


Molecular Classifiers in Skin Cancers: Challenges and Promises

Abstract
:Simple Summary
Abstract
1. Introduction
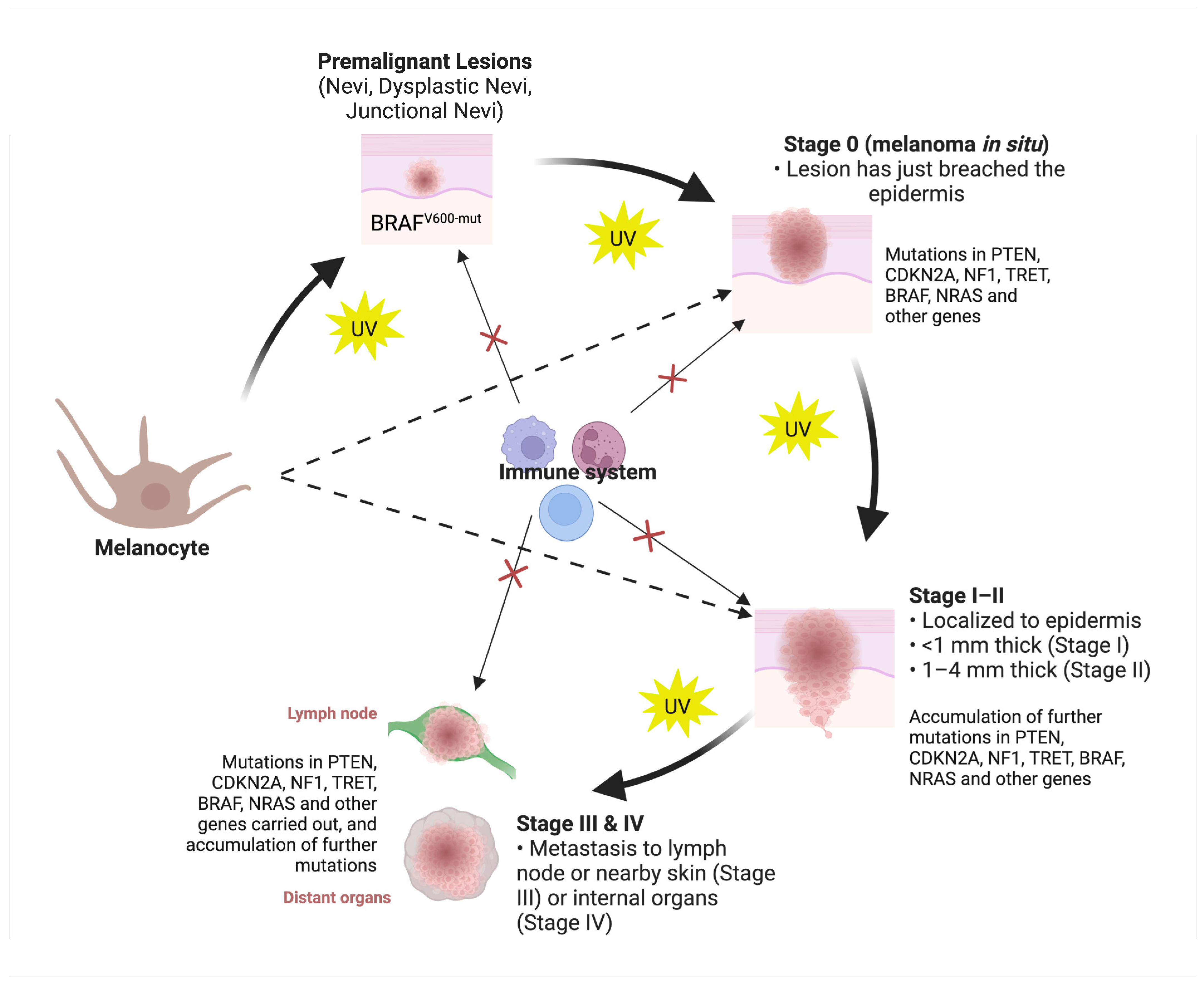
2. Classification of Skin Cancers
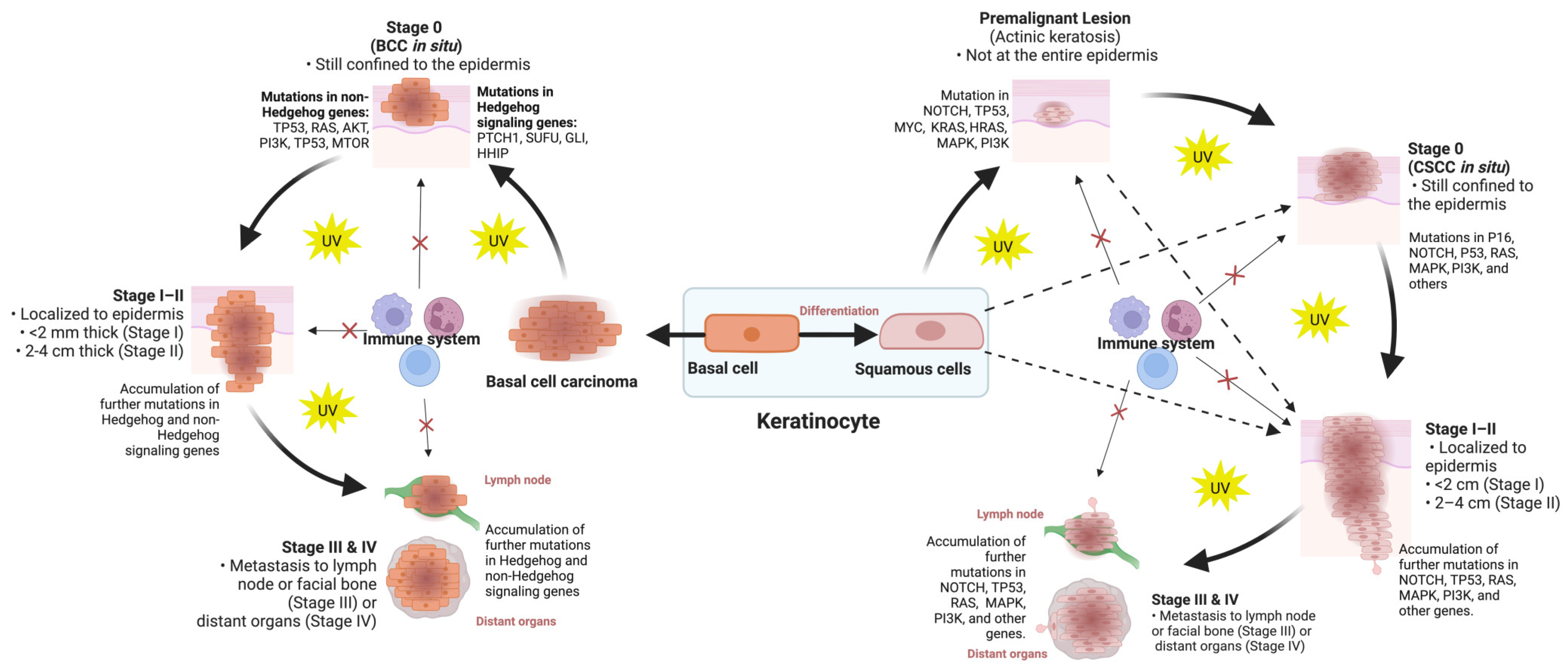
3. Molecular Classification of Skin Cancers
3.1. Genomic and Transcriptomic Classification in Skin Cancers
3.1.1. Cutaneous Melanoma
3.1.2. Cutaneous Squamous-Cell Carcinoma
3.1.3. Basal-Cell Carcinoma
3.2. Proteomic Classification in Skin Cancers
3.2.1. Cutaneous Melanoma
3.2.2. Cutaneous Squamous-Cell Carcinoma
3.2.3. Basal-Cell Carcinoma
4. Molecular Classifier for Therapeutic Stratification in Skin Cancers
4.1. Cutaneous Melanoma
4.2. Cutaneous Squamous-Cell Carcinoma
4.3. Basal-Cell Carcinoma
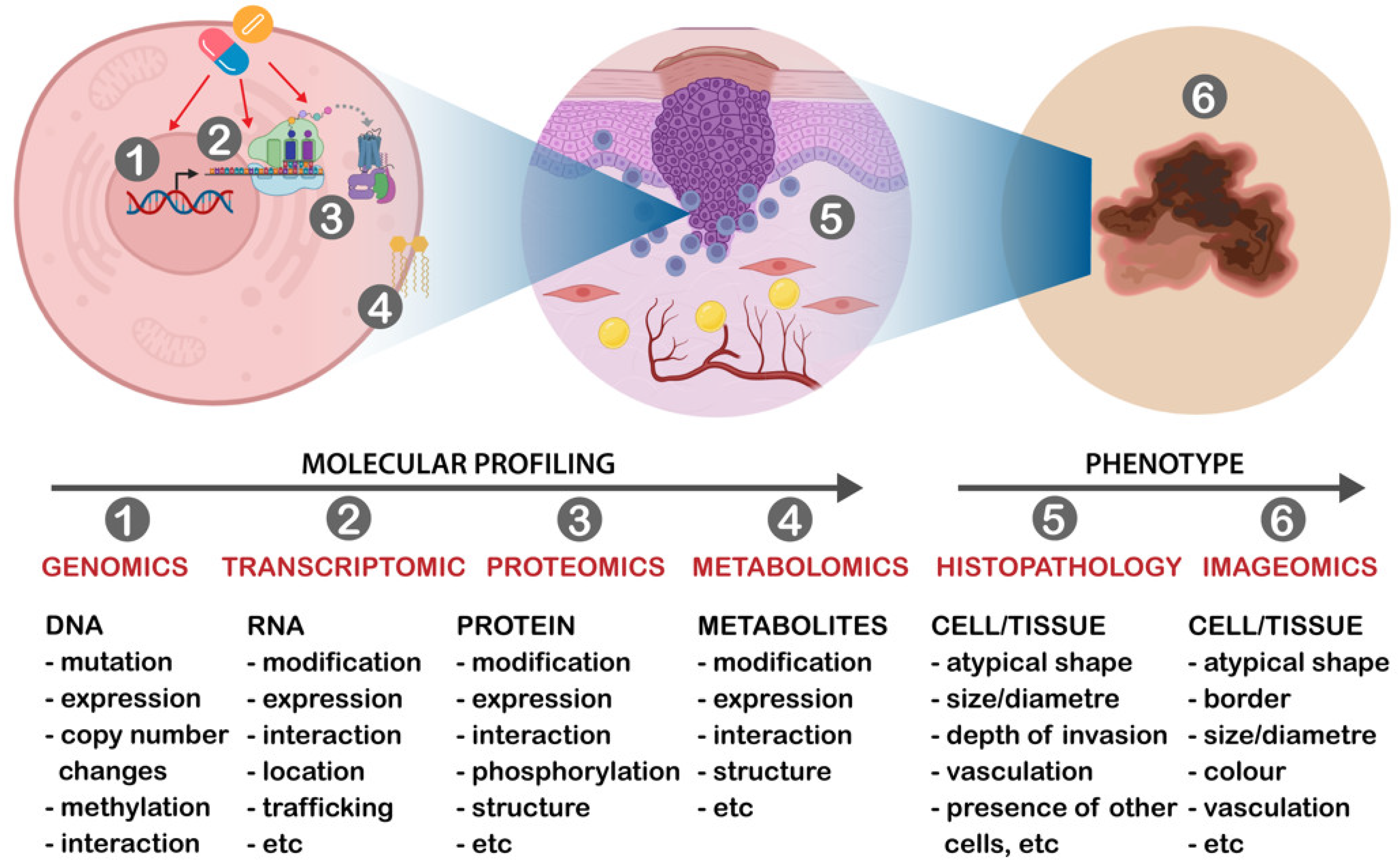
5. Multi-Omics Classifier in Skin Cancers
6. Challenges and Opportunities with Molecular Classifiers
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bakos, R.M.; Blumetti, T.P.; Roldán-Marín, R.; Salerni, G. Noninvasive Imaging Tools in the Diagnosis and Treatment of Skin Cancers. Am. J. Clin. Dermatol. 2018, 19, 3–14. [Google Scholar] [CrossRef]
- Giuffrida, R.; Conforti, C.; Di Meo, N.; Deinlein, T.; Guida, S.; Zalaudek, I. Use of noninvasive imaging in the management of skin cancer. Curr. Opin. Oncol. 2020, 32, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, V.; Apalla, Z.; Sotiriou, E.; Papageorgiou, C.; Lazaridou, E.; Vakirlis, S.; Ioannides, D.; Lallas, A. The limitations of dermoscopy: False-positive and false-negative tumours. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Lallas, A.; Argenziano, G.; Moscarella, E.; Longo, C.; Simonetti, V.; Zalaudek, I. Diagnosis and management of facial pigmented macules. Clin. Dermatol. 2014, 32, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Terushkin, V.; Warycha, M.; Levy, M.; Kopf, A.W.; Cohen, D.E.; Polsky, D. Analysis of the benign to malignant ratio of lesions biopsied by a general dermatologist before and after the adoption of dermoscopy. Arch. Dermatol. 2010, 146, 343–344. [Google Scholar] [CrossRef] [PubMed]
- Esdaile, B.; Mahmud, I.; Palmer, A.; Bowling, J. Diagnosing melanoma: How do we assess how good we are? Clin. Exp. Dermatol. 2014, 39, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Skvara, H.; Teban, L.; Fiebiger, M.; Binder, M.; Kittler, H. Limitations of dermoscopy in the recognition of melanoma. Arch. Dermatol. 2005, 141, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Ryu, T.H.; Kye, H.; Choi, J.E.; Ahn, H.H.; Kye, Y.C.; Seo, S.H. Features Causing Confusion between Basal Cell Carcinoma and Squamous Cell Carcinoma in Clinical Diagnosis. Ann. Dermatol. 2018, 30, 64–70. [Google Scholar] [CrossRef]
- Urbancek, S.; Fedorcova, P.; Tomkova, J.; Sutka, R. Misdiagnosis of Melanoma: A 7 Year Single-Center Analysis. Pigment. Disord. 2015, 2, 1000208. [Google Scholar]
- Ibrahim, S.F.; Kasprzak, J.M.; Hall, M.A.; Fitzgerald, A.L.; Siegel, J.J.; Kurley, S.J.; Covington, K.R.; Goldberg, M.S.; Farberg, A.S.; Trotter, S.C.; et al. Enhanced metastatic risk assessment in cutaneous squamous cell carcinoma with the 40-gene expression profile test. Future Oncol. 2022, 18, 833–847. [Google Scholar] [CrossRef]
- Genders, R.E.; Kuizinga, M.C.; Teune, T.M.; van der Kruijk, M.; van Rengen, A. Does biopsy accurately assess basal cell carcinoma (BCC) subtype? J. Am. Acad. Dermatol. 2016, 74, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Roozeboom, M.; Mosterd, K.; Winnepenninckx, V.; Nelemans, P.J.; Kelleners-Smeets, N. Agreement between histological subtype on punch biopsy and surgical excision in primary basal cell carcinoma. J. Eur. Acad. Dermatol. Venereol. JEADV 2012, 27, 894–898. [Google Scholar] [CrossRef]
- Elmore, J.G.; Barnhill, R.L.; Elder, D.E.; Longton, G.M.; Pepe, M.S.; Reisch, L.M.; Carney, P.A.; Titus, L.J.; Nelson, H.D.; Onega, T.; et al. Pathologists’ diagnosis of invasive melanoma and melanocytic proliferations: Observer accuracy and reproducibility study. BMJ 2017, 357, j2813. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; Lacouture, M.E.; Postow, M.A.; Wolchok, J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol. 2015, 26, 2375–2391. [Google Scholar] [CrossRef] [PubMed]
- Hughes, B.G.M.; Munoz-Couselo, E.; Mortier, L.; Bratland, Å.; Gutzmer, R.; Roshdy, O.; González Mendoza, R.; Schachter, J.; Arance, A.; Grange, F.; et al. Pembrolizumab for locally advanced and recurrent/metastatic cutaneous squamous cell carcinoma (KEYNOTE-629 study): An open-label, nonrandomized, multicenter, phase II trial. Ann. Oncol. 2021, 32, 1276–1285. [Google Scholar] [CrossRef]
- Azimi, A.; Kaufman, K.L.; Ali, M.; Arthur, J.; Kossard, S.; Fernandez-Penas, P. Differential proteomic analysis of actinic keratosis, Bowen’s disease and cutaneous squamous cell carcinoma by label-free LC-MS/MS. J. Dermatol. Sci. 2018, 91, 69–78. [Google Scholar] [CrossRef]
- Azimi, A.; Yang, P.; Ali, M.; Howard, V.; Mann, G.J.; Kaufman, K.L.; Fernandez-Penas, P. Data independent acquisition proteomic analysis can discriminate between actinic keratosis, Bowen’s disease and cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2019, 140, 212–222. [Google Scholar] [CrossRef]
- Deacon, D.C.; Smith, E.A.; Judson-Torres, R.L. Molecular Biomarkers for Melanoma Screening, Diagnosis and Prognosis: Current State and Future Prospects. Front. Med. 2021, 8, 642380. [Google Scholar] [CrossRef]
- Hessler, M.; Jalilian, E.; Xu, Q.; Reddy, S.; Horton, L.; Elkin, K.; Manwar, R.; Tsoukas, M.; Mehregan, D.; Avanaki, K. Melanoma Biomarkers and Their Potential Application for In Vivo Diagnostic Imaging Modalities. Int. J. Mol. Sci. 2020, 21, 9583. [Google Scholar] [CrossRef]
- Taylor, N.J.; Gaynanova, I.; Eschrich, S.A.; Welsh, E.A.; Garrett, T.J.; Beecher, C.; Sharma, R.; Koomen, J.M.; Smalley, K.S.; Messina, J.L. Metabolomics of primary cutaneous melanoma and matched adjacent extratumoral microenvironment. PLoS ONE 2020, 15, e0240849. [Google Scholar] [CrossRef]
- Mei, L.; Ying, L.; Wang, H.; Xu, G.; Ye, X.; Yang, G. 1H NMR-based metabolomics of skin squamous cell carcinoma and peri-tumoral region tissues. J. Pharm. Biomed. Anal. 2022, 212, 114643. [Google Scholar] [CrossRef] [PubMed]
- Malciu, A.M.; Lupu, M.; Voiculescu, V.M. Artificial Intelligence-Based Approaches to Reflectance Confocal Microscopy Image Analysis in Dermatology. J. Clin. Med. 2022, 11, 429. [Google Scholar] [CrossRef]
- Brinker, T.J.; Hekler, A.; Enk, A.H.; Klode, J.; Hauschild, A.; Berking, C.; Schilling, B.; Haferkamp, S.; Schadendorf, D.; Fröhling, S.; et al. A convolutional neural network trained with dermoscopic images performed on par with 145 dermatologists in a clinical melanoma image classification task. Eur. J. Cancer 2019, 111, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Brinker, T.J.; Hekler, A.; Enk, A.H.; Klode, J.; Hauschild, A.; Berking, C.; Schilling, B.; Haferkamp, S.; Schadendorf, D.; Holland-Letz, T.; et al. Deep learning outperformed 136 of 157 dermatologists in a head-to-head dermoscopic melanoma image classification task. Eur. J. Cancer 2019, 113, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.; Linet, M.S.; Cerhan, J.R.; Haiman, C.A.; Schottenfeld, D. Cancer Epidemiology and Prevention; Oxford University Press: Oxford, UK, 2017. [Google Scholar]
- García-Sancha, N.; Corchado-Cobos, R.; Bellido-Hernández, L.; Román-Curto, C.; Cardeñoso-Álvarez, E.; Pérez-Losada, J.; Orfao, A.; Cañueto, J. Overcoming Resistance to Immunotherapy in Advanced Cutaneous Squamous Cell Carcinoma. Cancers 2021, 13, 5134. [Google Scholar] [CrossRef] [PubMed]
- Thomson, J.; Harwoood, C.; Strid, J.; Healy, E.; Wang, J. O02 Neoantigens from actinic keratosis are predicted to be more immunogenic than those from cutaneous squamous cell carcinoma—A strategy for immune escape? Br. J. Dermatol. 2023, 189, e4–e5. [Google Scholar] [CrossRef]
- Azimi, A.; Kaufman, K.L.; Kim, J.; Ali, M.; Mann, G.J.; Fernandez-Penas, P. Proteomics: An emerging approach for the diagnosis and classification of cutaneous squamous cell carcinoma and its precursors. J. Dermatol. Sci. 2020, 99, 9–16. [Google Scholar] [CrossRef]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef]
- Rohani, N.; Eslahchi, C. Classifying Breast Cancer Molecular Subtypes by Using Deep Clustering Approach. Front. Genet. 2020, 11, 553587. [Google Scholar] [CrossRef] [PubMed]
- Kommoss, S.; McConechy, M.; Kommoss, F.; Leung, S.; Bunz, A.; Magrill, J.; Britton, H.; Grevenkamp, F.; Karnezis, A.; Yang, W. Final validation of the ProMisE molecular classifier for endometrial carcinoma in a large population-based case series. Ann. Oncol. 2018, 29, 1180–1188. [Google Scholar] [CrossRef]
- Kamoun, A.; de Reyniès, A.; Allory, Y.; Sjödahl, G.; Robertson, A.G.; Seiler, R.; Hoadley, K.A.; Groeneveld, C.S.; Al-Ahmadie, H.; Choi, W.; et al. A Consensus Molecular Classification of Muscle-invasive Bladder Cancer. Eur. Urol. 2020, 77, 420–433. [Google Scholar] [CrossRef] [PubMed]
- Birkeland, E.; Zhang, S.; Poduval, D.; Geisler, J.; Nakken, S.; Vodak, D.; Meza-Zepeda, L.A.; Hovig, E.; Myklebost, O.; Knappskog, S.; et al. Patterns of genomic evolution in advanced melanoma. Nat. Commun. 2018, 9, 2665. [Google Scholar] [CrossRef] [PubMed]
- Wachsman, W.; Morhenn, V.; Palmer, T.; Walls, L.; Hata, T.; Zalla, J.; Scheinberg, R.; Sofen, H.; Mraz, S.; Gross, K.; et al. Noninvasive genomic detection of melanoma. Br. J. Dermatol. 2011, 164, 797–806. [Google Scholar] [CrossRef]
- Amos, C.I.; Wang, L.-E.; Lee, J.E.; Gershenwald, J.E.; Chen, W.V.; Fang, S.; Kosoy, R.; Zhang, M.; Qureshi, A.A.; Vattathil, S.; et al. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma. Hum. Mol. Genet. 2011, 20, 5012–5023. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Cheng, P.; Jiang, J.; Ren, Y.; Wu, D.; Xue, D. Epigenomic and genomic analysis of transcriptome modulation in skin cutaneous melanoma. Aging 2020, 12, 12703–12725. [Google Scholar] [CrossRef]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef] [PubMed]
- Elder, D.E.; Massi, D.; Scolyer, R.A.; Willemze, R. WHO Classification of Skin Tumours; International Agency for Research on Cancer: Lyon, France, 2018. [Google Scholar]
- Ferrara, G.; Argenziano, G. The WHO 2018 Classification of Cutaneous Melanocytic Neoplasms: Suggestions From Routine Practice. Front Oncol. 2021, 11, 675296. [Google Scholar] [CrossRef]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B. Genomic classification of cutaneous melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef]
- Zager, J.S.; Gastman, B.R.; Leachman, S.; Gonzalez, R.C.; Fleming, M.D.; Ferris, L.K.; Ho, J.; Miller, A.R.; Cook, R.W.; Covington, K.R.; et al. Performance of a prognostic 31-gene expression profile in an independent cohort of 523 cutaneous melanoma patients. BMC Cancer 2018, 18, 130. [Google Scholar] [CrossRef]
- Jönsson, G.; Busch, C.; Knappskog, S.; Geisler, J.; Miletic, H.; Ringnér, M.; Lillehaug, J.R.; Borg, Å.; Lønning, P.E. Gene Expression Profiling–Based Identification of Molecular Subtypes in Stage IV Melanomas with Different Clinical Outcome. Clin. Cancer Res. 2010, 16, 3356–3367. [Google Scholar] [CrossRef]
- Szadai, L.; Velasquez, E.; Szeitz, B.; Almeida, N.P.D.; Domont, G.; Betancourt, L.H.; Gil, J.; Marko-Varga, M.; Oskolas, H.; Jánosi, Á.J.; et al. Deep Proteomic Analysis on Biobanked Paraffine-Archived Melanoma with Prognostic/Predictive Biomarker Read-Out. Cancers 2021, 13, 6105. [Google Scholar] [CrossRef] [PubMed]
- Newell, F.; Pires da Silva, I.; Johansson, P.A.; Menzies, A.M.; Wilmott, J.S.; Addala, V.; Carlino, M.S.; Rizos, H.; Nones, K.; Edwards, J.J.; et al. Multiomic profiling of checkpoint inhibitor-treated melanoma: Identifying predictors of response and resistance, and markers of biological discordance. Cancer Cell 2022, 40, 88–102.e107. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, S.; Kiyohara, Y.; Otsuka, M.; Kondou, R.; Nonomura, C.; Miyata, H.; Iizuka, A.; Ohshima, K.; Urakami, K.; Nagashima, T.; et al. Multi-omics Profiling of Patients with Melanoma Treated with Nivolumab in Project HOPE. Anticancer Res. 2017, 37, 1321–1328. [Google Scholar] [PubMed]
- Qendro, V.; Lundgren, D.H.; Rezaul, K.; Mahony, F.; Ferrell, N.; Bi, A.; Latifi, A.; Chowdhury, D.; Gygi, S.; Haas, W.; et al. Large-Scale Proteomic Characterization of Melanoma Expressed Proteins Reveals Nestin and Vimentin as Biomarkers That Can Potentially Distinguish Melanoma Subtypes. J. Proteome Res. 2014, 13, 5031–5040. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Thakur, R.; Laye, J.P.; Lauss, M.; Diaz, J.M.S.; O’Shea, S.J.; Poźniak, J.; Filia, A.; Harland, M.; Gascoyne, J.; Randerson-Moor, J.A.; et al. Transcriptomic Analysis Reveals Prognostic Molecular Signatures of Stage I Melanoma. Clin. Cancer Res. 2019, 25, 7424–7435. [Google Scholar] [CrossRef]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef]
- Wysong, A.; Newman, J.G.; Covington, K.R.; Kurley, S.J.; Ibrahim, S.F.; Farberg, A.S.; Bar, A.; Cleaver, N.J.; Somani, A.K.; Panther, D.; et al. Validation of a 40-gene expression profile test to predict metastatic risk in localized high-risk cutaneous squamous cell carcinoma. J. Am. Acad. Dermatol. 2021, 84, 361–369. [Google Scholar] [CrossRef]
- Shapanis, A.; Lai, C.; Smith, S.; Coltart, G.; Sommerlad, M.; Schofield, J.; Parkinson, E.; Skipp, P.; Healy, E. Identification of proteins associated with development of metastasis from cutaneous squamous cell carcinomas (cSCCs) via proteomic analysis of primary cSCCs. Br. J. Dermatol. 2021, 184, 709–721. [Google Scholar] [CrossRef]
- Jee, B.A.; Lim, H.; Kwon, S.M.; Jo, Y.; Park, M.C.; Lee, I.J.; Woo, H.G. Molecular classification of basal cell carcinoma of skin by gene expression profiling. Mol. Carcinog. 2015, 54, 1605–1612. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Xie, P.; Gunn, S.; Sasseville, D.; Lefrançois, P. The transcriptional landscape analysis of basal cell carcinomas reveals novel signalling pathways and actionable targets. Life Sci Alliance 2021, 4, 651. [Google Scholar] [CrossRef] [PubMed]
- Yerly, L.; Pich-Bavastro, C.; Di Domizio, J.; Wyss, T.; Tissot-Renaud, S.; Cangkrama, M.; Gilliet, M.; Werner, S.; Kuonen, F. Integrated multi-omics reveals cellular and molecular interactions governing the invasive niche of basal cell carcinoma. Nat. Commun. 2022, 13, 4897. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.; et al. Smoothened Variants Explain the Majority of Drug Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Yeh, I.; Bastian, B.C. Melanoma pathology: New approaches and classification. Br. J. Dermatol. 2021, 185, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.-M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Sarin, K.Y.; Lin, Y.; Daneshjou, R.; Ziyatdinov, A.; Thorleifsson, G.; Rubin, A.; Pardo, L.M.; Wu, W.; Khavari, P.A.; Uitterlinden, A.; et al. Genome-wide meta-analysis identifies eight new susceptibility loci for cutaneous squamous cell carcinoma. Nat. Commun. 2020, 11, 820. [Google Scholar] [CrossRef]
- Nan, H.; Xu, M.; Kraft, P.; Qureshi, A.A.; Chen, C.; Guo, Q.; Hu, F.B.; Curhan, G.; Amos, C.I.; Wang, L.-E.; et al. Genome-wide association study identifies novel alleles associated with risk of cutaneous basal cell carcinoma and squamous cell carcinoma. Hum. Mol. Genet. 2011, 20, 3718–3724. [Google Scholar] [CrossRef]
- Zhang, M.; Liang, L.; Morar, N.; Dixon, A.L.; Lathrop, G.M.; Ding, J.; Moffatt, M.F.; Cookson, W.O.C.; Kraft, P.; Qureshi, A.A.; et al. Integrating pathway analysis and genetics of gene expression for genome-wide association study of basal cell carcinoma. Hum. Genet. 2012, 131, 615–623. [Google Scholar] [CrossRef]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstråhle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514.e422. [Google Scholar] [CrossRef]
- Rodríguez-Paredes, M.; Bormann, F.; Raddatz, G.; Gutekunst, J.; Lucena-Porcel, C.; Köhler, F.; Wurzer, E.; Schmidt, K.; Gallinat, S.; Wenck, H.; et al. Methylation profiling identifies two subclasses of squamous cell carcinoma related to distinct cells of origin. Nat. Commun. 2018, 9, 577. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Cohen, P.R.; Boichard, A.; Frampton, G.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Genomic landscape of advanced basal cell carcinoma: Implications for precision treatment with targeted and immune therapies. Oncoimmunology 2018, 7, e1404217. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, L.; McMorrow, J.; Doolan, P.; McKiernan, E.; Mehta, J.P.; Ryan, E.; Gammell, P.; Joyce, H.; O’Donovan, N.; Walsh, N.; et al. Investigation of the molecular profile of basal cell carcinoma using whole genome microarrays. Mol. Cancer 2006, 5, 74. [Google Scholar] [CrossRef]
- Heller, E.R.; Gor, A.; Wang, D.; Hu, Q.; Lucchese, A.; Kanduc, D.; Katdare, M.; Liu, S.; Sinha, A.A. Molecular signatures of basal cell carcinoma susceptibility and pathogenesis: A genomic approach. Int. J. Oncol 2013, 42, 583–596. [Google Scholar] [CrossRef]
- Villani, R.; Murigneux, V.; Alexis, J.; Sim, S.-L.; Wagels, M.; Saunders, N.; Soyer, H.P.; Parmentier, L.; Nikolaev, S.; Fink, J.L.; et al. Subtype-Specific Analyses Reveal Infiltrative Basal Cell Carcinomas Are Highly Interactive with their Environment. J. Investig. Dermatol. 2021, 141, 2380–2390. [Google Scholar] [CrossRef] [PubMed]
- Di Nardo, L.; Pellegrini, C.; Di Stefani, A.; Ricci, F.; Fossati, B.; Del Regno, L.; Carbone, C.; Piro, G.; Corbo, V.; Delfino, P.; et al. Molecular alterations in basal cell carcinoma subtypes. Sci. Rep. 2021, 11, 13206. [Google Scholar] [CrossRef]
- Yu, M.; Zloty, D.; Cowan, B.; Shapiro, J.; Haegert, A.; Bell, R.H.; Warshawski, L.; Carr, N.; McElwee, K.J. Superficial, nodular, and morpheiform basal-cell carcinomas exhibit distinct gene expression profiles. J. Investig. Dermatol. 2008, 128, 1797–1805. [Google Scholar] [CrossRef]
- Celis, J.E.; Gromov, P. Proteomics in translational cancer research: Toward an integrated approach. Cancer Cell 2003, 3, 9–15. [Google Scholar] [CrossRef]
- Lazova, R.; Seeley, E.H. Proteomic mass spectrometry imaging for skin cancer diagnosis. Dermatol. Clin. 2017, 35, 513–519. [Google Scholar] [CrossRef]
- Lazova, R.; Yang, Z.; El Habr, C.; Lim, Y.; Choate, K.A.; Seeley, E.H.; Caprioli, R.M.; Yangqun, L. Mass Spectrometry Imaging Can Distinguish on a Proteomic Level between Proliferative Nodules within a Benign Congenital Nevus and Malignant Melanoma. Am. J. Dermatopathol. 2017, 39, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Casadonte, R.; Kriegsmann, M.; Kriegsmann, K.; Hauk, I.; Meliß, R.R.; Müller, C.S.L.; Kriegsmann, J. Imaging Mass Spectrometry-Based Proteomic Analysis to Differentiate Melanocytic Nevi and Malignant Melanoma. Cancers 2021, 13, 3197. [Google Scholar] [CrossRef]
- Betancourt, L.H.; Gil, J.; Kim, Y.; Doma, V.; Çakır, U.; Sanchez, A.; Murillo, J.R.; Kuras, M.; Parada, I.P.; Sugihara, Y.; et al. The human melanoma proteome atlas—Defining the molecular pathology. Clin. Transl. Med. 2021, 11, e473. [Google Scholar] [CrossRef] [PubMed]
- Azimi, A.; Lo, K.; Kim, J.; Fernandez-Penas, P. Investigating proteome changes between primary and metastatic cutaneous squamous cell carcinoma using SWATH mass spectrometry. J. Dermatol. Sci. 2020, 99, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, K.D.; Neittaanmäki, N.; Zaar, O.; Angerer, T.B.; Paoli, J.; Fletcher, J.S. TOF-SIMS imaging reveals tumor heterogeneity and inflammatory response markers in the microenvironment of basal cell carcinoma. Biointerphases 2020, 15, 041012. [Google Scholar] [CrossRef]
- Vukadin, S.; Khaznadar, F.; Kizivat, T.; Vcev, A.; Smolic, M. Molecular Mechanisms of Resistance to Immune Checkpoint Inhibitors in Melanoma Treatment: An Update. Biomedicines 2021, 9, 835. [Google Scholar] [CrossRef]
- Beck, L.; Harel, M.; Yu, S.; Markovits, E.; Boursi, B.; Markel, G.; Geiger, T. Clinical Proteomics of Metastatic Melanoma Reveals Profiles of Organ Specificity and Treatment Resistance. Clin. Cancer Res. 2021, 27, 2074. [Google Scholar] [CrossRef]
- Reuben, A.; Spencer, C.N.; Prieto, P.A.; Gopalakrishnan, V.; Reddy, S.M.; Miller, J.P.; Mao, X.; De Macedo, M.P.; Chen, J.; Song, X.; et al. Genomic and immune heterogeneity are associated with differential responses to therapy in melanoma. NPJ Genom. Med. 2017, 2, 10. [Google Scholar] [CrossRef] [PubMed]
- Guminski, A.D.; Lim, A.M.L.; Khushalani, N.I.; Schmults, C.D.; Hernandez-Aya, L.F.; Modi, B.; Dunn, L.; Hughes, B.G.M.; Chang, A.L.S.; Hauschild, A. Phase 2 Study of Cemiplimab, a Human Monoclonal Anti-PD-1, in Patients (pts) with Metastatic Cutaneous Squamous Cell Carcinoma (mCSCC; Group 1): 12-Month Follow-Up; American Society of Clinical Oncology: Alexandria, VA, USA, 2019. [Google Scholar]
- Trodello, C.; Pepper, J.-P.; Wong, M.; Wysong, A. Cisplatin and Cetuximab Treatment for Metastatic Cutaneous Squamous Cell Carcinoma: A Systematic Review. Dermatol. Surg. 2017, 43, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Veness, M.J.; Morgan, G.J.; Palme, C.E.; Gebski, V. Surgery and adjuvant radiotherapy in patients with cutaneous head and neck squamous cell carcinoma metastatic to lymph nodes: Combined treatment should be considered best practice. Laryngoscope 2005, 115, 870–875. [Google Scholar] [CrossRef]
- Migden, M.R.; Chang, A.L.S.; Dirix, L.; Stratigos, A.J.; Lear, J.T. Emerging trends in the treatment of advanced basal cell carcinoma. Cancer Treat. Rev. 2018, 64, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tiosano, A.; Ben-Ishai, M.; Fenig, E.; Ben Simon, G.J.; Yassur, I. The initial rate of tumour response to vismodegib treatment, can predict a complete response outcome for periocular LA-BCC. Eye 2023, 37, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Gracia-Cazaña, T.; Mascaraque, M.; Lucena, S.R.; Vera-Álvarez, J.; González, S.; Juarranz, Á.; Gilaberte, Y. Biomarkers of basal cell carcinoma resistance to methyl-aminolevulinate photodynamic therapy. PLoS ONE 2019, 14, e0215537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Sun, K.; Yang, X.; Dai, C.; Guo, Y. Integrated Multi-omics Analysis Using Variational Autoencoders: Application to Pan-cancer Classification. In Proceedings of the 2019 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), San Diego, CA, USA, 18–21 November 2019; pp. 765–769. [Google Scholar]
- Qi, L.; Wang, W.; Wu, T.; Zhu, L.; He, L.; Wang, X. Multi-Omics Data Fusion for Cancer Molecular Subtyping Using Sparse Canonical Correlation Analysis. Front. Genet. 2021, 12, 7817. [Google Scholar] [CrossRef] [PubMed]
- Valle, F.; Osella, M.; Caselle, M. Multi-omics Topic Modeling for Breast Cancer Classification. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mo, Q.; Wan, L.; Schell, M.J.; Jim, H.; Tworoger, S.S.; Peng, G. Integrative Analysis Identifies Multi-Omics Signatures That Drive Molecular Classification of Uveal Melanoma. Cancers 2021, 13, 6168. [Google Scholar] [CrossRef]
- Pham, T.-C.; Luong, C.-M.; Hoang, V.-D.; Doucet, A. AI outperformed every dermatologist in dermoscopic melanoma diagnosis, using an optimized deep-CNN architecture with custom mini-batch logic and loss function. Sci. Rep. 2021, 11, 17485. [Google Scholar] [CrossRef]
- Bera, K.; Schalper, K.A.; Rimm, D.L.; Velcheti, V.; Madabhushi, A. Artificial intelligence in digital pathology—New tools for diagnosis and precision oncology. Nat. Rev. Clin. Oncol. 2019, 16, 703–715. [Google Scholar] [CrossRef]
- Brinker, T.J.; Schmitt, M.; Krieghoff-Henning, E.I.; Barnhill, R.; Beltraminelli, H.; Braun, S.A.; Carr, R.; Fernandez-Figueras, M.-T.; Ferrara, G.; Fraitag, S. Diagnostic performance of artificial intelligence for histologic melanoma recognition compared to 18 international expert pathologists. J. Am. Acad. Dermatol. 2021, 86, 640–642. [Google Scholar] [CrossRef]
- Campanella, G.; Hanna, M.G.; Geneslaw, L.; Miraflor, A.; Werneck Krauss Silva, V.; Busam, K.J.; Brogi, E.; Reuter, V.E.; Klimstra, D.S.; Fuchs, T.J. Clinical-grade computational pathology using weakly supervised deep learning on whole slide images. Nat. Med. 2019, 25, 1301–1309. [Google Scholar] [CrossRef]
- Gertych, A.; Swiderska-Chadaj, Z.; Ma, Z.; Markiewicz, T.; Cierniak, S.; Salemi, H.; Guzman, S.; Walts, A.E.; Knudsen, B.S. Convolutional neural networks can accurately distinguish four histologic growth patterns of lung adenocarcinoma in digital slides. Sci. Rep. 2019, 9, 1483. [Google Scholar] [CrossRef] [PubMed]
- Kratz, J.R.; Haro, G.J.; Cook, N.R.; He, J.; Van Den Eeden, S.K.; Woodard, G.A.; Gubens, M.A.; Jahan, T.M.; Jones, K.D.; Kim, I.J.; et al. Incorporation of a Molecular Prognostic Classifier Improves Conventional Non-Small Cell Lung Cancer Staging. J. Thorac. Oncol. 2019, 14, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Purcell, R.V.; Schmeier, S.; Lau, Y.C.; Pearson, J.F.; Frizelle, F.A. Molecular subtyping improves prognostication of Stage 2 colorectal cancer. BMC Cancer 2019, 19, 1155. [Google Scholar] [CrossRef] [PubMed]
- ten Hoorn, S.; de Back, T.R.; Sommeijer, D.W.; Vermeulen, L. Clinical Value of Consensus Molecular Subtypes in Colorectal Cancer: A Systematic Review and Meta-Analysis. JNCI J. Natl. Cancer Inst. 2021, 114, 503–516. [Google Scholar] [CrossRef]
- Zhang, T.; Tan, T.; Han, L.; Appelman, L.; Veltman, J.; Wessels, R.; Duvivier, K.M.; Loo, C.; Gao, Y.; Wang, X.; et al. Predicting breast cancer types on and beyond molecular level in a multi-modal fashion. NPJ Breast Cancer 2023, 9, 16. [Google Scholar] [CrossRef]
- Paietta, E.; Roberts, K.G.; Wang, V.; Gu, Z.; Buck, G.A.N.; Pei, D.; Cheng, C.; Levine, R.L.; Abdel-Wahab, O.; Cheng, Z.; et al. Molecular classification improves risk assessment in adult BCR-ABL1–negative B-ALL. Blood 2021, 138, 948–958. [Google Scholar] [CrossRef]
- Robert, C.; Gautheret, D. Multi-omics prediction in melanoma immunotherapy: A new brick in the wall. Cancer Cell 2022, 40, 14–16. [Google Scholar] [CrossRef]
- Yoshida, H.; Kiyuna, T. Requirements for implementation of artificial intelligence in the practice of gastrointestinal pathology. World J. Gastroenterol. 2021, 27, 2818–2833. [Google Scholar] [CrossRef]
- Tarazona, S.; Arzalluz-Luque, A.; Conesa, A. Undisclosed, unmet and neglected challenges in multi-omics studies. Nat. Comput. Sci. 2021, 1, 395–402. [Google Scholar] [CrossRef]
- Cai, Z.; Poulos, R.C.; Liu, J.; Zhong, Q. Machine learning for multi-omics data integration in cancer. iScience 2022, 25, 103798. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| a. Melanoma | ||||
| No | Features Attributing to the Classes/or “Omics” Analysis Used | Classes | Main Findings and Significance | Ref |
| 1 | Genetic mutation information (major contributor), cumulative solar damage (CSD) and histology | 1. Pathway I: superficial spreading melanoma (CSD, BRAF p.V600 mutations) 2. Pathway II: lentigo malignant melanoma (CSD; NF1, NRAS, non-p.V600E BRAF mutations) 3. Pathway III: desmoplastic melanoma (CSD; inactivating NF1, promoting NFKBIE, and activating MAPK pathway mutations) 4. Pathway IV: spitz melanoma (no CSD; HRAS mutation, kinase fusions in ROS1, NTRK1, NTRK3, ALK, BRAF, MET, and RET; CDKN2A deletion, promoting TERT mutations) 5. Pathway V: acral melanoma (no CSD, CCND1, KIT, and TERT amplifications; BRAF, NRAS, and KIT mutations) 6. Pathway VI: mucosal melanoma (no CSD, copy number variations; KIT and NRAS mutations) 7. Pathway VII: melanoma arising in congenital nevi (no CSD (NRAS mutation in large congenital nevi; BRAF mutation in small to medium congenital nevi) 8. Pathway VIII: melanoma arising in blue nevi (no CSD (GNAQ, CYSLTR2, GNA11 and PLCB4 mutations; copy number aberrations in SF3B1 and EIF1AX) 9. Pathway IX: uveal melanoma (no CSD, GNAQ, GNA11, PLCB4, CYSLTR2, BAP1, SF3B1, and EIF1AX mutations) | Within this classification framework, reduced significance has been attributed to the clinical and histopathological factors, underscoring the elevated prominence of molecular criteria within the realm of melanoma classification and subsequent management of the tumor. This paradigm shift highlights the greater emphasis on discerning and utilizing molecular markers to inform the classification and comprehensive management of melanoma cases. | [38,39] |
| 2 | Whole-exome sequencing, DNA copy-number profiling, DNA methylation profiling and protein array expression profiling analysis | 1. Mutant BRAF 2. Mutant RAS 3. Mutant NF1 4. Triple-WT (wild-type) | This study introduces a structured framework for genomic classification, identifying four distinct subtypes determined by the prevailing pattern of mutated genes. | [40] |
| 3 | Transcriptomic analysis | 1. Immune: overexpression of immune-related genes) 2. Keratin: overexpression of genes associated with keratins 3. MITF-low: decreased expression of pigmentation and epithelial expression genes. | Regionally metastatic tumors in the “immune” subclass show more favorable and the “keratin” subclass less favorable post-accession survival, suggesting that transcript expression analysis will improve patient stratification. | [40] |
| 4 | Genomic analysis | 1. Low risk of recurrence-free and distant metastasis-free survival 2. High risk of recurrence-free and distant metastasis-free survival | The risk of metastasis can be accurately predicted in 70% of stage I and II melanomas using 30-gene expression analysis, offering a useful tool for estimating individual’s risk of recurrence and for considering adjuvant therapy. | [41] |
| 5 | Genomic analysis | 1. Immune response subtype 2. Pigmentation differentiation subtype 3. Proliferative subtype 4. Stromal composition subtype | There had been significant differences in mutations between the subtypes stage III and IV melanomas studied, with the proliferative subtype having a poor prognosis. Low expression of defined gene set associated with immune response was also found to be associated with poor outcome, highlighting the importance of genome-based subtype classification for personalized management of melanoma. | [42] |
| 6 | Proteomic analysis | Six clusters of melanomas based on their distinct proteomic profile showing different survival. | The study identified that proteins like TRAF6 and ARMC10 are linked to shorter survival, while AIFI1 is linked to longer survival. In the immunotherapy and targeted therapy groups, certain pathways and processes were linked to better patient outcomes, potentially aiding precision medicine. | [43] |
| 7 | Whole-genome, transcriptome, methylome and immune cell infiltrate analysis | 1. Class 1: Respondents to anti-PD-1 therapy, with or without anti-CTLA-4 2. Class 2: Non-respondents to anti-PD-1 therapy, with or without anti-CTLA-4 | Analysis of patients with advanced cutaneous melanoma undergoing anti-PD-1 therapy, with or without anti-CTLA-4 showed that response to immunotherapy is associated with high tumor mutation burden, neoantigen load, expression of IFNγ-related genes, programmed death ligand expression, low PSMB8 methylation and presence of T cells in the tumor microenvironment. A combined model involving tumor mutation burden and IFNγ-related gene expression predicted the response at AUC 0.79. | [44] |
| 8 | Whole-exome sequencing and gene expression profiling analysis | 1. Class 1: Good responders to anti-PD-1 therapy. 2. Class 2: Non-responders to anti-PD-1 therapy. | Using integrative whole-exome sequencing and gene expression profiling analysis, melanoma patients with PD-L1 upregulation were found to be good responders to anti-PD-1 therapy. | [45] |
| 9 | Proteomic analysis | Classes of melanoma with different levels of aggressiveness | The expression of proteins such as nestin and vimentin could predict melanoma aggressiveness in different melanoma subgroups, allowing risk molecular stratification. | [46] |
| 10 | Genomic and transcriptomic analysis | 1. Class 1: increased response to anti-PD-1 therapies 2. Class 2: increased resistance to anti-PD-1 therapies | Cactors such as high BRCA2 gene mutational loads are associated with increased response and upregulation of genes associated with mesenchymal transition, extracellular matrix remodeling and angiogenesis with increased resistance to anti-PD-1 therapy in metastatic melanomas. | [47] |
| 11 | Transcriptomic analysis | A total of 687 primary melanoma were categorized as classes 1 to 6, where classes 1 and 5 were typically thin and nonulcerated, classes 2 and 4 exhibited thicker characteristics. Class 3 and 6 tumors were the thickest and most frequently ulcerated. These six classes were significantly linked to mutation status: BRAF mutations were common in classes 1, 5, and 6, while NRAS mutations were frequent in classes 2, 3, and 4. | The performance of transcriptomic signatures in stage I melanoma showed similar indicator of prognosis when compared with sentinel node biopsy. | [48] |
| b: Cutaneous squamous-cell carcinoma | ||||
| No | Features Attributing to the Classes/or “Omics” Analysis Used | Classes or Molecular Sub-Groups | Main Findings and Significance | Ref |
| 12 | Whole-exome sequencing analysis | The study identified signatures of well-differentiated (six genes including SULF1, ZNF528, NRCAM and FAT1) and moderately/poorly differentiated (16 genes including TMEM51, GRHL2, ZZEF1 and GMDS) tumors. | This research elucidates the intricate molecular makeup of cSCC, uncovering driver genes, pathways, and mechanisms linked to the formation of well-differentiated and moderately/poorly differentiated tumors. | [37] |
| 13 | Targeted genomic analysis | This study identifies metastatic cSCC patients with overall good or poor survival. | Substantiates the connection between mutations in chromatin-modifying genes or mutations involving chromatin modifiers in combination with RAS/RTK/PI3K and unfavorable outcomes. | [49] |
| 14 | Genomic analysis | 1. Class 1: patients with low risk of metastasis 2. Class 2: patients with high risk of metastasis 3. Class 3: patients with highest risk of metastasis | Using a 40-gene expression test, the risk of metastasis can be predicted in primary cSCC patients, complementing current staging systems for high-risk patients. | [50] |
| 15 | Proteomic analysis | Class 1: patients with high risk of metastasis Class 2: patients with low risk of metastasis | Primary cSCC lesions with higher levels of ANXA5 and DDOST proteins is associated with reduced time to metastasis. A prediction model based on these proteins showed a classification performance with an accuracy of 91.2% and higher sensitivity and specificity compared to the existing clinical cSCC staging systems. | [51] |
| c: Basal-cell Carcinoma | ||||
| No | Features Attributing to the Classesor “Omics” Analysis Used | Classes | Main Findings and Significance | Ref |
| 16 | Transcriptomic analysis | 1. Class 1: classical BCC 2. Class 2: SCC-like BCC, 3. Class 3: normal-like BCC | Every subgroup exhibited specific molecular traits, offering distinct understanding into the diverse features of these lesions. For instance, the classical BCC subtype demonstrated heightened engagement of Wnt and Hedgehog signaling pathways, whereas the SCC-like BCC subtype displayed enrichment in genes tied to immune responses and oxidative stress. Additionally, the classical BCC subtype exhibited marked activation of metabolic pathways, with a notable emphasis on fatty acid metabolism. | [52] |
| 17 | Hierarchical clustering analysis of BCC samples based on RNA expression levels has also found a mixed cluster of high-risk and low-risk tumors with moderate upregulation of genes such as SPHK1, MTHFD1 and BMS1P20 . When clustering advanced versus non-advanced BCCs, a third group of lesions with no clear clustering with advanced and non-advanced tumors with moderate to highly moderate upregulation of genes including COL1A1, COL1A2 and COL3A1 were found. | [53] | ||
| 18 | Single-cell and spatial transcriptomics analysis | The authors identify the tumor nodular, tumor infiltrative, stroma nodular and stroma infiltrative areas of interests in BCC, each with a distinct genomic profile. | This study reveals distinct gene expression differences between tumor and stroma cells in infiltrative and nodular BCC samples, and notes that invasive edge tumor cells exhibit collective migration phenotype, while nearby fibroblasts remodel the extracellular matrix. | [54] |
| 19 | Transcriptomic and whole exome analysis | Class 1: BCCs resistant to vismodegib treatment Class 2: BCCs sensitive to vismodegib treatment | The study discovered SMO mutations in half of the resistant BCCs, demonstrating their role in sustaining Hedgehog signaling despite SMO inhibitor (vismodegib) treatment. These findings highlight SMO gene mutations as significant contributors to resistance. Consequently, the research suggests that screening for genetic mutations in BCC samples could serve as a useful method for predicting drug resistance in patients. | [55] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azimi, A.; Fernandez-Peñas, P. Molecular Classifiers in Skin Cancers: Challenges and Promises. Cancers 2023, 15, 4463. https://doi.org/10.3390/cancers15184463
Azimi A, Fernandez-Peñas P. Molecular Classifiers in Skin Cancers: Challenges and Promises. Cancers. 2023; 15(18):4463. https://doi.org/10.3390/cancers15184463
Chicago/Turabian StyleAzimi, Ali, and Pablo Fernandez-Peñas. 2023. "Molecular Classifiers in Skin Cancers: Challenges and Promises" Cancers 15, no. 18: 4463. https://doi.org/10.3390/cancers15184463