

Mesenchymal Chondrosarcoma from Diagnosis to Clinical Trials
 , , , , ,
, , , , ,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Chondrosarcoma Diagnostics
2.1. Clinical Presentation
2.2. Symptoms and Signs
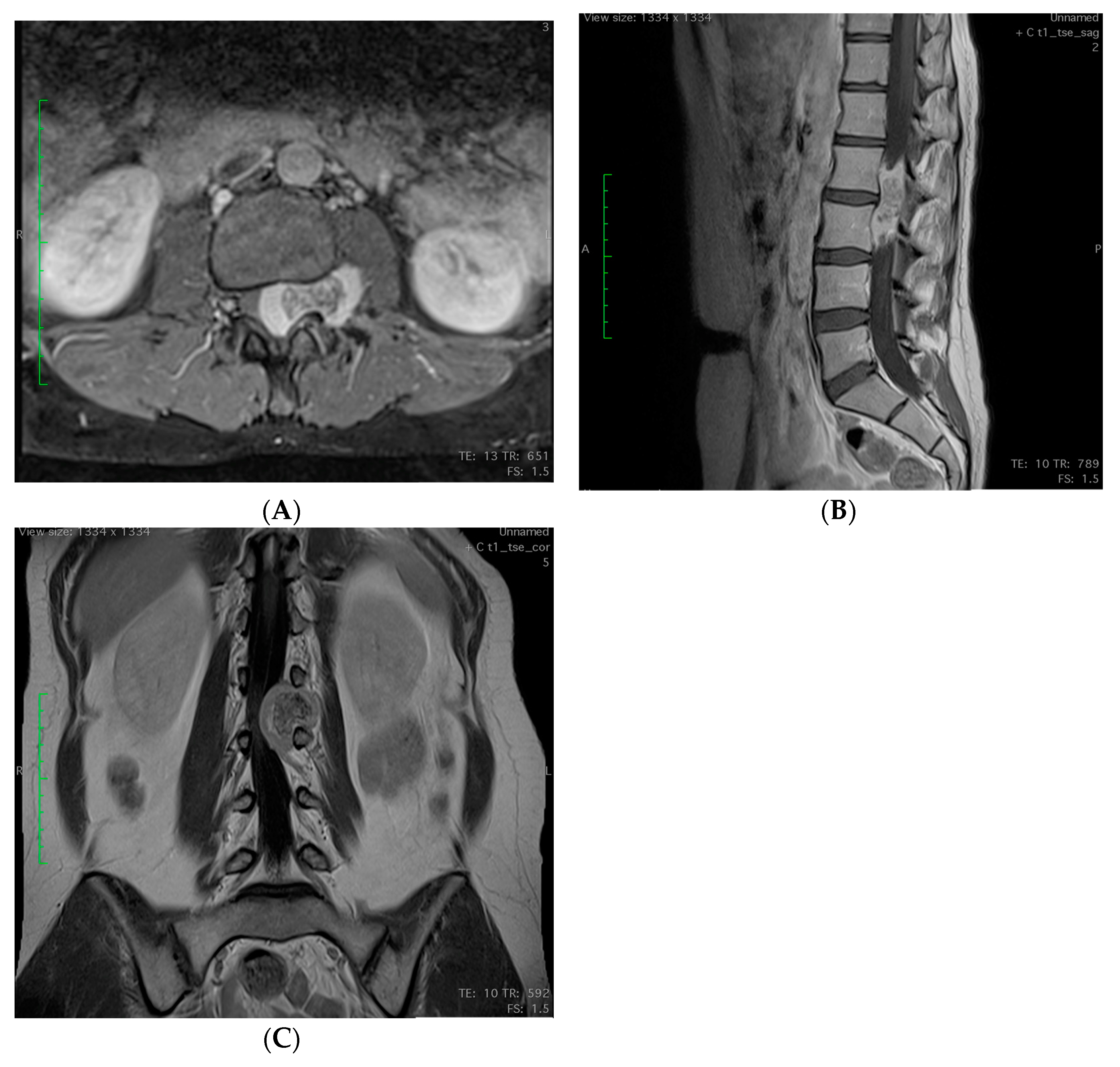
2.3. Imagining
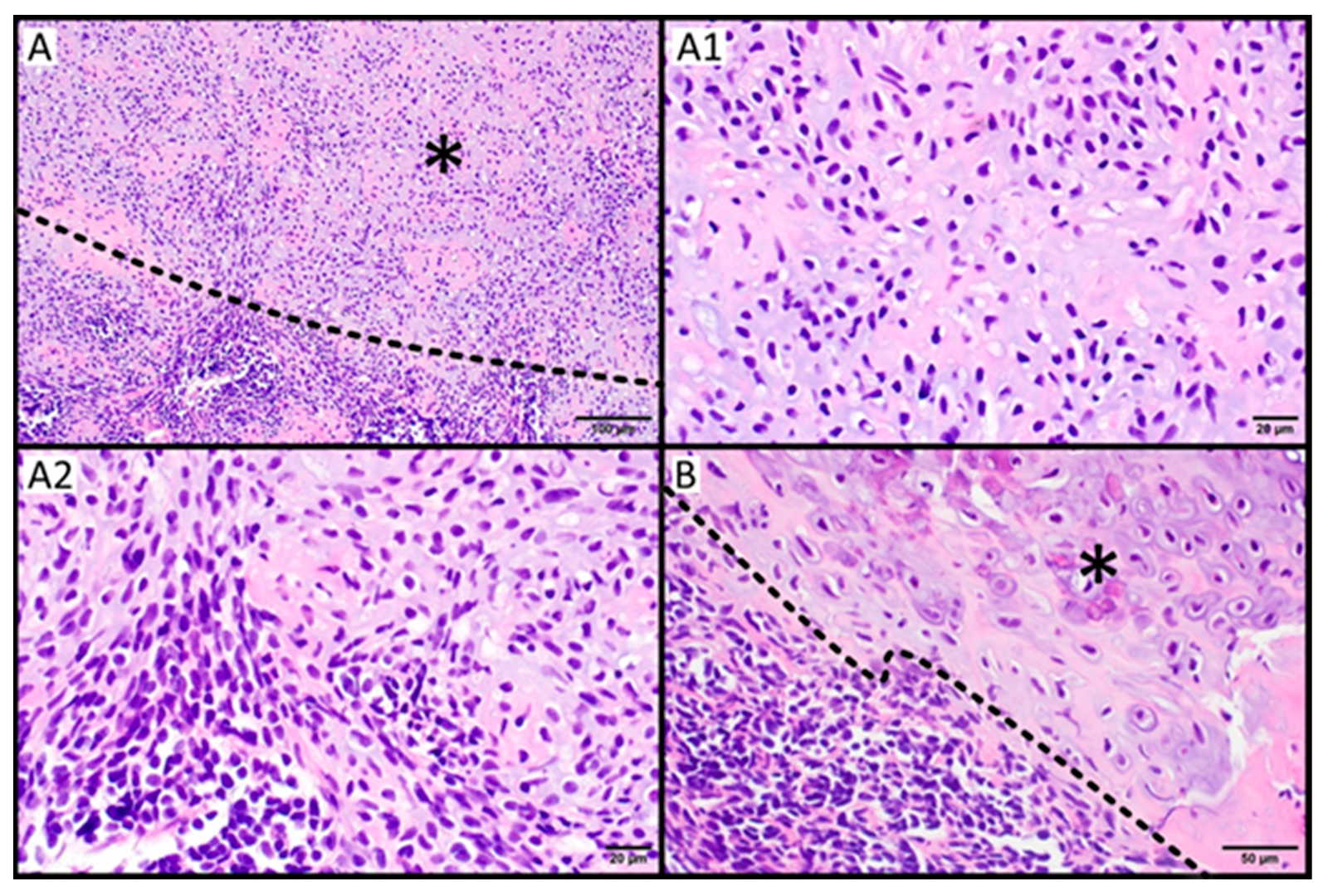
2.4. Histopathology
3. Genetic Alterations and Potential Targets for Novel Therapies
3.1. HEY1-NCOA2 Fusion and Related Signaling Pathways
3.2. IRF2BP2-CDX Fusion and Other Cytogenetic Changes
3.3. Other Reported Genetic Changes
3.4. Diagnostic Genetic Markers
4. Radical Surgical Treatment
5. Radiation Therapy
6. Treatment of Locally Advanced Disease
6.1. Systemic Therapy in an Unresectable and Metastatic Setting
6.2. Drugs in Development
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, Y.; Meng, X.; Liu, W.; Wang, H.; Xin, T. Rare malignant primary spinal intradural extramedullary mesenchymal chondrosarcoma: A case report and literature review. Transl. Cancer Res. 2022, 11, 3371–3378. [Google Scholar] [CrossRef] [PubMed]
- Bishop, M.W.; Somerville, J.M.; Bahrami, A.; Kaste, S.C.; Interiano, R.B.; Wu, J.; Mao, S.; Boop, F.A.; Williams, R.F.; Pappo, A.S.; et al. Mesenchymal Chondrosarcoma in Children and Young Adults: A Single Institution Retrospective Review. Sarcoma 2015, 2015, 608279. [Google Scholar] [CrossRef] [PubMed]
- Bovée, J.V.M.G. WHO Classification of Tumours: Soft Tissue and Bone Tumours; WHO Classification of Tumours Editorial Board: Geneva, Switzerland, 2020; pp. 370–390. [Google Scholar]
- Chen, J.J.; Chou, C.W. A Rare Case Report of Mesenchymal Chondrosarcoma with Pancreatic Metastasis. Medicina 2022, 58, 639. [Google Scholar] [CrossRef] [PubMed]
- Dantonello, T.M.; Int-Veen, C.; Leuschner, I.; Schuck, A.; Furtwaengler, R.; Claviez, A.; Schneider, D.T.; Klingebiel, T.; Bielack, S.S.; Koscielniak, E.; et al. Mesenchymal chondrosarcoma of soft tissues and bone in children, adolescents, and young adults: Experiences of the CWS and COSS study groups. Cancer 2008, 112, 2424–2431. [Google Scholar] [CrossRef]
- Xu, J.; Li, D.; Xie, L.; Tang, S.; Guo, W. Mesenchymal chondrosarcoma of bone and soft tissue: A systematic review of 107 patients in the past 20 years. PLoS ONE 2015, 10, e0122216. [Google Scholar] [CrossRef]
- Strach, M.C.; Grimison, P.S.; Hong, A.; Boyle, R.; Stalley, P.; Karim, R.; Connolly, E.A.; Bae, S.; Desai, J.; Crowe, P.; et al. Mesenchymal chondrosarcoma: An Australian multi-centre cohort study. Cancer Med. 2022, 12, 368–378. [Google Scholar] [CrossRef]
- Amer, K.M.; Munn, M.; Congiusta, D.; Abraham, J.A.; Basu Mallick, A. Survival and Prognosis of Chondrosarcoma Subtypes: SEER Database Analysis. J. Orthop. Res. 2020, 38, 311–319. [Google Scholar] [CrossRef]
- Frezza, A.M.; Cesari, M.; Baumhoer, D.; Biau, D.; Bielack, S.; Campanacci, D.A.; Casanova, J.; Esler, C.; Ferrari, S.; Funovics, P.T.; et al. Mesenchymal chondrosarcoma: Prognostic factors and outcome in 113 patients. A European Musculoskeletal Oncology Society study. Eur. J. Cancer 2015, 51, 374–381. [Google Scholar] [CrossRef]
- Lee, Y.; Choi, S.; Kim, H.S. Extraskeletal Mesenchymal Chondrosarcoma of the Uterus. Diagnostics 2022, 12, 643. [Google Scholar] [CrossRef]
- Esfahani, M.M.; Mirazimi, S.M.A.; Azadbakht, J.; Dashti, F. Retroperitoneal mesenchymal chondrosarcoma with metastasis to iliac vein: A rare case report and review of the literature. Clin. Case Rep. 2022, 10, e6633. [Google Scholar] [CrossRef]
- Goldenberg, M.; Ramgopal, A.A.; Salgado, C.M.; Reyes-Mugica, M.; Malek, M.M.; Tersak, J.M. Mesenchymal chondrosarcoma of the chest wall in an adolescent patient: A case report and brief review of the literature. Cancer Rep. 2022, 5, e1453. [Google Scholar] [CrossRef] [PubMed]
- Ghafoor, S.; Hameed, M.R.; Tap, W.D.; Hwang, S. Mesenchymal chondrosarcoma: Imaging features and clinical findings. Skelet. Radiol. 2021, 50, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Zhou, H.; Zhang, X.; Hu, Y.; Guo, T.; Shi, J. Primary mesenchymal chondrosarcoma of the adult lumbar spine: A case report and review of the literature. Transl. Cancer Res. 2022, 11, 3363–3370. [Google Scholar] [CrossRef]
- Chen, M.; Lai, Q. Primary intra- and extradural extramedullary mesenchymal chondrosarcoma with isolated punctate calcification: Case report and literature review. BMC Neurol. 2022, 22, 112. [Google Scholar] [CrossRef]
- Ranjan, R.; Shah, P.K.; Giratkar, S.; Ramachandran, S.; Venkatapathy, N. Primary intraocular mesenchymal chondrosarcoma: A histopathological surprise in an enucleated eye. Can. J. Ophthalmol. 2020, 55, e104–e107. [Google Scholar] [CrossRef]
- Nachawi, N.; Lew, M.; Konopka, K.; Sandouk, Z. A challenging case of Mesenchymal Chondrosarcoma involving the thyroid and special considerations for diagnosis. Clin. Diabetes Endocrinol. 2020, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.Y.; Han, Q.; Cheng, P.; Yang, H.J.; Zhao, Y. Rare case report and literature review of intracranial mesenchymal chondrosarcoma. Ann. Palliat. Med. 2021, 10, 12012–12017. [Google Scholar] [CrossRef]
- Chu, J.; Ma, H.; Wang, Y.; Li, K.; Liao, C.; Ding, Y. CT and MRI findings of intracranial extraskeletal mesenchymal chondrosarcoma-a case report and literature review. Transl. Cancer Res. 2022, 11, 3409–3415. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, S.K. Classification of Chondrosarcoma: From Characteristic to Challenging Imaging Findings. Cancers 2023, 15, 1703. [Google Scholar] [CrossRef]
- Arora, K.; Riddle, N.D. Extraskeletal Mesenchymal Chondrosarcoma. Arch. Pathol. Lab. Med. 2018, 142, 1421–1424. [Google Scholar] [CrossRef]
- Lightenstein, L.a.D.B. Unusual benign and malignant chondroid tumors of bone. A survey of some mesenchymal cartilage tumors and malignant chondroblastic tumors, including a few multicentric ones, as well as many atypical benign chondroblastomas and chondromyxoid fibromas. Cancer 1959, 12, 1142–1157. [Google Scholar] [CrossRef] [PubMed]
- Shakked, R.J.; Geller, D.S.; Gorlick, R.; Dorfman, H.D. Mesenchymal chondrosarcoma: Clinicopathologic study of 20 cases. Arch. Pathol. Lab. Med. 2012, 136, 61–75. [Google Scholar] [CrossRef]
- Yao, K.; Duan, Z.; Yang, S.; Du, Z.; Wang, Y.; Qi, X. OLIG2 Immunolabeling of Mesenchymal Chondrosarcoma: Report of 14 Cases. J. Neuropathol. Exp. Neurol. 2020, 79, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Gorunova, L.; Bjerkehagen, B.; Boye, K.; Heim, S. Chromosome aberrations and HEY1-NCOA2 fusion gene in a mesenchymal chondrosarcoma. Oncol. Rep. 2014, 32, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Uneda, A.; Kurozumi, K.; Fujimura, A.; Kamiya, A.; Hirose, T.; Yanai, H.; Date, I. Intracranial Mesenchymal Chondrosarcoma Lacking the Typical Histopathological Features Diagnosed by HEY1-NCOA2 Gene Fusion. NMC Case Rep. J. 2020, 7, 47–52. [Google Scholar] [CrossRef]
- Bhardwaj, S.; Sharma, A.; Sharma, A. Mesenchymal Chondrosarcoma of the Brain with Metastasis: A Case Report with Literature Review. J. Neurosci. Rural Pract. 2020, 11, 344–348. [Google Scholar] [CrossRef]
- Salas, S.; de Pinieux, G.; Gomez-Brouchet, A.; Larrousserie, F.; Leroy, X.; Aubert, S.; Decouvelaere, A.V.; Giorgi, R.; Fernandez, C.; Bouvier, C. Ezrin immunohistochemical expression in cartilaginous tumours: A useful tool for differential diagnosis between chondroblastic osteosarcoma and chondrosarcoma. Virchows Arch. 2009, 454, 81–87. [Google Scholar] [CrossRef]
- Zając, A.E.; Kopeć, S.; Szostakowski, B.; Spałek, M.J.; Fiedorowicz, M.; Bylina, E.; Filipowicz, P.; Szumera-Ciećkiewicz, A.; Tysarowski, A.; Czarnecka, A.M.; et al. Chondrosarcoma-from Molecular Pathology to Novel Therapies. Cancers 2021, 13, 2390. [Google Scholar] [CrossRef]
- Syed, M.; Mushtaq, S.; Loya, A.; Hassan, U. NKX3.1 a useful marker for mesenchymal chondrosarcoma: An immunohistochemical study. Ann. Diagn. Pathol. 2021, 50, 151660. [Google Scholar] [CrossRef]
- Park, Y.K.; Park, H.R.; Chi, S.G.; Kim, C.J.; Sohn, K.R.; Koh, J.S.; Kim, C.W.; Yang, W.I.; Ro, J.Y.; Ahn, K.W.; et al. Overexpression of p53 and rare genetic mutation in mesenchymal chondrosarcoma. Oncol. Rep. 2000, 7, 1041–1047. [Google Scholar] [CrossRef]
- Meijer, D.; de Jong, D.; Pansuriya, T.C.; van den Akker, B.E.; Picci, P.; Szuhai, K.; Bovee, J.V. Genetic characterization of mesenchymal, clear cell, and dedifferentiated chondrosarcoma. Genes. Chromosomes Cancer 2012, 51, 899–909. [Google Scholar] [CrossRef]
- Van Oosterwijk, J.G.; Meijer, D.; van Ruler, M.A.; van den Akker, B.E.; Oosting, J.; Krenács, T.; Picci, P.; Flanagan, A.M.; Liegl-Atzwanger, B.; Leithner, A.; et al. Screening for potential targets for therapy in mesenchymal, clear cell, and dedifferentiated chondrosarcoma reveals Bcl-2 family members and TGFbeta as potential targets. Am. J. Pathol. 2013, 182, 1347–1356. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; de Graaff, M.A.; Ingram, D.R.; Lim, S.; Lev, D.C.; Briaire-de Bruijn, I.H.; Somaiah, N.; Bovee, J.V.; Lazar, A.J.; Nielsen, T.O. NY-ESO-1 (CTAG1B) expression in mesenchymal tumors. Mod. Pathol. 2015, 28, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.F.; Hayes, M.M.; Lebrun, D.; Espinosa, I.; Nielsen, G.P.; Rosenberg, A.E.; Lee, C.H. FLI-1 distinguishes Ewing sarcoma from small cell osteosarcoma and mesenchymal chondrosarcoma. Appl. Immunohistochem. Mol. Morphol. 2011, 19, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Fanburg-Smith, J.C.; Auerbach, A.; Marwaha, J.S.; Wang, Z.; Santi, M.; Judkins, A.R.; Rushing, E.J. Immunoprofile of mesenchymal chondrosarcoma: Aberrant desmin and EMA expression, retention of INI1, and negative estrogen receptor in 22 female-predominant central nervous system and musculoskeletal cases. Ann. Diagn. Pathol. 2010, 14, 8–14. [Google Scholar] [CrossRef]
- Hung, Y.P.; Fletcher, C.D.; Hornick, J.L. Evaluation of NKX2-2 expression in round cell sarcomas and other tumors with EWSR1 rearrangement: Imperfect specificity for Ewing sarcoma. Mod. Pathol. 2016, 29, 370–380. [Google Scholar] [CrossRef]
- Kawakami, F.; Nambu, J.; Hirose, T.; Sasayama, T.; Itoh, T. Central neurocytoma with ependymoma-like glial component. Brain Tumor Pathol. 2015, 32, 119–123. [Google Scholar] [CrossRef]
- Trepant, A.L.; Trépant, A.L.; Bouchart, C.; Rorive, S.; Sauvage, S.; Decaestecker, C.; Demetter, P.; Salmon, I. Identification of OLIG2 as the most specific glioblastoma stem cell marker starting from comparative analysis of data from similar DNA chip microarray platforms. Tumour Biol. 2015, 36, 1943–1953. [Google Scholar] [CrossRef]
- Wegner, M. Expression of transcription factors during oligodendroglial development. Microsc. Res. Tech. 2001, 52, 746–752. [Google Scholar] [CrossRef]
- Matsumura, N.; Yokoo, H.; Mao, Y.; Yin, W.; Nakazato, Y. Olig2-positive cells in glioneuronal tumors show both glial and neuronal characters: The implication of a common progenitor cell? Neuropathology 2013, 33, 246–255. [Google Scholar] [CrossRef]
- Zhang, L.; He, X.; Liu, X.; Zhang, F.; Huang, L.F.; Potter, A.S.; Xu, L.; Zhou, W.; Zheng, T.; Luo, Z.; et al. Single-Cell Transcriptomics in Medulloblastoma Reveals Tumor-Initiating Progenitors and Oncogenic Cascades during Tumorigenesis and Relapse. Cancer Cell 2019, 36, 302–318.e307. [Google Scholar] [CrossRef] [PubMed]
- Esain, V.; Postlethwait, J.H.; Charnay, P.; Ghislain, J. FGF-receptor signalling controls neural cell diversity in the zebrafish hindbrain by regulating olig2 and sox9. Development 2010, 137, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.I.; Machado, I.; Motoi, T.; Parafioriti, A.; Lacambra, M.; Ichikawa, H.; Kawai, A.; Antonescu, C.R.; Yoshida, A. NKX3-1 Is a Useful Immunohistochemical Marker of EWSR1-NFATC2 Sarcoma and Mesenchymal Chondrosarcoma. Am. J. Surg. Pathol. 2020, 44, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Hornick, J.L.; Fletcher, C.D.M. NKX3.1 immunoreactivity is not identified in mesenchymal chondrosarcoma: A 25-case cohort study. Histopathology 2021, 78, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Motoi, T.; Khanin, R.; Olshen, A.; Mertens, F.; Bridge, J.; Dal Cin, P.; Antonescu, C.R.; Singer, S.; Hameed, M.; et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer 2012, 51, 127–139. [Google Scholar] [CrossRef]
- Nyquist, K.B.; Panagopoulos, I.; Thorsen, J.; Haugom, L.; Gorunova, L.; Bjerkehagen, B.; Fosså, A.; Guriby, M.; Nome, T.; Lothe, R.A.; et al. Whole-transcriptome sequencing identifies novel IRF2BP2-CDX1 fusion gene brought about by translocation t(1;5)(q42;q32) in mesenchymal chondrosarcoma. PLoS ONE 2012, 7, e49705. [Google Scholar] [CrossRef]
- Sandberg, A.A.; Bridge, J.A. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: Chondrosarcoma and other cartilaginous neoplasms. Cancer Genet. Cytogenet. 2003, 143, 1–31. [Google Scholar] [CrossRef]
- Dong, C.; Wu, J.; Chen, Y.; Nie, J.; Chen, C. Activation of PI3K/AKT/mTOR Pathway Causes Drug Resistance in Breast Cancer. Front. Pharmacol. 2021, 12, 628690. [Google Scholar] [CrossRef]
- Shariati, M.; Meric-Bernstam, F. Targeting AKT for cancer therapy. Expert Opin. Investig. Drugs 2019, 28, 977–988. [Google Scholar] [CrossRef]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef]
- Roberts, A.W. Therapeutic development and current uses of BCL-2 inhibition. Hematol. Am. Soc. Hematol. Educ. Program. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Tan, K.; Dong, Y.; Lu, W.; Liu, F.; Mei, Y.; Huang, H.; Zhao, K.; Lv, Z.; Ye, Y.; et al. Therapeutic targeting the oncogenic driver EWSR1::FLI1 in Ewing sarcoma through inhibition of the FACT complex. Oncogene 2023, 42, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.H.; Lee, T.G.; Ko, Y.J.; Kim, S.Y.; Kim, H.R.; Kim, H.; Kim, C.H. Anti-tumor effect of CDK inhibitors on CDKN2A-defective squamous cell lung cancer cells. Cell Oncol. 2018, 41, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Le Cesne, A.; Bellera, C.; Piperno-Neumann, S.; Duffaud, F.; Penel, N.; Cassier, P.; Domont, J.; Takebe, N.; Kind, M.; et al. GDC-0449 in patients with advanced chondrosarcomas: A French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann. Oncol. 2013, 24, 2922–2926. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Liu, Y.; Yang, S.; Wu, X.; Li, H.; Wang, Q. MEK inhibitors for the treatment of non-small cell lung cancer. J. Hematol. Oncol. 2021, 14, 1. [Google Scholar] [CrossRef]
- Qi, W.; Rosikiewicz, W.; Yin, Z.; Xu, B.; Jiang, H.; Wan, S.; Fan, Y.; Wu, G.; Wang, L. Genomic profiling identifies genes and pathways dysregulated by HEY1-NCOA2 fusion and shines a light on mesenchymal chondrosarcoma tumorigenesis. J. Pathol. 2022, 257, 579–592. [Google Scholar] [CrossRef]
- El Beaino, M.; Roszik, J.; Livingston, J.A.; Wang, W.L.; Lazar, A.J.; Amini, B.; Subbiah, V.; Lewis, V.; Conley, A.P. Mesenchymal Chondrosarcoma: A Review with Emphasis on its Fusion-Driven Biology. Curr. Oncol. Rep. 2018, 20, 37. [Google Scholar] [CrossRef]
- Tanaka, M.; Homme, M.; Teramura, Y.; Kumegawa, K.; Yamazaki, Y.; Yamashita, K.; Osato, M.; Maruyama, R.; Nakamura, T. HEY1-NCOA2 expression modulates chondrogenic differentiation and induces mesenchymal chondrosarcoma in mice. JCI Insight 2023, 8, e160279. [Google Scholar] [CrossRef]
- Iso, T.; Kedes, L.; Hamamori, Y. HES and HERP families: Multiple effectors of the Notch signaling pathway. J. Cell Physiol. 2003, 194, 237–255. [Google Scholar] [CrossRef]
- Brown, R.E.; Boyle, J.L. Mesenchymal chondrosarcoma: Molecular characterization by a proteomic approach, with morphogenic and therapeutic implications. Ann. Clin. Lab. Sci. 2003, 33, 131–141. [Google Scholar]
- de Jong, Y.; van Maldegem, A.M.; Marino-Enriquez, A.; de Jong, D.; Suijker, J.; Briaire-de Bruijn, I.H.; Kruisselbrink, A.B.; Cleton-Jansen, A.M.; Szuhai, K.; Gelderblom, H.; et al. Inhibition of Bcl-2 family members sensitizes mesenchymal chondrosarcoma to conventional chemotherapy: Report on a novel mesenchymal chondrosarcoma cell line. Lab. Investig. 2016, 96, 1128–1137. [Google Scholar] [CrossRef] [PubMed]
- Lohnes, D. The Cdx1 homeodomain protein: An integrator of posterior signaling in the mouse. Bioessays 2003, 25, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Downing, J.R.; Head, D.R.; Parham, D.M.; Douglass, E.C.; Hulshof, M.G.; Link, M.P.; Motroni, T.A.; Grier, H.E.; Curcio-Brint, A.M.; Shapiro, D.N. Detection of the (11;22)(q24;q12) translocation of Ewing’s sarcoma and peripheral neuroectodermal tumor by reverse transcription polymerase chain reaction. Am. J. Pathol. 1993, 143, 1294–1300. [Google Scholar]
- Sainati, L.; Scapinello, A.; Montaldi, A.; Bolcato, S.; Ninfo, V.; Carli, M.; Basso, G. A mesenchymal chondrosarcoma of a child with the reciprocal translocation (11;22)(q24;q12). Cancer Genet. Cytogenet. 1993, 71, 144–147. [Google Scholar] [CrossRef]
- Gamberi, G.; Cocchi, S.; Benini, S.; Magagnoli, G.; Morandi, L.; Kreshak, J.; Gambarotti, M.; Picci, P.; Zanella, L.; Alberghini, M. Molecular diagnosis in Ewing family tumors: The Rizzoli experience--222 consecutive cases in four years. J. Mol. Diagn. 2011, 13, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Naumann, S.; Krallman, P.A.; Unni, K.K.; Fidler, M.E.; Neff, J.R.; Bridge, J.A. Translocation der(13;21)(q10;q10) in skeletal and extraskeletal mesenchymal chondrosarcoma. Mod. Pathol. 2002, 15, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Dobin, S.M.; Donner, L.R.; Speights, V.O., Jr. Mesenchymal chondrosarcoma. A cytogenetic, immunohistochemical and ultrastructural study. Cancer Genet. Cytogenet. 1995, 83, 56–60. [Google Scholar] [CrossRef]
- Lukasik, P.; Baranowska-Bosiacka, I.; Kulczycka, K.; Gutowska, I. Inhibitors of Cyclin-Dependent Kinases: Types and Their Mechanism of Action. Int. J. Mol. Sci. 2021, 22, 2806. [Google Scholar] [CrossRef]
- Nishikawa, S.; Iwakuma, T. Drugs Targeting p53 Mutations with FDA Approval and in Clinical Trials. Cancers 2023, 15, 429. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Bovee, J.V.; Hogendoorn, P.C.; Wunder, J.S.; Alman, B.A. Cartilage tumours and bone development: Molecular pathology and possible therapeutic targets. Nat. Rev. Cancer 2010, 10, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Zito, P.M.; Nassereddin, A.; Scharf, R. Vismodegib. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Hubbard, S.R. The insulin receptor: Both a prototypical and atypical receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol. 2013, 5, a008946. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Katayama, S.; Onuma, M.; Rikiishi, T.; Hosaka, M.; Watanabe, M.; Hasegawa, T.; Sasahara, Y.; Kure, S. Mesenchymal chondrosarcoma diagnosed on FISH for HEY1-NCOA2 fusion gene. Pediatr. Int. 2014, 56, e55–e57. [Google Scholar] [CrossRef] [PubMed]
- Toki, S.; Motoi, T.; Miyake, M.; Kobayashi, E.; Kawai, A.; Yoshida, A. Minute mesenchymal chondrosarcoma within osteochondroma: An unexpected diagnosis confirmed by HEY1-NCOA2 fusion. Hum. Pathol. 2018, 81, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Rooper, L.M.; Dermawan, J.K.; Zhang, Y.; Suurmeijer, A.J.H.; Dickson, B.C.; Demicco, E.G.; Antonescu, C.R. Mesenchymal chondrosarcoma of the head and neck with HEY1::NCOA2 fusion: A clinicopathologic and molecular study of 13 cases with emphasis on diagnostic pitfalls. Genes Chromosomes Cancer 2022, 61, 670–677. [Google Scholar] [CrossRef]
- Mendenhall, W.M.; Reith, J.D.; Scarborough, M.T.; Stechmiller, B.K.; Mendenhall, N.P. Mesenchymal Chondrosarcoma. Int. J. Part Ther. 2016, 3, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Derenda, M.; Borof, D.; Kowalina, I.; Wesołowski, W.; Kloc, W.; Iżycka-Świeszewska, E. Primary Spinal Intradural Mesenchymal Chondrosarcoma with Several Local Regrowths Treated with Osteoplastic Laminotomies: A Case Report. Surg. J. 2017, 3, e117–e123. [Google Scholar] [CrossRef]
- Rushing, E.J.; Armonda, R.A.; Ansari, Q.; Mena, H. Mesenchymal chondrosarcoma: A clinicopathologic and flow cytometric study of 13 cases presenting in the central nervous system. Cancer 1996, 77, 1884–1891. [Google Scholar] [CrossRef]
- Kawaguchi, S.; Weiss, I.; Lin, P.P.; Huh, W.W.; Lewis, V.O. Radiation Therapy Is Associated With Fewer Recurrences in Mesenchymal Chondrosarcoma. Clin. Orthop. Relat. Res. 2014, 472, 856–864. [Google Scholar] [CrossRef]
- Mody, M.G.; Rao, G.; Rhines, L.D. Surgical management of spinal mesenchymal tumors. Curr. Oncol. Rep. 2006, 8, 297–304. [Google Scholar] [CrossRef]
- Dehneh, Y.; Aldabbas, M.; Elfarissi, M.A.; Khoulali, M.; Oulali, N.; Moufid, F. Spinal mesenchymal chondrosarcoma: A case report of a rare malignant tumor. Surg. Neurol. Int. 2023, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Prevedello, D.M.-S.; Cordeiro, J.G.; Koerbel, A.; Ditzel, L.F.d.S.; Araújo, J.C. Management of primary spinal chondrosarcoma: Report of two cases causing cord compression. Arq. Neuro-Psiquiatr. 2004, 62, 875–878. [Google Scholar] [CrossRef]
- Wang, M.; Song, Y.; Liu, S.; Sun, W. Effect of surgery and radiotherapy on overall survival in patients with chondrosarcoma: A SEER-based study. J. Orthop. Surg. 2022, 30, 10225536221086319. [Google Scholar] [CrossRef] [PubMed]
- De Amorim Bernstein, K.; Liebsch, N.; Chen, Y.L.; Niemierko, A.; Schwab, J.H.; Raskin, K.; Lozano-Calderon, S.A.; Cote, G.; Harmon, D.C.; Choy, E.; et al. Clinical outcomes for patients after surgery and radiation therapy for mesenchymal chondrosarcomas. J. Surg. Oncol. 2016, 114, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, A.; Tudor, M.; Montanari, J.; Commenchail, K.; Savu, D.I.; Lesueur, P.; Chevalier, F. Chondrosarcoma Resistance to Radiation Therapy: Origins and Potential Therapeutic Solutions. Cancers 2023, 15, 1962. [Google Scholar] [CrossRef] [PubMed]
- Zasadziński, K.; Spałek, M.J. The role of stereotactic body radiotherapy in the management of oligometastatic soft tissue and bone sarcomas. Biul. Pol. Tow. Onkol. Nowotw. 2023, 8, 32–37. [Google Scholar]
- Radziunas, A. Hypofractionated Gamma Knife Stereotactic Radiosurgery as a Tool for Intracranial Mesenchymal Chondrosarcoma Local Treatment: Case Report and Literature Review. Biomed. J. Sci. Tech. Res. 2021, 35, 27836–27842. [Google Scholar] [CrossRef]
- Sallabanda, M.; Garcia, R.; Lorenzana, L.; Santaolalla, I.; Abarca, J.; Sallabanda, K. Treatment of Chordomas and Chondrosarcomas With CyberKnife Robotic Hypofractionated Radiosurgery: A Single Institution Experience. Cureus 2021, 13, e17012. [Google Scholar] [CrossRef]
- Fuji, H.; Nakasu, Y.; Ishida, Y.; Horiguchi, S.; Mitsuya, K.; Kashiwagi, H.; Murayama, S. Feasibility of proton beam therapy for chordoma and chondrosarcoma of the skull base. Skull Base 2011, 21, 201–206. [Google Scholar] [CrossRef]
- Ioakeim-Ioannidou, M.; Kim, D.W.; Goldberg, S.I.; Nielsen, P.; Caruso, P.; Adams, J.A.; Fullerton, B.C.; Tarbell, N.J.; Liebsch, N.J.; MacDonald, S. Proton Radiation Therapy for Pediatric Skull Base Chondrosarcomas. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, e179. [Google Scholar] [CrossRef]
- Rutkowski, P.; Owczarek, W.; Nejc, D.; Jeziorski, A.; Wysocki, W.M.; Słowińska, M.; Dudzisz-Śledź, M.; Wiśniewski, P.; Tchórzewska-Korba, H.; Szumera-Ciećkiewicz, A.; et al. Postępowanie diagnostyczno-terapeutyczne u chorych na mięsaki kości—Zalecenia ekspertów. Onkol. Prakt. Klin.—Eduk. 2022, 8, 125–148. [Google Scholar]
- Rutkowski, E.P.; Świtaj, T. Bone sarcomas. Oncol. Clin. Pract. 2018, 14, 115–128. [Google Scholar]
- Goryń, T.; Szostakowski, B.; Pieńkowski, A. Advances in bone reconstructions after sarcoma resection. Oncol. Clin. Pract. 2018, 14, 331–340. [Google Scholar] [CrossRef]
- Rutkowski, P.; Śpiewankiewicz, B.; Koseła, H.; Świtaj, T.; Osuch, B.; Wiater, K.; Falkowski, S.; Maździarz, A. Sarcoma of the uterus: Diagnostic and therapeutic recommendations. Curr. Gynecol. Oncol. 2013, 11, 24–32. [Google Scholar] [CrossRef]
- Martin-Broto, J.; Hindi, N.; Cruz, J.; Martinez-Trufero, J.; Valverde, C.; De Sande, L.M.; Sala, A.; Bellido, L.; De Juan, A.; Rubio-Casadevall, J.; et al. Relevance of Reference Centers in Sarcoma Care and Quality Item Evaluation: Results from the Prospective Registry of the Spanish Group for Research in Sarcoma (GEIS). Oncologist 2019, 24, e338–e346. [Google Scholar] [CrossRef]
- Rutkowski, P.; Koseła-Paterczyk, H.; Kozak, K.; Ługowska, I.; Fijuth, J.; Jeziorski, A.; Ryś, J.; Spałek, M.; Borkowska, A.; Wągrodzki, M.; et al. Postępowanie diagnostyczno-terapeutyczne u chorych na mięsaki tkanek miękkich u dorosłych—Zalecenia ekspertów. Onkol. Prakt. Klin.—Eduk. 2023, 9, 149–180. [Google Scholar]
- Rutkowski, P.; Świtaj, T.; Koseła-Paterczyk, H.; Kotrych, D.; Goryń, T.; Mazurkiewicz, T.; Fijuth, J.; Grzesiakowska, U.; Borkowska, A.; Spałek, M.; et al. Postępowanie diagnostyczno-terapeutyczne u chorych na mięsaki kości—Zalecenia ekspertów. Onkol. Prakt. Klin.—Eduk. 2023, 9, 181–200. [Google Scholar]
- Gazendam, A.; Popovic, S.; Parasu, N.; Ghert, M. Chondrosarcoma: A Clinical Review. J. Clin. Med. 2023, 12, 2506. [Google Scholar] [CrossRef]
- Casali, P.G.; Bielack, S.; Abecassis, N.; Aro, H.T.; Bauer, S.; Biagini, R.; Bonvalot, S.; Boukovinas, I.; Bovee, J.; Brennan, B.; et al. Bone sarcomas: ESMO-PaedCan-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv79–iv95. [Google Scholar] [CrossRef]
- Redondo, A.; Bague, S.; Bernabeu, D.; Ortiz-Cruz, E.; Valverde, C.; Alvarez, R.; Martinez-Trufero, J.; Lopez-Martin, J.A.; Correa, R.; Cruz, J.; et al. Malignant bone tumors (other than Ewing’s): Clinical practice guidelines for diagnosis, treatment and follow-up by Spanish Group for Research on Sarcomas (GEIS). Cancer Chemother. Pharmacol. 2017, 80, 1113–1131. [Google Scholar] [CrossRef]
- Wood, G.E.; Graves, L.A.; Rubin, E.M.; Reed, D.R.; Riedel, R.F.; Strauss, S.J. Bad to the Bone: Emerging Approaches to Aggressive Bone Sarcomas. Am. Soc. Clin. Oncol. Educ. Book 2023, 43, e390306. [Google Scholar] [CrossRef] [PubMed]
- Cesari, M.; Bertoni, F.; Bacchini, P.; Mercuri, M.; Palmerini, E.; Ferrari, S. Mesenchymal chondrosarcoma. An analysis of patients treated at a single institution. Tumori 2007, 93, 423–427. [Google Scholar] [CrossRef]
- Gelderblom, H.; Hogendoorn, P.C.; Dijkstra, S.D.; van Rijswijk, C.S.; Krol, A.D.; Taminiau, A.H.; Bovee, J.V. The clinical approach towards chondrosarcoma. Oncologist 2008, 13, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Rock, A.; Ali, S.; Chow, W.A. Systemic Therapy for Chondrosarcoma. Curr. Treat. Options Oncol. 2022, 23, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Tansir, G.; Rastogi, S.; Barwad, A.; Dhamija, E. Long lasting response with trabectedin monotherapy in relapsed metastatic mesenchymal chondrosarcoma. Clin. Sarcoma Res. 2020, 10, 16. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Honoki, K.; Kido, A.; Fujii, H.; Enomoto, Y.; Ohbayashi, C.; Tanaka, Y. Chemotherapy improved prognosis of mesenchymal chondrosarcoma with rare metastasis to the pancreas. Case Rep. Oncol. Med. 2014, 2014, 249757. [Google Scholar] [CrossRef]
- Le Cesne, A.; Cresta, S.; Maki, R.G.; Blay, J.Y.; Verweij, J.; Poveda, A.; Casali, P.G.; Balana, C.; Schoffski, P.; Grosso, F.; et al. A retrospective analysis of antitumour activity with trabectedin in translocation-related sarcomas. Eur. J. Cancer 2012, 48, 3036–3044. [Google Scholar] [CrossRef]
- Araki, N.; Takahashi, S.; Sugiura, H.; Ueda, T.; Yonemoto, T.; Takahashi, M.; Morioka, H.; Hiraga, H.; Hiruma, T.; Kunisada, T.; et al. Retrospective inter- and intra-patient evaluation of trabectedin after best supportive care for patients with advanced translocation-related sarcoma after failure of standard chemotherapy. Eur. J. Cancer 2016, 56, 122–130. [Google Scholar] [CrossRef]
- Kawai, A.; Araki, N.; Sugiura, H.; Ueda, T.; Yonemoto, T.; Takahashi, M.; Morioka, H.; Hiraga, H.; Hiruma, T.; Kunisada, T.; et al. Trabectedin monotherapy after standard chemotherapy versus best supportive care in patients with advanced, translocation-related sarcoma: A randomised, open-label, phase 2 study. Lancet Oncol. 2015, 16, 406–416. [Google Scholar] [CrossRef]
- Morioka, H.; Takahashi, S.; Araki, N.; Sugiura, H.; Ueda, T.; Takahashi, M.; Yonemoto, T.; Hiraga, H.; Hiruma, T.; Kunisada, T.; et al. Results of sub-analysis of a phase 2 study on trabectedin treatment for extraskeletal myxoid chondrosarcoma and mesenchymal chondrosarcoma. BMC Cancer 2016, 16, 479. [Google Scholar] [CrossRef]
- Chow, W.; Frankel, P.; Ruel, C.; Araujo, D.M.; Milhem, M.; Okuno, S.; Hartner, L.; Undevia, S.; Staddon, A. Results of a prospective phase 2 study of pazopanib in patients with surgically unresectable or metastatic chondrosarcoma. Cancer 2020, 126, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Koseła-Paterczyk, H. Immunotherapy in sarcoma. Nowotwory J. Oncol. 2020, 70, 296–302. [Google Scholar] [CrossRef]
- Paoluzzi, L.; Cacavio, A.; Ghesani, M.; Karambelkar, A.; Rapkiewicz, A.; Weber, J.; Rosen, G. Response to anti-PD1 therapy with nivolumab in metastatic sarcomas. Clin. Sarcoma Res. 2016, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Blum, V.; Andrei, V.; Ameline, B.; Hofer, S.; Fuchs, B.; Strobel, K.; Allemann, A.; Bode, B.; Baumhoer, D. Metastatic mesenchymal chondrosarcoma showing a sustained response to cabozantinib: A case report. Front. Oncol. 2022, 12, 1086677. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Genetic Alteration Identified in Mesenchymal Chondrosarcoma | Related Processes and Activated Pathways | Potential Therapy | Clinical Status of Chondrosarcoma | Examples of Approved Application | References |
|---|---|---|---|---|---|
| HEY1-NCOA2 fusion | PDGF/PI3K/AKT | PI3K/AKT inhibitors | TQB3525 for advanced bone sarcomas with PI3KA mutations (NCT04690725) | Alpelisib in treatment of HR+/HER2− metastatic breast/Capivasertib in treatment of prostate cancer and solid tumors | [49,50] |
| chromatin remodeling | HDACi | Belinostat in combination with guadecitabine or cedazuridine for locally advanced, metastatic and unresectable chondrosarcoma (NCT04340843) | Belinostat in in treatment of soft tissue sarcoma; Romidepsin in treatment of sarcoma et al. | [51] | |
| antiapoptotic activity | BCL2 inhibitors | - | Venetoclax used in treatment of hematological malignancies | [52] | |
| EWSR1-FLI1 fusion | chromatin remodeling | FACT inhibitor drug, e.g., CBL0137 | - | - | [53] |
| loss of CDKN2A/p16 | pRB pathway | CDK inhibitors | Abemaciclib for bone and soft tissue sarcoma, including chondrosarcoma, with CDK pathway alteration (NCT04040205) | Palbociclib 1, Ribo-ciclib 2 and Abemaciclib 3 in treatment of ER+/HER2− breast cancer | [54] |
| PTCH1 point mutations | Hh pathway | Hh inhibitors | Vismodegib for advanced chondrosarcoma (NCT01267955) | Vismodegib in treatment of basal cell carcinoma treatment | [55] |
| INSR point mutations | Ras/MAPK | MEK inhibitors | - | Cobimetinib Selumetinib, or Binimetinib, Trametinib in treatment of melanoma or non-small-cell lung cancer | [56] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dudzisz-Śledź, M.; Kondracka, M.; Rudzińska, M.; Zając, A.E.; Firlej, W.; Sulejczak, D.; Borkowska, A.; Szostakowski, B.; Szumera-Ciećkiewicz, A.; Piątkowski, J.; et al. Mesenchymal Chondrosarcoma from Diagnosis to Clinical Trials. Cancers 2023, 15, 4581. https://doi.org/10.3390/cancers15184581
Dudzisz-Śledź M, Kondracka M, Rudzińska M, Zając AE, Firlej W, Sulejczak D, Borkowska A, Szostakowski B, Szumera-Ciećkiewicz A, Piątkowski J, et al. Mesenchymal Chondrosarcoma from Diagnosis to Clinical Trials. Cancers. 2023; 15(18):4581. https://doi.org/10.3390/cancers15184581
Chicago/Turabian StyleDudzisz-Śledź, Monika, Monika Kondracka, Monika Rudzińska, Agnieszka E. Zając, Wiktoria Firlej, Dorota Sulejczak, Aneta Borkowska, Bartłomiej Szostakowski, Anna Szumera-Ciećkiewicz, Jakub Piątkowski, and et al. 2023. "Mesenchymal Chondrosarcoma from Diagnosis to Clinical Trials" Cancers 15, no. 18: 4581. https://doi.org/10.3390/cancers15184581
APA StyleDudzisz-Śledź, M., Kondracka, M., Rudzińska, M., Zając, A. E., Firlej, W., Sulejczak, D., Borkowska, A., Szostakowski, B., Szumera-Ciećkiewicz, A., Piątkowski, J., Rutkowski, P., & Czarnecka, A. M. (2023). Mesenchymal Chondrosarcoma from Diagnosis to Clinical Trials. Cancers, 15(18), 4581. https://doi.org/10.3390/cancers15184581