxia telangiectasia mutated (ATM) gene in childhood Hodgkin disease. Blood 2004, 103, 283–290. [Google Scholar] [CrossRef]
- Villaruz, L.C.; Jones, H.; Dacic, S.; Abberbock, S.; Kurland, B.F.; Stabile, L.P.; Siegfried, J.M.; Conrads, T.P.; Smith, N.R.; O’Connor, M.J.; et al. ATM protein is deficient in over 40% of lung adenocarcinomas. Oncotarget 2016, 7, 57714–57725. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.N.; Rooney, C.; Griffin, N.; Roudier, M.; Young, L.A.; Garcia-Trinidad, A.; Hughes, G.D.; Whiteaker, J.R.; Wilson, Z.; Odedra, R.; et al. pRAD50: A novel and clinically applicable pharmacodynamic biomarker of both ATM and ATR inhibition identified using mass spectrometry and immunohistochemistry. Br. J. Cancer 2018, 119, 1233–1243. [Google Scholar] [CrossRef]
- Graeser, M.; McCarthy, A.; Lord, C.J.; Savage, K.; Hills, M.; Salter, J.; Orr, N.; Parton, M.; Smith, I.E.; Reis-Filho, J.S.; et al. A marker of homologous recombination predicts pathologic complete response to neoadjuvant chemotherapy in primary breast cancer. Clin. Cancer Res. 2010, 16, 6159–6168. [Google Scholar] [CrossRef]
- Pellegrino, B.; Herencia-Ropero, A.; Llop-Guevara, A.; Pedretti, F.; Moles-Fernandez, A.; Viaplana, C.; Villacampa, G.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Preclinical In Vivo Validation of the RAD51 Test for Identification of Homologous Recombination-Deficient Tumors and Patient Stratification. Cancer Res. 2022, 82, 1646–1657. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for metastatic castration-resistant prostate cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Bang, Y.J.; Xu, R.H.; Chin, K.; Lee, K.W.; Park, S.H.; Rha, S.Y.; Shen, L.; Qin, S.; Xu, N.; Im, S.A.; et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1637–1651. [Google Scholar] [CrossRef]
- Yap, T.A.; Tan, D.S.P.; Terbuch, A.; Caldwell, R.; Guo, C.; Goh, B.C.; Heong, V.; Haris, N.R.M.; Bashir, S.; Drew, Y.; et al. First-in-human trial of the oral ataxia telangiectasia and RAD3-related (ATR) inhibitor BAY 1895344 in patients with advanced solid tumors. Cancer Discov. 2021, 11, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Tan, D.S.; Stathis, A.; Shapiro, G.I.; Iwasa, S.; Joerger, M.; Zhang, J.; Plummer, R.; Sawyer, M.; Tan, A.C.; et al. Phase Ib expansion trial of the safety and efficacy of the oral ataxia telangiectasia and Rad3-related (ATR) inhibitor elimusertib in advanced solid tumors with DNA damage response (DDR) defects (Abstract CT006). Cancer Res. 2022, 82, CT006. [Google Scholar] [CrossRef]
- Yap, T.A.; Fontana, E.; Lee, E.K.; Spigel, D.R.; Hojgaard, M.; Lheureux, S.; Mettu, N.B.; Carneiro, B.A.; Carter, L.; Plummer, R.; et al. Camonsertib in DNA damage response-deficient advanced solid tumors: Phase 1 trial results. Nat. Med. 2023, 29, 1400–1411. [Google Scholar] [CrossRef] [PubMed]
- Jamal, K.; Galbiati, A.; Armenia, J.; Illuzzi, G.; Hall, J.; Bentouati, S.; Barrell, D.; Ahdesmäki, M.; Functional Genomics Centre Group; O’Connor, M.J.; et al. Drug–gene Interaction Screens Coupled to Tumor Data Analyses Identify the Most Clinically Relevant Cancer Vulnerabilities Driving Sensitivity to PARP Inhibition. Cancer Res. Commun. 2022, 2, 1244–1254. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Study Investigating DNA-Damage Response Agents in Molecularly Altered Advanced Cancer. 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04564027 (accessed on 31 May 2023).

| Treatment Group | Comments |
|---|---|
| Vehicle control | |
| AZD6738, 12.5 mg/kg PO BID (14 days on, 14 days off) | BID dosed 8 h apart |
| AZD6738, 25 mg/kg PO BID (14 days on, 14 days off) | BID dosed 8 h apart |
| AZD7648, 100 mg/kg PO QD continuous | |
| Olaparib, 100 mg/kg PO QD continuous | |
| Olaparib, mono 100 mg/kg PO BID | |
| AZD7648, 100 mg/kg PO QD continuous | |
| AZD6738, 12.5 mg/kg PO BID (14 days on, 14 days off) + olaparib 100 mg/kg PO QD continuous | AZD6738 dosed 1 h after olaparib dose |
| AZD6738, 12.5 mg/kg PO BID (14 days on, 14 days off) + AZD7648, 100 mg/kg PO QD | AZD6738 dosed 1 h after AZD7648 dose |
| AZD7648, 100 mg/kg PO QD + olaparib, 100 mg/kg PO QD | Olaparib dosed 1 h after AZD7648 dose |
| Model ID | Tumour Type | Protein Change | Mutation Type | ATM VAF Original (a) | ATM mRNA Expression [log2(Value + 1)] | tATM Mean IHC H Score | pRAD50 Mean IHC H Score | pATM/Vinc WB Signal (b) | pKAP1/Vinc WB Signal (b) |
|---|---|---|---|---|---|---|---|---|---|
| CR2506 | Colorectal (ADC) | R3008H | Missense | 1 (1) | 10.28 | 22.2 | 2.45 | 0.002 (0.13) | 0.001 (1.5) |
| CR3424 | Colorectal (ADC) | K2811fs | Frameshift | 1 (0.85) | NA | 0.38 | 9.97 | 0.002 (0.11) | 0.004 (5.4) |
| OV2029 | Ovarian | R250 * | Nonsense | 1 (1) | NA | 0.05 | 1.70 | 0 (0) | 0.001 (1.9) |
| BN2276 | Glioblastoma | K2811fs | Frameshift | 0.63 (0.4) | NA | 0.05 | 19.90 | 0.64 (39) | 0.007 (10.7) |
| LU6473 | Lung | E1199 * | Nonsense | 0.53 (0.2) | 10.93 | 0.76 | 1.98 | 0.12 (6.6) | 0.019 (27.3) |
| GA2254 | Gastric | Y2514 * | Nonsense | 0.89 (0.98) | NA | 0.06 | 11.63 | 0.002 (0.11) | 0.001 (2) |
| GA6275 | Gastric (ADC) | K1773fs | Frameshift | 0.57 (0.5) | 11.33 | 0.05 | 0.64 | 0 (0) | 0.005 (7.2) |
| PA1221 | Pancreatic (ADC) | R2443 * | Nonsense | 0.97 (0.98) | 9.84 | 0.69 | 0.87 | 0 (0) | 0.001 (0.9) |
| PA3023 | Pancreatic (ADC) | N1000fs | Frameshift | 0.95 (0.97) | NA | 0.27 | 0.58 | 0 (0) | 0.011 (16) |
| LI6622 | Liver | I709I | Splice region | 1 (1) | 10.73 | 94.87 | 3.92 | 1.64 (100) | 0.069 (100) |
| CR3280 | Colorectal (ADC) | E2444K | Missense | 1 (0.98) | 10.55 | 12.68 | 2.60 | 0.016 (1) | 0.012 (16.8) |
| CTG 0828 | Large-cell lung (ADC) | E473 * | Nonsense | 1 | 1 | 2.16 | 10.6 | NA | NA |
| CTG 1140 | Head and neck (SCC) | R35 * | Nonsense | 0.73 | NA | 80.91 | NA | NA | NA |
| CTG 0166 | Lung (SCC) | W3055 * | Nonsense | 0.44 | NA | 55.49 | 10.4 | NA | NA |
| CTG 0198 | Small-cell lung | W1858 * | Nonsense | 0.42 | NA | 78.82 | NA | NA | NA |
| CTG 0149 | Head and neck | NA | NA | NA | 100 | 71.93 | NA | NA | NA |
| CTG 0776 | Head and neck | NA | NA | NA | 100 | 51.47 | NA | NA | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karmokar, A.; Sargeant, R.; Hughes, A.M.; Baakza, H.; Wilson, Z.; Talbot, S.; Bloomfield, S.; Leo, E.; Jones, G.N.; Likhatcheva, M.; et al. Relevance of ATM Status in Driving Sensitivity to DNA Damage Response Inhibitors in Patient-Derived Xenograft Models. Cancers 2023, 15, 4195. https://doi.org/10.3390/cancers15164195
Karmokar A, Sargeant R, Hughes AM, Baakza H, Wilson Z, Talbot S, Bloomfield S, Leo E, Jones GN, Likhatcheva M, et al. Relevance of ATM Status in Driving Sensitivity to DNA Damage Response Inhibitors in Patient-Derived Xenograft Models. Cancers. 2023; 15(16):4195. https://doi.org/10.3390/cancers15164195
Chicago/Turabian StyleKarmokar, Ankur, Rebecca Sargeant, Adina M. Hughes, Hana Baakza, Zena Wilson, Sara Talbot, Sarah Bloomfield, Elisabetta Leo, Gemma N. Jones, Maria Likhatcheva, and et al. 2023. "Relevance of ATM Status in Driving Sensitivity to DNA Damage Response Inhibitors in Patient-Derived Xenograft Models" Cancers 15, no. 16: 4195. https://doi.org/10.3390/cancers15164195
APA StyleKarmokar, A., Sargeant, R., Hughes, A. M., Baakza, H., Wilson, Z., Talbot, S., Bloomfield, S., Leo, E., Jones, G. N., Likhatcheva, M., Tobalina, L., Dean, E., Cadogan, E. B., & Lau, A. (2023). Relevance of ATM Status in Driving Sensitivity to DNA Damage Response Inhibitors in Patient-Derived Xenograft Models. Cancers, 15(16), 4195. https://doi.org/10.3390/cancers15164195