
Dysregulation of Type I Interferon (IFN-I) Signaling: A Potential Contributor to Racial Disparity in Hepatocellular Carcinoma (HCC)

 , ,
, ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Chemicals
2.2. Analysis of TCGA RNA-sequencing (RNA-seq) Data
2.3. RNA Sequencing (RNA-seq) of Tumor Samples
2.4. Total RNA Extraction, cDNA Preparation and Quantitative Real Time PCR of Tumor Samples
2.5. RNA in situ Hybridization (RNAscope) Assay
2.6. Immunohistochemistry
2.7. Ethanolic Extraction of Ginger
2.8. Measurement of Cell Proliferation by MTT Assay
2.9. Western Blot Analysis
2.10. Measurement of ISG Expression by Real-Time Polymerase Chain Reaction (RT-PCR)
2.11. Statistical Analysis
3. Results
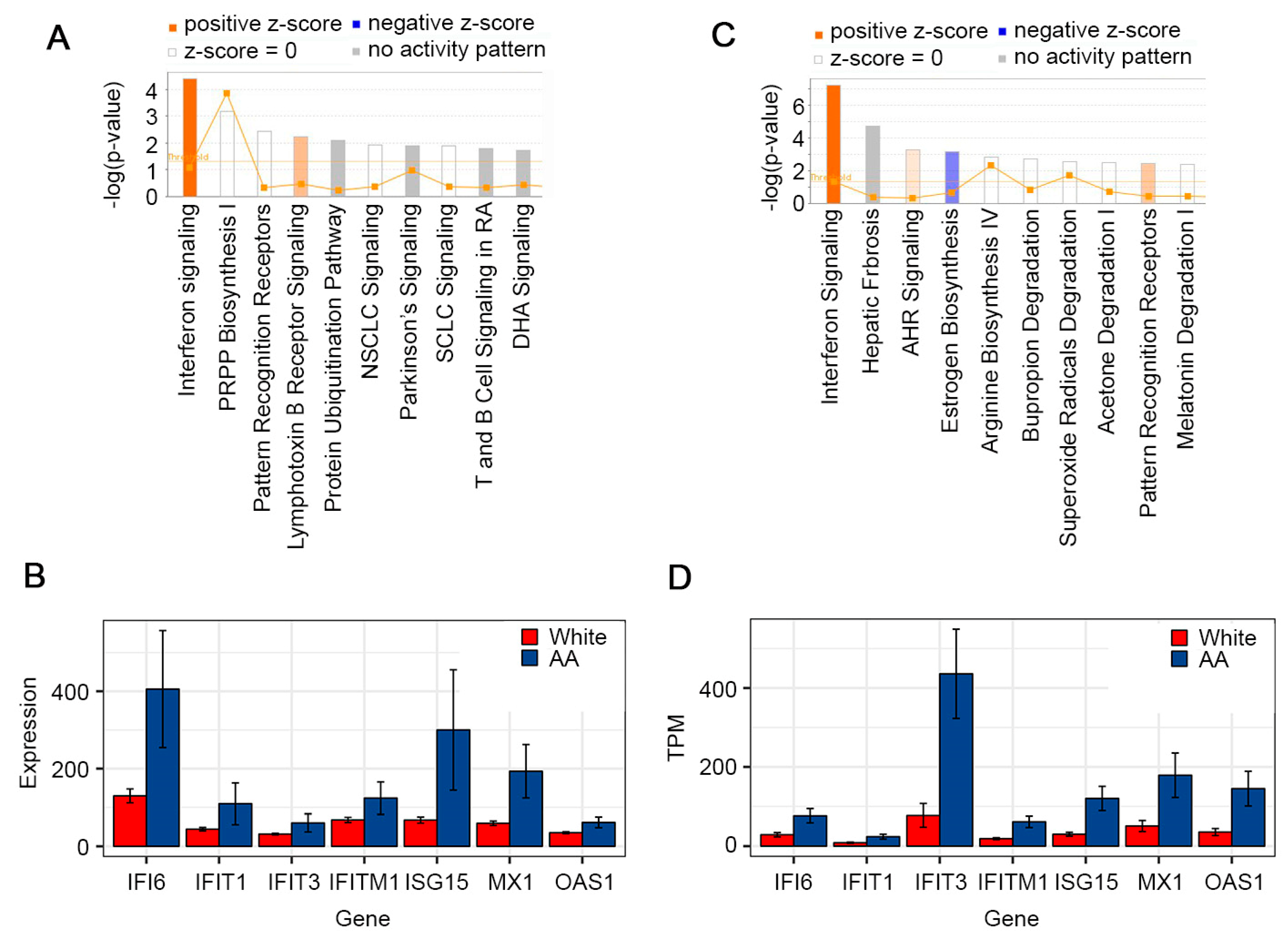
3.1. Analysis of TCGA Database
3.2. In-House RNA-seq and Analysis of White vs. AA/Black HCC Samples
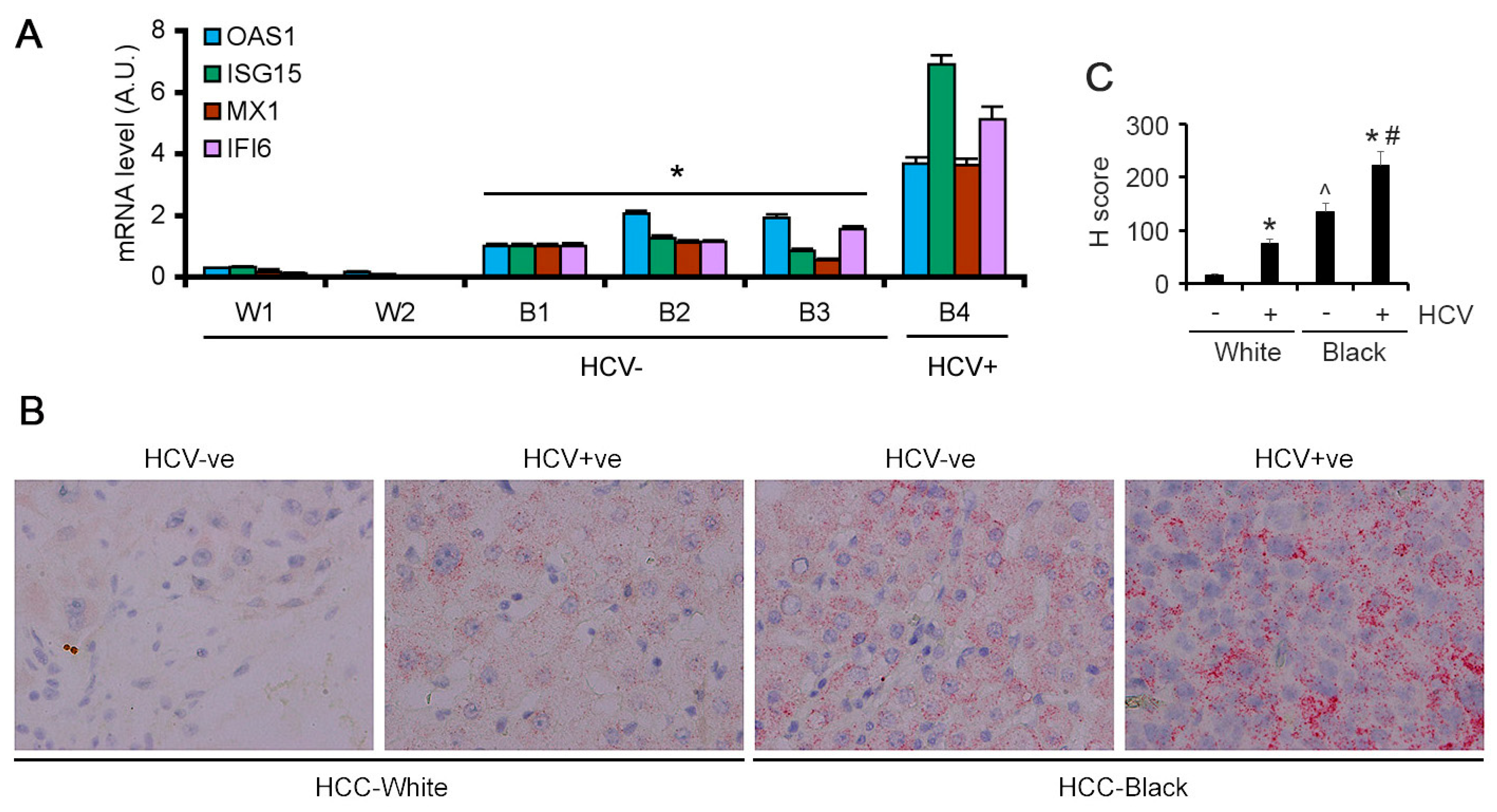
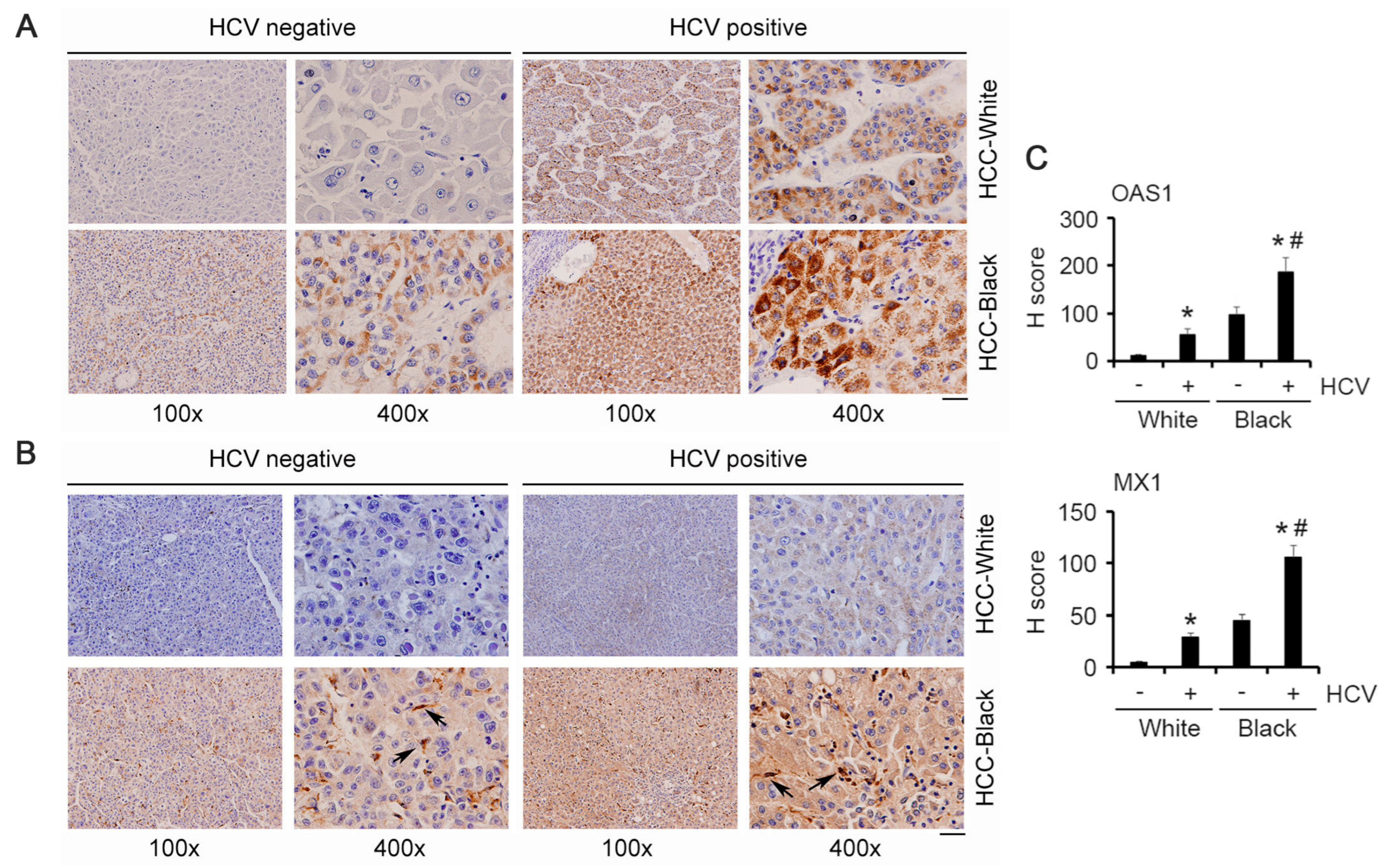
3.3. Validation in Patient Samples
3.4. Effect of Ginger Extract (GE) on the Viability of HCC Cell Lines
3.5. Effect of GE Treatment on JAK/STAT Signaling Pathway
3.6. Expression Level of ISGs after GE Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Burroughs, A.; Bruix, J. Hepatocellular carcinoma. Lancet 2003, 362, 1907–1917. [Google Scholar] [CrossRef]
- Lee, M.S.; Ryoo, B.Y.; Hsu, C.H.; Numata, K.; Stein, S.; Verret, W.; Hack, S.P.; Spahn, J.; Liu, B.; Abdullah, H.; et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): An open-label, multicentre, phase 1b study. Lancet Oncol. 2020, 21, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Casak, S.J.; Donoghue, M.; Fashoyin-Aje, L.; Jiang, X.; Rodriguez, L.; Shen, Y.L.; Xu, Y.; Jiang, X.; Liu, J.; Zhao, H.; et al. FDA Approval Summary: Atezolizumab Plus Bevacizumab for the Treatment of Patients with Advanced Unresectable or Metastatic Hepatocellular Carcinoma. Clin. Cancer Res. 2021, 27, 1836–1841. [Google Scholar] [CrossRef]
- Islami, F.; Miller, K.D.; Siegel, R.L.; Fedewa, S.A.; Ward, E.M.; Jemal, A. Disparities in liver cancer occurrence in the United States by race/ethnicity and state. CA A Cancer J. Clin. 2017, 67, 273–289. [Google Scholar] [CrossRef]
- Rich, N.E.; Hester, C.; Odewole, M.; Murphy, C.C.; Parikh, N.D.; Marrero, J.A.; Yopp, A.C.; Singal, A.G. Racial and Ethnic Differences in Presentation and Outcomes of Hepatocellular Carcinoma. Clin. Gastroenterol. Hepatol. 2018, 17, 551–559. [Google Scholar] [CrossRef]
- Harrison, L.E.; Reichman, T.; Koneru, B.; Fisher, A.; Wilson, D.; Dela Torre, A.; Samanta, A.; Korogodsky, M. Racial discrepancies in the outcome of patients with hepatocellular carcinoma. Arch. Surg. 2004, 139, 992–996. [Google Scholar] [CrossRef]
- Li, J.; Hansen, B.E.; Peppelenbosch, M.P.; De Man, R.A.; Pan, Q.; Sprengers, D. Factors associated with ethnical disparity in overall survival for patients with hepatocellular carcinoma. Oncotarget 2017, 8, 15193–15204. [Google Scholar] [CrossRef]
- Robbins, A.S.; Cox, D.D.; Johnson, L.B.; Ward, E.M. Persistent disparities in liver transplantation for patients with hepatocellular carcinoma in the United States, 1998 through 2007. Cancer 2011, 117, 4531–4539. [Google Scholar] [CrossRef]
- Siegel, A.B.; McBride, R.B.; El-Serag, H.B.; Hershman, D.L.; Brown, R.S., Jr.; Renz, J.F.; Emond, J.; Neugut, A.I. Racial disparities in utilization of liver transplantation for hepatocellular carcinoma in the United States, 1998-2002. Am. J. Gastroenterol. 2008, 103, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.M.; Liu, L.I.; Abraham, I.E.; Uy, A.B.; Dudek, A.Z. Efficacy and Safety of Sorafenib in a Racially Diverse Patient Population with Advanced Hepatocellular Carcinoma. Anticancer Res. 2018, 38, 4027–4034. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar] [CrossRef]
- Di Poto, C.; He, S.; Varghese, R.S.; Zhao, Y.; Ferrarini, A.; Su, S.; Karabala, A.; Redi, M.; Mamo, H.; Rangnekar, A.S.; et al. Identification of race-associated metabolite biomarkers for hepatocellular carcinoma in patients with liver cirrhosis and hepatitis C virus infection. PLoS ONE 2018, 13, e0192748. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://foodinsight.org/wp-content/uploads/2019/10/Gen-X-Special-Report_AICR-IFIC-.pdf (accessed on 1 January 2020).
- Panji, M.; Behmard, V.; Zare, Z.; Malekpour, M.; Nejadbiglari, H.; Yavari, S.; Nayerpour Dizaj, T.; Safaeian, A.; Bakhshi, A.; Abazari, O.; et al. Synergistic effects of green tea extract and paclitaxel in the induction of mitochondrial apoptosis in ovarian cancer cell lines. Gene 2021, 787, 145638. [Google Scholar] [CrossRef]
- Kamat, A.M.; Tharakan, S.T.; Sung, B.; Aggarwal, B.B. Curcumin potentiates the antitumor effects of Bacillus Calmette-Guerin against bladder cancer through the downregulation of NF-kappaB and upregulation of TRAIL receptors. Cancer Res. 2009, 69, 8958–8966. [Google Scholar] [CrossRef]
- Zadorozhna, M.; Mangieri, D. Mechanisms of Chemopreventive and Therapeutic Proprieties of Ginger Extracts in Cancer. Int. J. Mol. Sci. 2021, 22, 6599. [Google Scholar] [CrossRef]
- Hamza, A.A.; Heeba, G.H.; Hamza, S.; Abdalla, A.; Amin, A. Standardized extract of ginger ameliorates liver cancer by reducing proliferation and inducing apoptosis through inhibition oxidative stress/ inflammation pathway. Biomed. Pharmacother. 2021, 134, 111102. [Google Scholar] [CrossRef]
- Li, H.; Rafie, R.; Xu, Z.; Siddiqui, R.A. Phytochemical profile and anti-oxidation activity changes during ginger (Zingiber officinale) harvest: Baby ginger attenuates lipid accumulation and ameliorates glucose uptake in HepG2 cells. Food Sci. Nutr. 2022, 10, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, J.; Robertson, C.L.; Rajasekaran, D.; Gredler, R.; Siddiq, A.; Emdad, L.; Mukhopadhyay, N.D.; Ghosh, S.; Hylemon, P.B.; Gil, G.; et al. AEG-1 Regulates Retinoid X Receptor and Inhibits Retinoid Signaling. Cancer Res. 2014, 74, 4364–4377. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, Article3. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. Royal Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 May 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Bullard, J.H.; Purdom, E.; Hansen, K.D.; Dudoit, S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinform. 2010, 11, 94. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Robertson, C.L.; Srivastava, J.; Siddiq, A.; Gredler, R.; Emdad, L.; Rajasekaran, D.; Akiel, M.; Shen, X.N.; Guo, C.; Giashuddin, S.; et al. Genetic deletion of AEG-1 prevents hepatocarcinogenesis. Cancer Res. 2014, 74, 6184–6193. [Google Scholar] [CrossRef]
- Snell, L.M.; McGaha, T.L.; Brooks, D.G. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017, 38, 542–557. [Google Scholar] [CrossRef]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. 2016, 16, 131–144. [Google Scholar] [CrossRef]
- El-Serag, H.B. Hepatocellular carcinoma. N. Engl. J. Med. 2011, 365, 1118–1127. [Google Scholar] [CrossRef] [PubMed]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.B.; Kacha, A.K.; Kline, J.; Woo, S.R.; Kranz, D.M.; Murphy, K.M.; Gajewski, T.F. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8α+ dendritic cells. J. Exp. Med. 2011, 208, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Obora, A.; Shiratori, Y.; Okuno, M.; Adachi, S.; Takano, Y.; Matsushima-Nishiwaki, R.; Yasuda, I.; Yamada, Y.; Akita, K.; Sano, T.; et al. Synergistic induction of apoptosis by acyclic retinoid and interferon-beta in human hepatocellular carcinoma cells. Hepatology 2002, 36, 1115–1124. [Google Scholar] [CrossRef]
- Harris, L.D.; Tabb, B.; Sodora, D.L.; Paiardini, M.; Klatt, N.R.; Douek, D.C.; Silvestri, G.; Muller-Trutwin, M.; Vasile-Pandrea, I.; Apetrei, C.; et al. Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J. Virol. 2010, 84, 7886–7891. [Google Scholar] [CrossRef]
- Jacquelin, B.; Mayau, V.; Targat, B.; Liovat, A.S.; Kunkel, D.; Petitjean, G.; Dillies, M.A.; Roques, P.; Butor, C.; Silvestri, G.; et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J. Clin. Investig. 2009, 119, 3544–3555. [Google Scholar] [CrossRef]
- Cunningham, C.R.; Champhekar, A.; Tullius, M.V.; Dillon, B.J.; Zhen, A.; de la Fuente, J.R.; Herskovitz, J.; Elsaesser, H.; Snell, L.M.; Wilson, E.B.; et al. Type I and Type II Interferon Coordinately Regulate Suppressive Dendritic Cell Fate and Function during Viral Persistence. PLoS Pathog. 2016, 12, e1005356. [Google Scholar] [CrossRef]
- Mueller, S.N.; Matloubian, M.; Clemens, D.M.; Sharpe, A.H.; Freeman, G.J.; Gangappa, S.; Larsen, C.P.; Ahmed, R. Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc. Natl. Acad. Sci. USA 2007, 104, 15430–15435. [Google Scholar] [CrossRef]
- Munn, D.H.; Mellor, A.L. IDO in the Tumor Microenvironment: Inflammation, Counter-Regulation, and Tolerance. Trends Immunol. 2016, 37, 193–207. [Google Scholar] [CrossRef]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef]
- Terawaki, S.; Chikuma, S.; Shibayama, S.; Hayashi, T.; Yoshida, T.; Okazaki, T.; Honjo, T. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J. Immunol. 2011, 186, 2772–2779. [Google Scholar] [CrossRef] [PubMed]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Riley, J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Quigley, M.; Pereyra, F.; Nilsson, B.; Porichis, F.; Fonseca, C.; Eichbaum, Q.; Julg, B.; Jesneck, J.L.; Brosnahan, K.; Imam, S.; et al. Transcriptional analysis of HIV-specific CD8+ T cells shows that PD-1 inhibits T cell function by upregulating BATF. Nat. Med. 2010, 16, 1147–1151. [Google Scholar] [CrossRef]
- Wei, F.; Zhong, S.; Ma, Z.; Kong, H.; Medvec, A.; Ahmed, R.; Freeman, G.J.; Krogsgaard, M.; Riley, J.L. Strength of PD-1 signaling differentially affects T-cell effector functions. Proc. Natl. Acad. Sci. USA 2013, 110, E2480–E2489. [Google Scholar] [CrossRef]
- Wilson, E.B.; Yamada, D.H.; Elsaesser, H.; Herskovitz, J.; Deng, J.; Cheng, G.; Aronow, B.J.; Karp, C.L.; Brooks, D.G. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013, 340, 202–207. [Google Scholar] [CrossRef]
- Helbig, K.J.; Lau, D.T.; Semendric, L.; Harley, H.A.; Beard, M.R. Analysis of ISG expression in chronic hepatitis C identifies viperin as a potential antiviral effector. Hepatology 2005, 42, 702–710. [Google Scholar] [CrossRef]
- Hornung, V.; Hartmann, R.; Ablasser, A.; Hopfner, K.P. OAS proteins and cGAS: Unifying concepts in sensing and responding to cytosolic nucleic acids. Nat. Rev. Immunol. 2014, 14, 521–528. [Google Scholar] [CrossRef]
- Marino, N.; Collins, J.W.; Shen, C.; Caplen, N.J.; Merchant, A.S.; Gokmen-Polar, Y.; Goswami, C.P.; Hoshino, T.; Qian, Y.; Sledge, G.W., Jr.; et al. Identification and validation of genes with expression patterns inverse to multiple metastasis suppressor genes in breast cancer cell lines. Clin. Exp. Metastasis 2014, 31, 771–786. [Google Scholar] [CrossRef]
- Kazma, R.; Mefford, J.A.; Cheng, I.; Plummer, S.J.; Levin, A.M.; Rybicki, B.A.; Casey, G.; Witte, J.S. Association of the innate immunity and inflammation pathway with advanced prostate cancer risk. PLoS ONE 2012, 7, e51680. [Google Scholar] [CrossRef]
- Mandal, S.; Abebe, F.; Chaudhary, J. 2′-5′ oligoadenylate synthetase 1 polymorphism is associated with prostate cancer. Cancer 2011, 117, 5509–5518. [Google Scholar] [CrossRef]
- Villarroya-Beltri, C.; Guerra, S.; Sanchez-Madrid, F. ISGylation—A key to lock the cell gates for preventing the spread of threats. J. Cell Sci. 2017, 130, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Desai, S.D.; Haas, A.L.; Wood, L.M.; Tsai, Y.C.; Pestka, S.; Rubin, E.H.; Saleem, A.; Nur, E.K.A.; Liu, L.F. Elevated expression of ISG15 in tumor cells interferes with the ubiquitin/26S proteasome pathway. Cancer Res. 2006, 66, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Lenschow, D.J.; Lai, C.; Frias-Staheli, N.; Giannakopoulos, N.V.; Lutz, A.; Wolff, T.; Osiak, A.; Levine, B.; Schmidt, R.E.; Garcia-Sastre, A.; et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. USA 2007, 104, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, J.; Zhang, H.; Zhu, M.; Chen, F.; Hu, Y.; Liu, H.; Zhu, H. Interferon-stimulated gene 15 (ISG15) is a trigger for tumorigenesis and metastasis of hepatocellular carcinoma. Oncotarget 2014, 5, 8429–8441. [Google Scholar] [CrossRef] [PubMed]
- Cheriyath, V.; Glaser, K.B.; Waring, J.F.; Baz, R.; Hussein, M.A.; Borden, E.C. G1P3, an IFN-induced survival factor, antagonizes TRAIL-induced apoptosis in human myeloma cells. J. Clin. Investig. 2007, 117, 3107–3117. [Google Scholar] [CrossRef]
- Cheriyath, V.; Kaur, J.; Davenport, A.; Khalel, A.; Chowdhury, N.; Gaddipati, L. G1P3 (IFI6), a mitochondrial localised antiapoptotic protein, promotes metastatic potential of breast cancer cells through mtROS. Br. J. Cancer 2018, 119, 52–64. [Google Scholar] [CrossRef]
- Cheriyath, V.; Kuhns, M.A.; Jacobs, B.S.; Evangelista, P.; Elson, P.; Downs-Kelly, E.; Tubbs, R.; Borden, E.C. G1P3, an interferon- and estrogen-induced survival protein contributes to hyperplasia, tamoxifen resistance and poor outcomes in breast cancer. Oncogene 2012, 31, 2222–2236. [Google Scholar] [CrossRef]
- Cheriyath, V.; Leaman, D.W.; Borden, E.C. Emerging roles of FAM14 family members (G1P3/ISG 6-16 and ISG12/IFI27) in innate immunity and cancer. J. Interferon Cytokine Res. 2011, 31, 173–181. [Google Scholar] [CrossRef]
- Parker, N.; Porter, A.C. Identification of a novel gene family that includes the interferon-inducible human genes 6-16 and ISG12. BMC Genom. 2004, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Forloni, M.; Bisserier, M.; Dogra, S.K.; Yang, Q.; Wajapeyee, N. Interferon alpha-inducible protein 6 regulates NRASQ61K-induced melanomagenesis and growth. Elife 2016, 5, e16432. [Google Scholar] [CrossRef] [PubMed]
- Verhelst, J.; Hulpiau, P.; Saelens, X. Mx proteins: Antiviral gatekeepers that restrain the uninvited. Microbiol. Mol. Biol. Rev. 2013, 77, 551–566. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Incidence Rate per 100,000, 2009 to 2013 | Death Rate per 100,000, 2010 to 2014 | 5-Year Survival (%), 2006 to 2012 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Race/Ethnicity | All | Male | Female | All | Male | Female | All | Male | Female |
| White | 6.3 | 9.7 | 3.3 | 5.5 | 8.0 | 3.3 | 20.1 | 20.2 | 19.8 |
| AA/Black | 10.2 * | 16.8 * | 5.0 * | 8.4 * | 13.3 * | 4.6 * | 16.3 * | 15.5 * | 18.8 |
| RNA Alias | Race | Gender | Age at Dx | HCV | HBV |
|---|---|---|---|---|---|
| VLI1 | Caucasian | Male | 70 | No | No |
| VLI5 | Caucasian | Male | 78 | No | No |
| VLI14 | Caucasian | Male | 74 | No | No |
| VLI15 | Caucasian | Male | 39 | No | No |
| VLI22 | Caucasian | Male | 19 | No | No |
| VLI28 | Caucasian | Male | 75 | No | No |
| VLI26 | Caucasian | Female | 83 | No | No |
| VLI27 | Caucasian | Female | 80 | No | No |
| VLI3 | Caucasian | Male | 54 | Yes * | No |
| VLI11 | Caucasian | Male | 58 | Yes * | No |
| VLI21 | Caucasian | Male | 68 | Yes * | No |
| VLI30 | Caucasian | Male | 65 | Yes | No |
| VLI31 | Caucasian | Male | 51 | Yes * | No |
| VLI18 | Caucasian | Female | 61 | Yes * | No |
| VLI8 | African American | Male | 73 | No | No |
| VLI25 | African American | Female | 62 | No | No |
| VLI4 | African American | Male | 56 | Yes | No |
| VLI6 | African American | Male | 61 | Yes | No |
| VLI13 | African American | Male | 60 | Yes * | No |
| VLI16 | African American | Male | 64 | Yes * | No |
| VLI20 | African American | Male | 52 | Yes * | No |
| VLI23 | African American | Male | 65 | Yes * | No |
| VLI24 | African American | Male | 64 | Yes * | No |
| VLI29 | African American | Male | 57 | Yes | No |
| VLI32 | African American | Male | 60 | Yes | No |
| VLI33 | African American | Male | 66 | Yes * | No |
| VLI7 | African American | Female | 58 | Yes | No |
| VLI9 | African American | Female | 65 | Yes * | No |
| VLI12 | African American | Female | 62 | Yes * | No |
| VLI19 | African American | Female | 51 | Yes * | No |
| VLI34 | African American | Female | 69 | Yes * | No |
| VLI35 | African American | Female | 74 | Yes | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reghupaty, S.C.; Kanwal, S.; Mendoza, R.G.; Davis, E.; Li, H.; Lai, Z.; Dozmorov, M.G.; Faison, M.O.; Siddiqui, R.A.; Sarkar, D. Dysregulation of Type I Interferon (IFN-I) Signaling: A Potential Contributor to Racial Disparity in Hepatocellular Carcinoma (HCC). Cancers 2023, 15, 4283. https://doi.org/10.3390/cancers15174283
Reghupaty SC, Kanwal S, Mendoza RG, Davis E, Li H, Lai Z, Dozmorov MG, Faison MO, Siddiqui RA, Sarkar D. Dysregulation of Type I Interferon (IFN-I) Signaling: A Potential Contributor to Racial Disparity in Hepatocellular Carcinoma (HCC). Cancers. 2023; 15(17):4283. https://doi.org/10.3390/cancers15174283
Chicago/Turabian StyleReghupaty, Saranya Chidambaranathan, Sadia Kanwal, Rachel G. Mendoza, Eva Davis, Haiwen Li, Zhao Lai, Mikhail G. Dozmorov, Milton Omar Faison, Rafat Ali Siddiqui, and Devanand Sarkar. 2023. "Dysregulation of Type I Interferon (IFN-I) Signaling: A Potential Contributor to Racial Disparity in Hepatocellular Carcinoma (HCC)" Cancers 15, no. 17: 4283. https://doi.org/10.3390/cancers15174283
APA StyleReghupaty, S. C., Kanwal, S., Mendoza, R. G., Davis, E., Li, H., Lai, Z., Dozmorov, M. G., Faison, M. O., Siddiqui, R. A., & Sarkar, D. (2023). Dysregulation of Type I Interferon (IFN-I) Signaling: A Potential Contributor to Racial Disparity in Hepatocellular Carcinoma (HCC). Cancers, 15(17), 4283. https://doi.org/10.3390/cancers15174283