

Generation of Novel Immunocompetent Mouse Cell Lines to Model Experimental Metastasis of High-Risk Neuroblastoma
 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
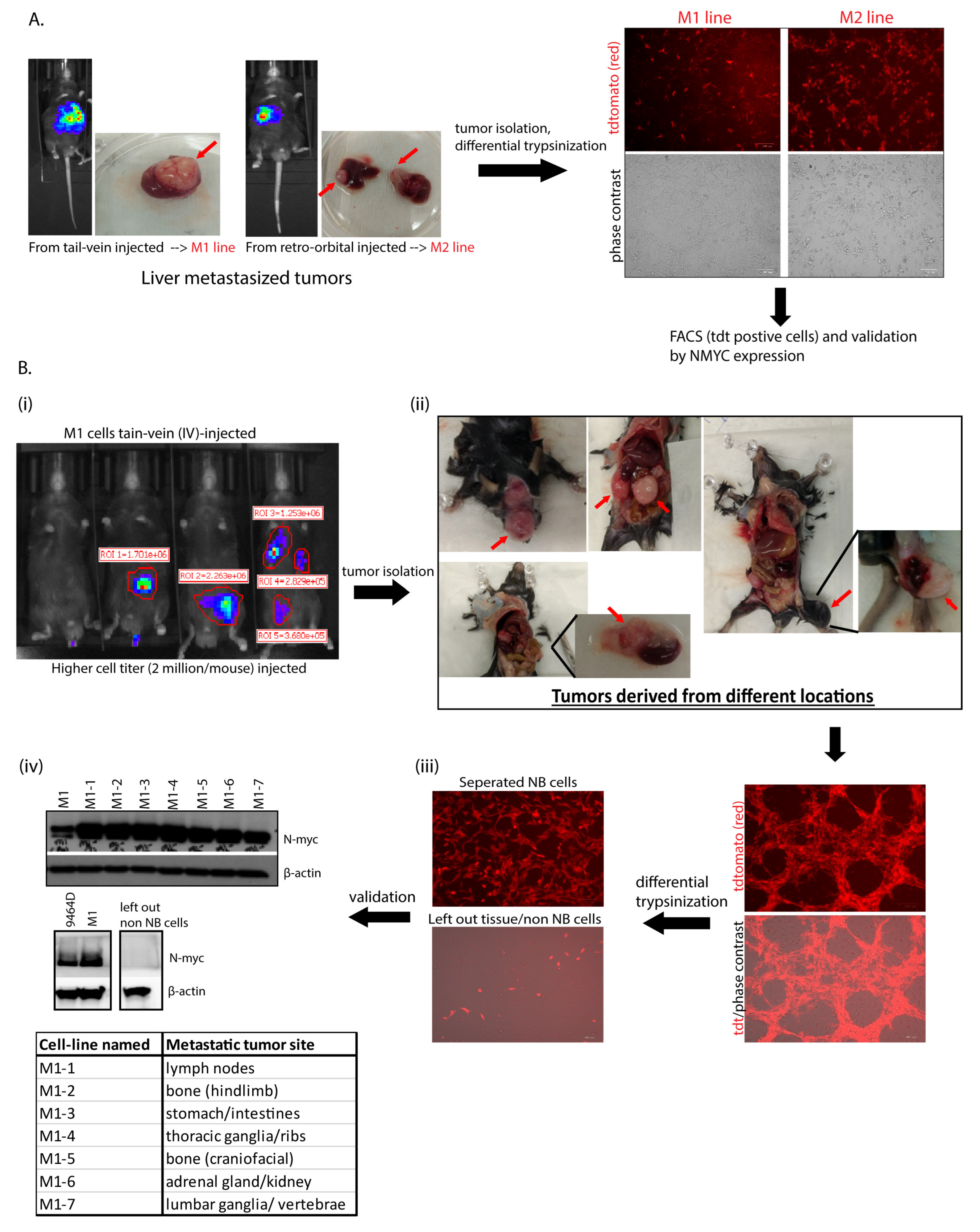
2.1.1. M1 and M2 Cell Line Generation
2.1.2. Generation of M1-Derivative Lines
2.2. In Vivo Characterization
2.3. Cell-Based In Vitro Functional Assays
2.3.1. IncuCyte Cell Proliferation and Migration Assays
2.3.2. Invasion Assay
2.3.3. Colony Formation Assay
2.3.4. Soft Agar Assay
2.4. Western Blot
2.5. Flow Cytometry and Surface Staining
2.6. RNA-Seq and Differential Gene Expression Analysis
2.6.1. Library Preparation and Sequencing of mRNA (Illumina Kit)
2.6.2. Differential Gene Expression and Pathway Analysis
2.7. Statistical Analysis
3. Results
3.1. Generation of M2- and M1-Derived Cell Lines
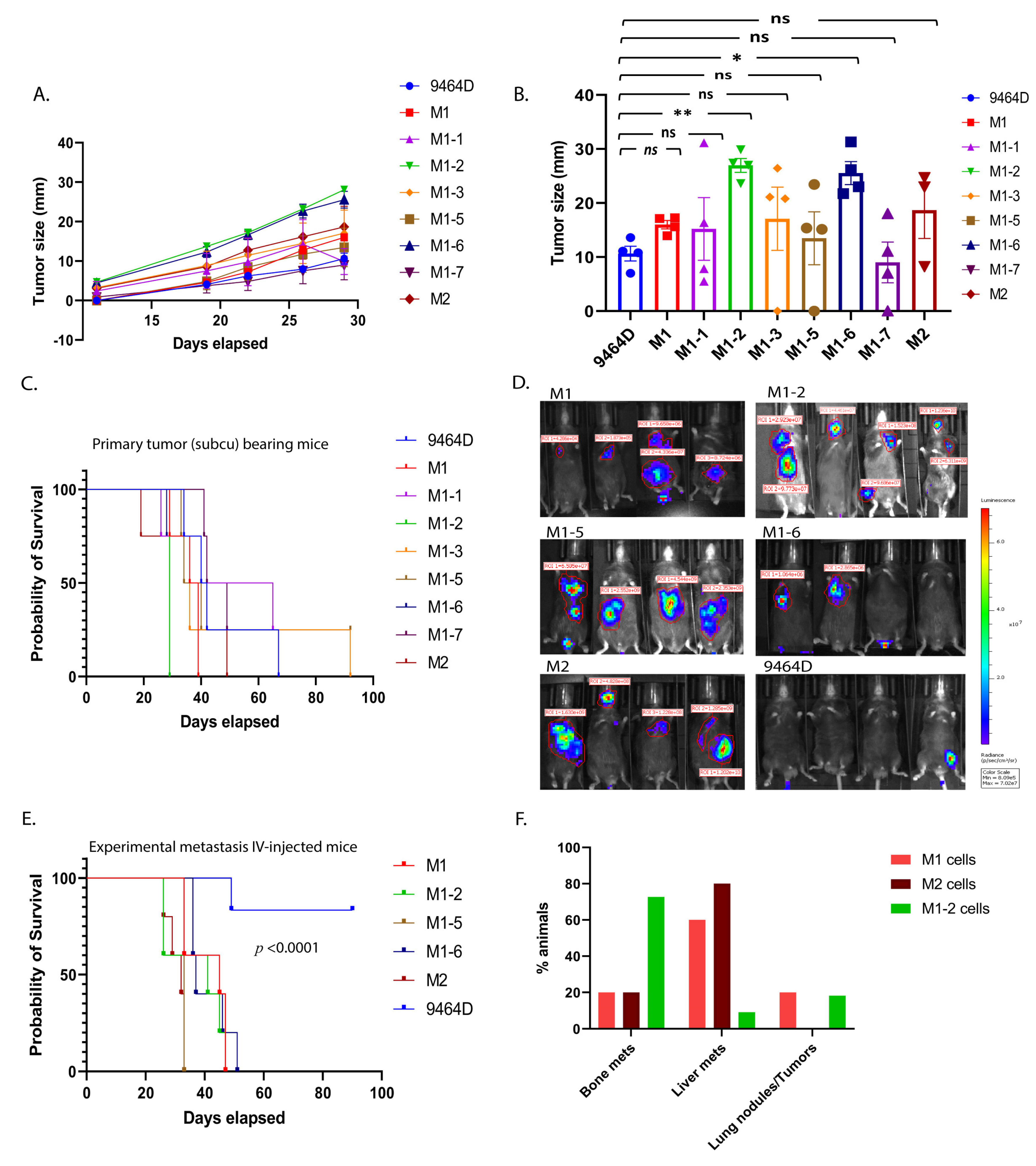
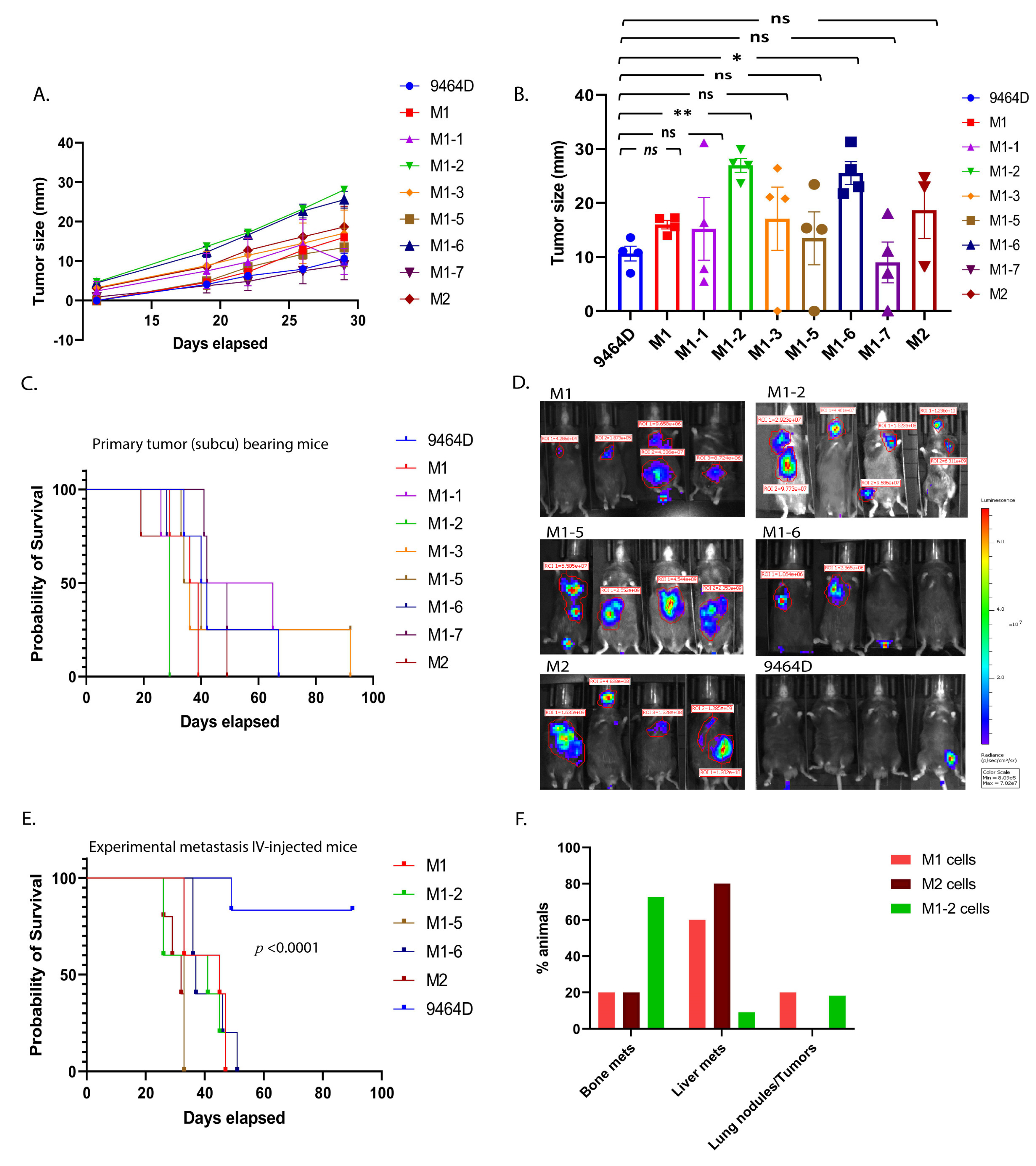
3.2. Primary Tumor Growth and Survival Kinetics Using Subcutaneous Model
3.3. M1 and M2 Cells and the M1 Derivatives M1-2 and M1-5 Show Increased Metastatic Ability
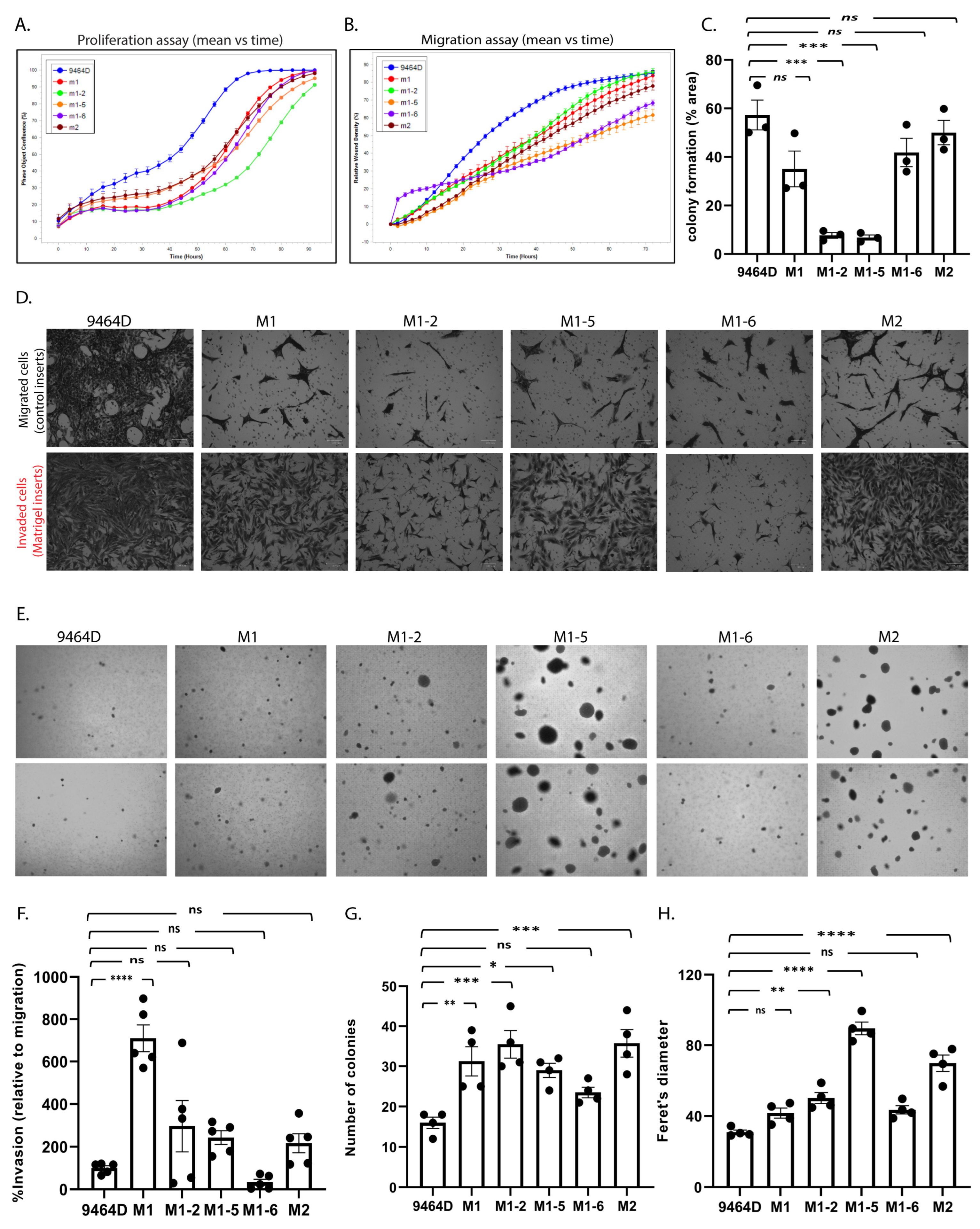
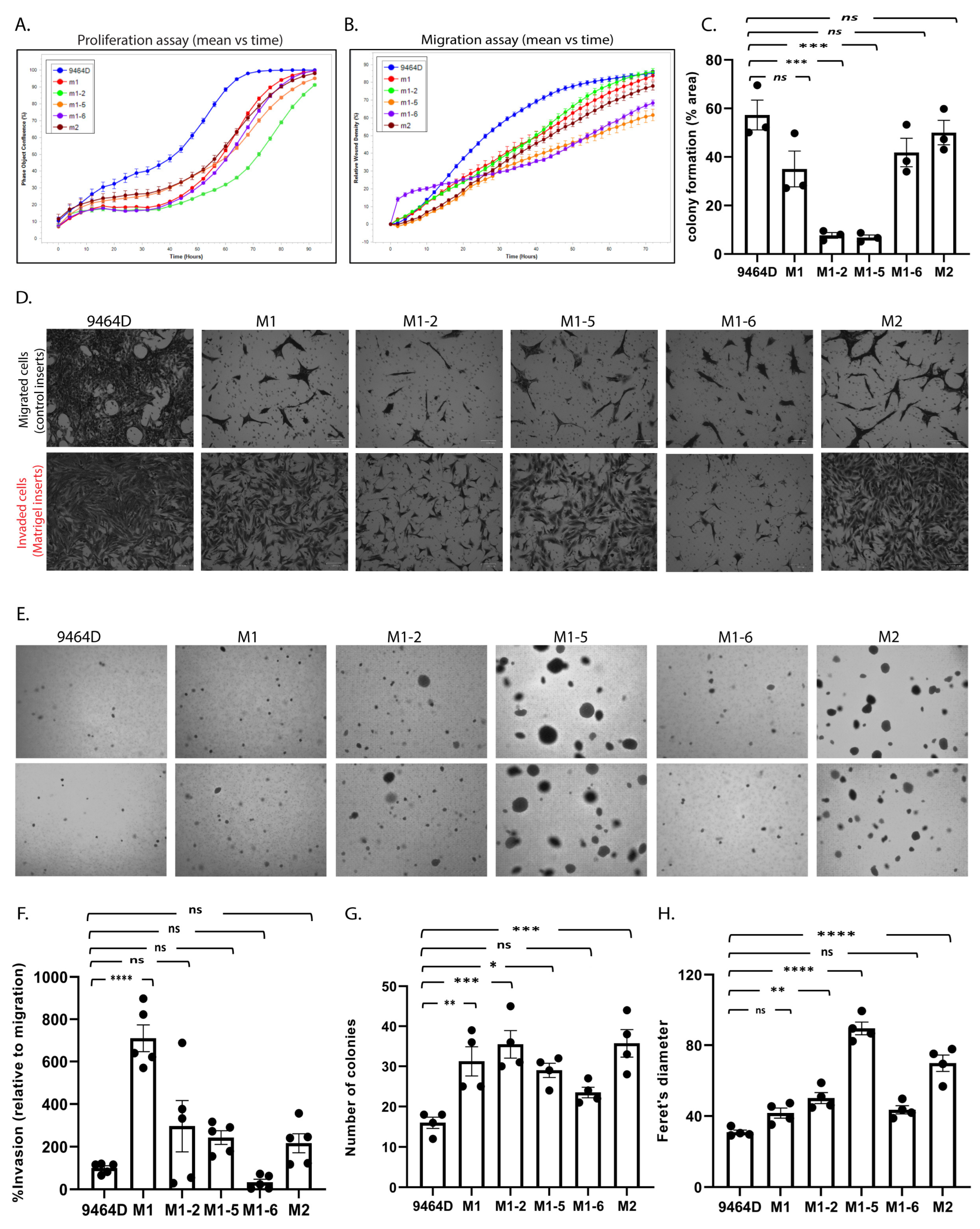
3.4. In Vitro/in-Culture Characterization of New Cell Lines
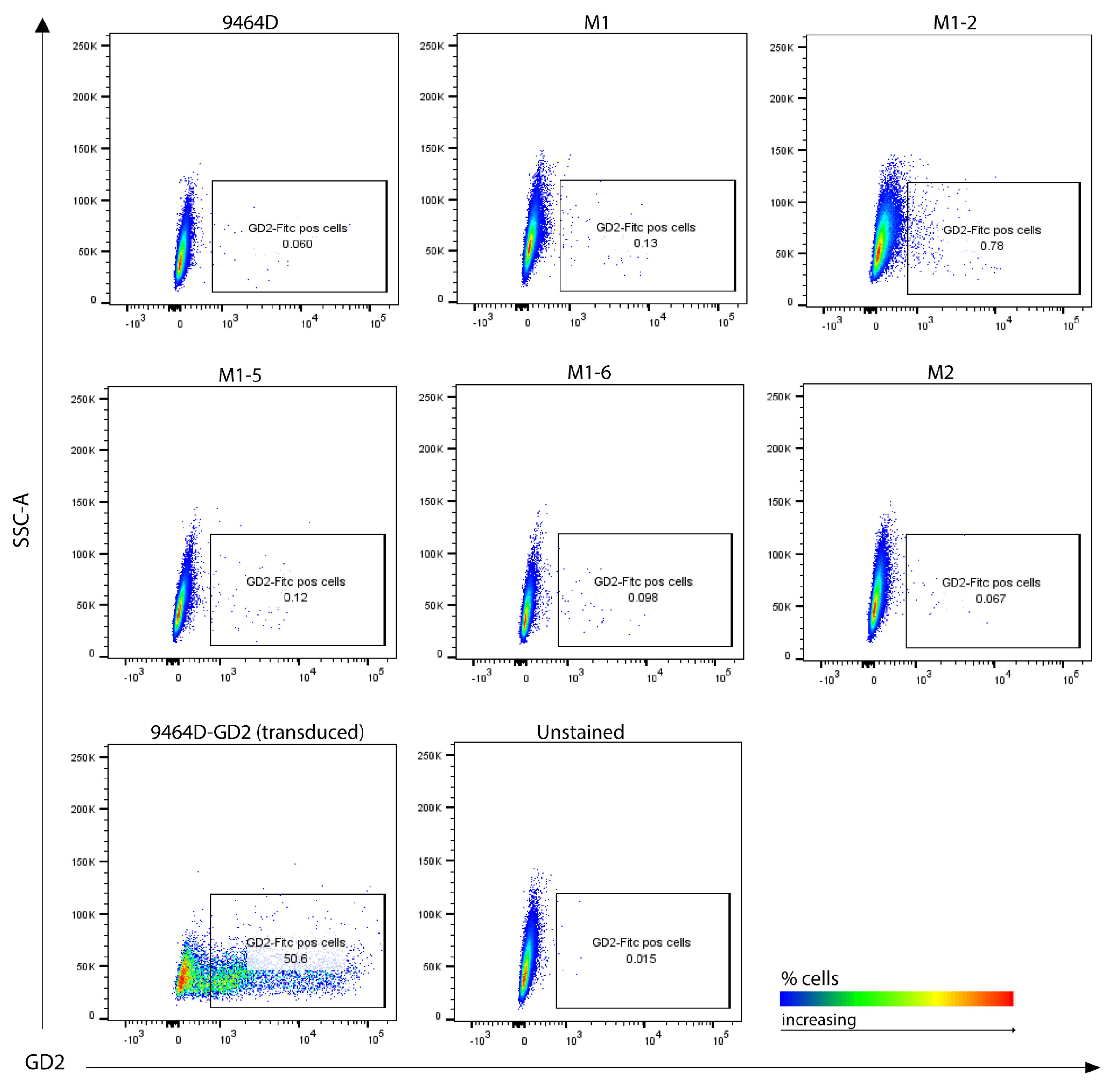
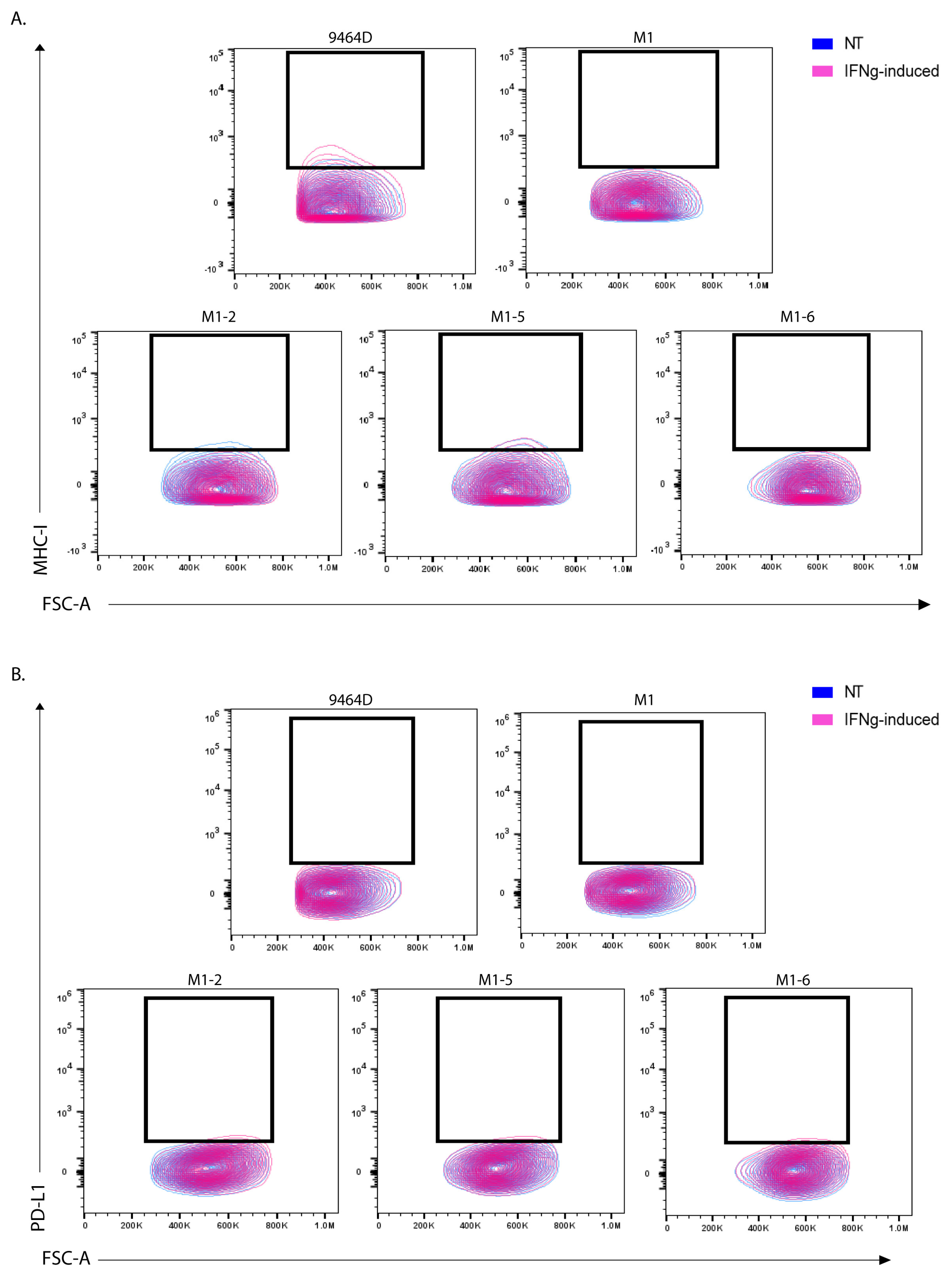
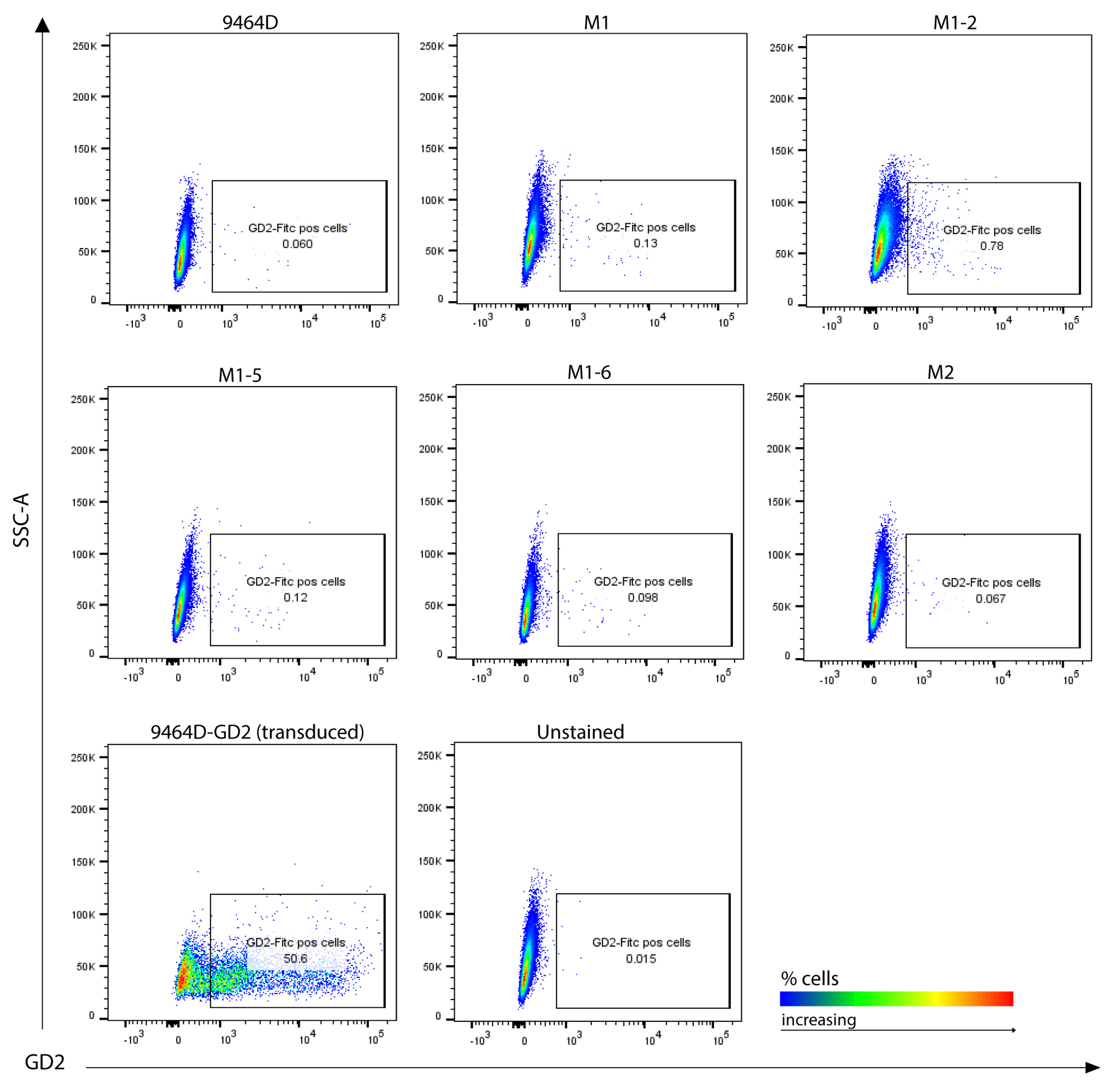
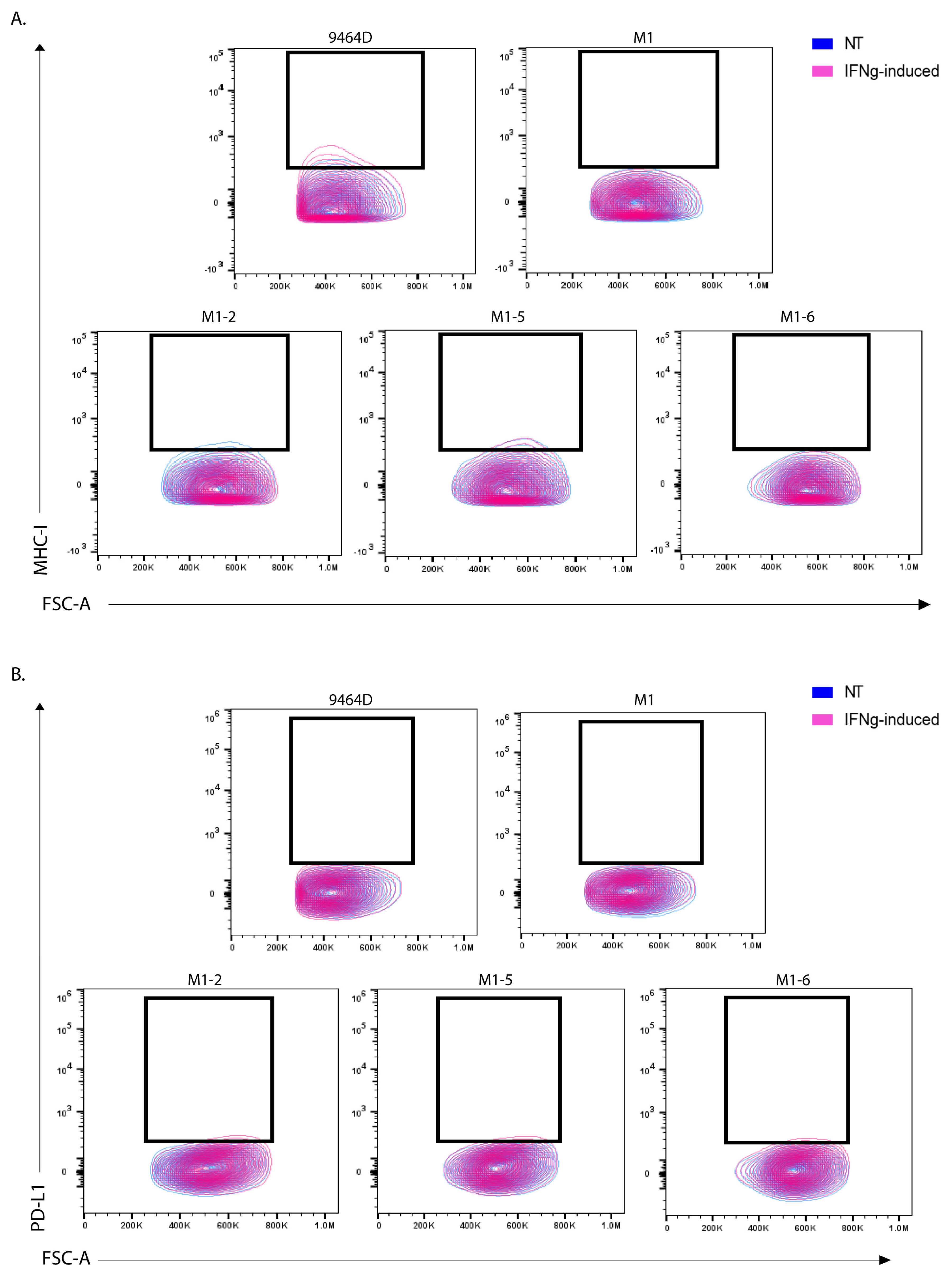
3.5. M1 Cells and Their Derived Variants Have Low Expression of GD2 and Are Resistant to MHC-I Induction via IFN-γ
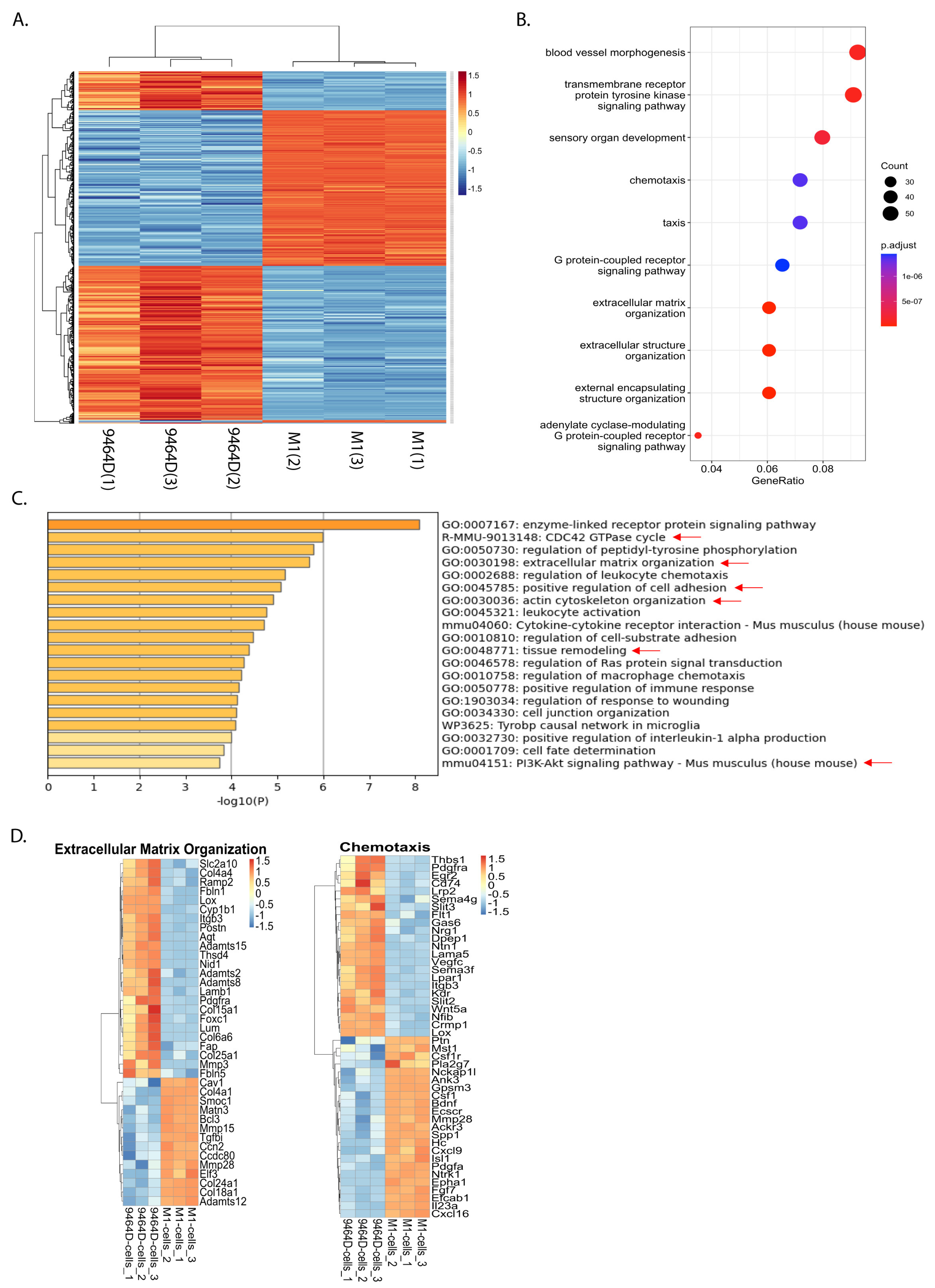
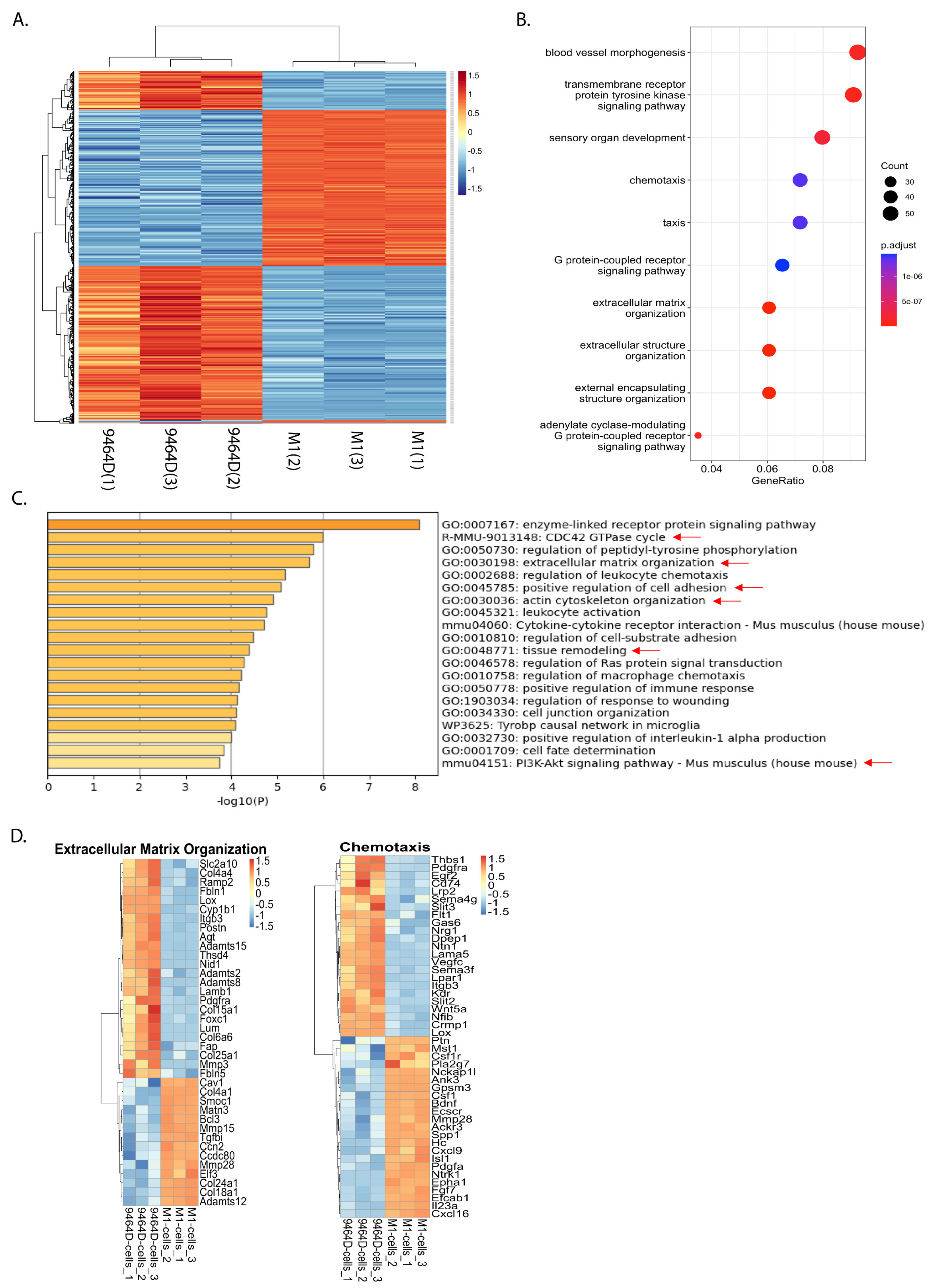
3.6. RNA-Seq Analysis Reveals a Differential Gene Expression Profile of M1 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.S.; Park, J.R. Neuroblastoma: Paradigm for precision medicine. Pediatr. Clin. N. Am. 2015, 62, 225–256. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Nakagawara, A.; Li, Y.; Izumi, H.; Muramori, K.; Inada, H.; Nishi, M. Neuroblastoma. Jpn. J. Clin. Oncol. 2018, 48, 214–241. [Google Scholar] [CrossRef]
- DuBois, S.G.; Macy, M.E.; Henderson, T.O. High-Risk and Relapsed Neuroblastoma: Toward More Cures and Better Outcomes. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 768–780. [Google Scholar] [CrossRef]
- London, W.B.; Bagatell, R.; Weigel, B.J.; Fox, E.; Guo, D.; Van Ryn, C.; Naranjo, A.; Park, J.R. Historical time to disease progression and progression-free survival in patients with recurrent/refractory neuroblastoma treated in the modern era on Children’s Oncology Group early-phase trials. Cancer 2017, 123, 4914–4923. [Google Scholar] [CrossRef]
- DuBois, S.G.; Kalika, Y.; Lukens, J.N.; Brodeur, G.M.; Seeger, R.C.; Atkinson, J.B.; Haase, G.M.; Black, C.T.; Perez, C.; Shimada, H.; et al. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J. Pediatr. Hematol. Oncol. 1999, 21, 181–189. [Google Scholar] [CrossRef]
- Liu, S.; Yin, W.; Lin, Y.; Huang, S.; Xue, S.; Sun, G.; Wang, C. Metastasis pattern and prognosis in children with neuroblastoma. World J. Surg. Oncol. 2023, 21, 130. [Google Scholar] [CrossRef]
- Kroesen, M.; Nierkens, S.; Ansems, M.; Wassink, M.; Orentas, R.J.; Boon, L.; den Brok, M.H.; Hoogerbrugge, P.M.; Adema, G.J. A transplantable TH-MYCN transgenic tumor model in C57Bl/6 mice for preclinical immunological studies in neuroblastoma. Int. J. Cancer 2014, 134, 1335–1345. [Google Scholar] [CrossRef]
- Kroesen, M.; Brok, I.C.; Reijnen, D.; van Hout-Kuijer, M.A.; Zeelenberg, I.S.; Den Brok, M.H.; Hoogerbrugge, P.M.; Adema, G.J. Intra-adrenal murine TH-MYCN neuroblastoma tumors grow more aggressive and exhibit a distinct tumor microenvironment relative to their subcutaneous equivalents. Cancer Immunol. Immunother. 2015, 64, 563–572. [Google Scholar] [CrossRef]
- Webb, E.R.; Lanati, S.; Wareham, C.; Easton, A.; Dunn, S.N.; Inzhelevskaya, T.; Sadler, F.M.; James, S.; Ashton-Key, M.; Cragg, M.S.; et al. Immune characterization of pre-clinical murine models of neuroblastoma. Sci. Rep. 2020, 10, 16695. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, J.K.; Orentas, R.J.; Lincoln, E.; Khan, T.; Salcedo, R.; Hixon, J.A.; Back, T.C.; Wei, J.S.; Patidar, R.; Song, Y.; et al. High-throughput molecular and histopathologic profiling of tumor tissue in a novel transplantable model of murine neuroblastoma: New tools for pediatric drug discovery. Cancer Investig. 2012, 30, 343–363. [Google Scholar] [CrossRef] [PubMed]
- Lode, H.N.; Xiang, R.; Varki, N.M.; Dolman, C.S.; Gillies, S.D.; Reisfeld, R.A. Targeted interleukin-2 therapy for spontaneous neuroblastoma metastases to bone marrow. J. Natl. Cancer Inst. 1997, 89, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Lode, H.N.; Moehler, T.; Xiang, R.; Jonczyk, A.; Gillies, S.D.; Cheresh, D.A.; Reisfeld, R.A. Synergy between an antiangiogenic integrin alphav antagonist and an antibody-cytokine fusion protein eradicates spontaneous tumor metastases. Proc. Natl. Acad. Sci. USA 1999, 96, 1591–1596. [Google Scholar] [CrossRef]
- Lode, H.N.; Xiang, R.; Dreier, T.; Varki, N.M.; Gillies, S.D.; Reisfeld, R.A. Natural killer cell-mediated eradication of neuroblastoma metastases to bone marrow by targeted interleukin-2 therapy. Blood 1998, 91, 1706–1715. [Google Scholar] [CrossRef]
- Voeller, J.; Erbe, A.K.; Slowinski, J.; Rasmussen, K.; Carlson, P.M.; Hoefges, A.; Van den Heuvel, S.; Stuckwisch, A.; Wang, X.; Gillies, S.D.; et al. Combined innate and adaptive immunotherapy overcomes resistance of immunologically cold syngeneic murine neuroblastoma to checkpoint inhibition. J. Immunother. Cancer 2019, 7, 344. [Google Scholar] [CrossRef]
- Weiss, W.A.; Aldape, K.; Mohapatra, G.; Feuerstein, B.G.; Bishop, J.M. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997, 16, 2985–2995. [Google Scholar] [CrossRef]
- Aiken, T.J.; Erbe, A.K.; Zebertavage, L.; Komjathy, D.; Feils, A.S.; Rodriguez, M.; Stuckwisch, A.; Gillies, S.D.; Morris, Z.S.; Birstler, J.; et al. Mechanism of effective combination radio-immunotherapy against 9464D-GD2, an immunologically cold murine neuroblastoma. J. Immunother. Cancer 2022, 10, e004834. [Google Scholar] [CrossRef]
- Dhamdhere, M.R.; Gowda, C.P.; Singh, V.; Liu, Z.; Carruthers, N.; Grant, C.N.; Sharma, A.; Dovat, S.; Sundstrom, J.M.; Wang, H.G.; et al. IGF2BP1 regulates the cargo of extracellular vesicles and promotes neuroblastoma metastasis. Oncogene 2023, 42, 1558–1571. [Google Scholar] [CrossRef]
- Borowicz, S.; Van Scoyk, M.; Avasarala, S.; Karuppusamy Rathinam, M.K.; Tauler, J.; Bikkavilli, R.K.; Winn, R.A. The soft agar colony formation assay. J. Vis. Exp. 2014, e51998. [Google Scholar] [CrossRef]
- Liu, X.; Wills, C.A.; Chen, L.; Zhang, J.; Zhao, Y.; Zhou, M.; Sundstrom, J.M.; Schell, T.; Spiegelman, V.S.; Young, M.M.; et al. Small extracellular vesicles induce resistance to anti-GD2 immunotherapy unveiling tipifarnib as an adjunct to neuroblastoma immunotherapy. J. Immunother. Cancer 2022, 10, e004399. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, D.J.; Smyth, G.K. Testing significance relative to a fold-change threshold is a TREAT. Bioinformatics 2009, 25, 765–771. [Google Scholar] [CrossRef]
- Gaujoux, R.; Seoighe, C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 2010, 11, 367. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Supek, F.; Bosnjak, M.; Skunca, N.; Smuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Harrell, M.I.; Iritani, B.M.; Ruddell, A. Lymph node mapping in the mouse. J. Immunol. Methods 2008, 332, 170–174. [Google Scholar] [CrossRef]
- Deng, Z.; Wang, H.; Liu, J.; Deng, Y.; Zhang, N. Comprehensive understanding of anchorage-independent survival and its implication in cancer metastasis. Cell Death Dis. 2021, 12, 629. [Google Scholar] [CrossRef]
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2796–2805. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.W.; Binte Hanafi, Z.; Chew, L.C.Y.; Mei, Y.; Liu, H. IL-1alpha Processing, Signaling and Its Role in Cancer Progression. Cells 2021, 10, 92. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Barajon, I.; Garlanda, C. IL-1 and IL-1 regulatory pathways in cancer progression and therapy. Immunol. Rev. 2018, 281, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Watari, K.; Shibata, T.; Kawahara, A.; Sata, K.; Nabeshima, H.; Shinoda, A.; Abe, H.; Azuma, K.; Murakami, Y.; Izumi, H.; et al. Tumor-derived interleukin-1 promotes lymphangiogenesis and lymph node metastasis through M2-type macrophages. PLoS ONE 2014, 9, e99568. [Google Scholar] [CrossRef]
- Leon, X.; Bothe, C.; Garcia, J.; Parreno, M.; Alcolea, S.; Quer, M.; Vila, L.; Camacho, M. Expression of IL-1alpha correlates with distant metastasis in patients with head and neck squamous cell carcinoma. Oncotarget 2015, 6, 37398–37409. [Google Scholar] [CrossRef]
- Tjomsland, V.; Spangeus, A.; Valila, J.; Sandstrom, P.; Borch, K.; Druid, H.; Falkmer, S.; Falkmer, U.; Messmer, D.; Larsson, M. Interleukin 1alpha sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia 2011, 13, 664–675. [Google Scholar] [CrossRef]
- Ma, J.; Liang, W.; Qiang, Y.; Li, L.; Du, J.; Pan, C.; Chen, B.; Zhang, C.; Chen, Y.; Wang, Q. Interleukin-1 receptor antagonist inhibits matastatic potential by down-regulating CXCL12/CXCR4 signaling axis in colorectal cancer. Cell Commun. Signal. 2021, 19, 122. [Google Scholar] [CrossRef]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011, 278, 16–27. [Google Scholar] [CrossRef]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef]
- Moro, N.; Mauch, C.; Zigrino, P. Metalloproteinases in melanoma. Eur. J. Cell Biol. 2014, 93, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Pittayapruek, P.; Meephansan, J.; Prapapan, O.; Komine, M.; Ohtsuki, M. Role of Matrix Metalloproteinases in Photoaging and Photocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 868. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zheng, X.; Feng, M.; Mo, Z.; Shan, Y.; Wang, Y.; Jin, J. Upregulated MMP28 in Hepatocellular Carcinoma Promotes Metastasis via Notch3 Signaling and Predicts Unfavorable Prognosis. Int. J. Biol. Sci. 2019, 15, 812–825. [Google Scholar] [CrossRef] [PubMed]
- Drury, J.; Rychahou, P.G.; Kelson, C.O.; Geisen, M.E.; Wu, Y.; He, D.; Wang, C.; Lee, E.Y.; Evers, B.M.; Zaytseva, Y.Y. Upregulation of CD36, a Fatty Acid Translocase, Promotes Colorectal Cancer Metastasis by Increasing MMP28 and Decreasing E-Cadherin Expression. Cancers 2022, 14, 252. [Google Scholar] [CrossRef]
- Zheng, S.; Wu, H.; Wang, F.; Lv, J.; Lu, J.; Fang, Q.; Wang, F.; Lu, Y.; Zhang, S.; Xu, Y.; et al. The oncoprotein HBXIP facilitates metastasis of hepatocellular carcinoma cells by activation of MMP15 expression. Cancer Manag. Res. 2019, 11, 4529–4540. [Google Scholar] [CrossRef]
- Wu, M.H.; Wu, P.R.; Hsieh, Y.H.; Lin, C.L.; Liu, C.J.; Ying, T.H. Silencing PROK2 Inhibits Invasion of Human Cervical Cancer Cells by Targeting MMP15 Expression. Int. J. Mol. Sci. 2020, 21, 6391. [Google Scholar] [CrossRef]
- Mochizuki, S.; Okada, Y. ADAMs in cancer cell proliferation and progression. Cancer Sci. 2007, 98, 621–628. [Google Scholar] [CrossRef]
- Kang, T.Z.E.; Zhu, L.; Yang, D.; Ding, D.; Zhu, X.; Wan, Y.C.E.; Liu, J.; Ramakrishnan, S.; Chan, L.L.; Chan, S.Y.; et al. The elevated transcription of ADAM19 by the oncohistone H2BE76K contributes to oncogenic properties in breast cancer. J. Biol. Chem. 2021, 296, 100374. [Google Scholar] [CrossRef]
- Sui, Y.; Jiang, H.; Kellogg, C.M.; Oh, S.; Janknecht, R. Promotion of colorectal cancer by transcription factor BHLHE40 involves upregulation of ADAM19 and KLF7. Front. Oncol. 2023, 13, 1122238. [Google Scholar] [CrossRef]
- Wildeboer, D.; Naus, S.; Amy Sang, Q.X.; Bartsch, J.W.; Pagenstecher, A. Metalloproteinase disintegrins ADAM8 and ADAM19 are highly regulated in human primary brain tumors and their expression levels and activities are associated with invasiveness. J. Neuropathol. Exp. Neurol. 2006, 65, 516–527. [Google Scholar] [CrossRef]
- Hou, Y.; Xu, Y.; Wu, D. ADAMTS12 acts as a tumor microenvironment related cancer promoter in gastric cancer. Sci. Rep. 2021, 11, 10996. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yuan, Z.; Tang, Z. ADAMTS12, a novel prognostic predictor, promotes cell proliferation, migration, and invasion in head and neck squamous cell carcinoma. Oral Dis. 2022, 10, 1111. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Luo, X.; Huang, B.; Wang, X.; Deng, Y.; Zhong, Z. ADAMTS12 acts as a cancer promoter in colorectal cancer via activating the Wnt/beta-catenin signaling pathway in vitro. Ann. Transl. Med. 2020, 8, 301. [Google Scholar] [CrossRef]
- Romer, A.M.A.; Thorseth, M.L.; Madsen, D.H. Immune Modulatory Properties of Collagen in Cancer. Front. Immunol. 2021, 12, 791453. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Xu, H.; Wang, W.; Li, S.; Li, H.; Li, T.; Zhang, W.; Yu, X.; Liu, L. The role of collagen in cancer: From bench to bedside. J. Transl. Med. 2019, 17, 309. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Jin, H.; Hu, J.; Li, X.; Ruan, H.; Xu, H.; Wei, L.; Dong, W.; Teng, F.; Gu, J.; et al. COL4A1 promotes the growth and metastasis of hepatocellular carcinoma cells by activating FAK-Src signaling. J. Exp. Clin. Cancer Res. 2020, 39, 148. [Google Scholar] [CrossRef]
- Yan, L.; Xu, F.; Dai, C. Overexpression of COL24A1 in Hepatocellular Carcinoma Predicts Poor Prognosis: A Study Based on Multiple Databases, Clinical Samples and Cell Lines. OncoTargets Ther. 2020, 13, 2819–2832. [Google Scholar] [CrossRef]
- Yuan, H.; Liu, F.; Ma, T.; Zeng, Z.; Zhang, N. miR-338-3p inhibits cell growth, invasion, and EMT process in neuroblastoma through targeting MMP-2. Open Life Sci. 2021, 16, 198–209. [Google Scholar] [CrossRef]
- Stefano, E.; Muscella, A.; Benedetti, M.; De Castro, F.; Fanizzi, F.P.; Marsigliante, S. Antitumor and antimigration effects of a new Pt compound on neuroblastoma cells. Biochem. Pharmacol. 2022, 202, 115124. [Google Scholar] [CrossRef]
- Li, X.Y.; Huang, G.H.; Liu, Q.K.; Yang, X.T.; Wang, K.; Luo, W.Z.; Liang, T.S.; Yuan, S.P.; Zhen, Y.W.; Yan, D.M. Porf-2 Inhibits Tumor Cell Migration Through the MMP-2/9 Signaling Pathway in Neuroblastoma and Glioma. Front. Oncol. 2020, 10, 975. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-gamma in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Castro, F.; Cardoso, A.P.; Goncalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef]
- Kaplan, D.H.; Shankaran, V.; Dighe, A.S.; Stockert, E.; Aguet, M.; Old, L.J.; Schreiber, R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7556–7561. [Google Scholar] [CrossRef]
- Yu, A.L.; Gilman, A.L.; Ozkaynak, M.F.; Naranjo, A.; Diccianni, M.B.; Gan, J.; Hank, J.A.; Batova, A.; London, W.B.; Tenney, S.C.; et al. Long-Term Follow-up of a Phase III Study of ch14.18 (Dinutuximab) + Cytokine Immunotherapy in Children with High-Risk Neuroblastoma: COG Study ANBL0032. Clin. Cancer Res. 2021, 27, 2179–2189. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhamdhere, M.R.; Spiegelman, D.V.; Schneper, L.; Erbe, A.K.; Sondel, P.M.; Spiegelman, V.S. Generation of Novel Immunocompetent Mouse Cell Lines to Model Experimental Metastasis of High-Risk Neuroblastoma. Cancers 2023, 15, 4693. https://doi.org/10.3390/cancers15194693
Dhamdhere MR, Spiegelman DV, Schneper L, Erbe AK, Sondel PM, Spiegelman VS. Generation of Novel Immunocompetent Mouse Cell Lines to Model Experimental Metastasis of High-Risk Neuroblastoma. Cancers. 2023; 15(19):4693. https://doi.org/10.3390/cancers15194693
Chicago/Turabian StyleDhamdhere, Mayura R., Dan V. Spiegelman, Lisa Schneper, Amy K. Erbe, Paul M. Sondel, and Vladimir S. Spiegelman. 2023. "Generation of Novel Immunocompetent Mouse Cell Lines to Model Experimental Metastasis of High-Risk Neuroblastoma" Cancers 15, no. 19: 4693. https://doi.org/10.3390/cancers15194693
APA StyleDhamdhere, M. R., Spiegelman, D. V., Schneper, L., Erbe, A. K., Sondel, P. M., & Spiegelman, V. S. (2023). Generation of Novel Immunocompetent Mouse Cell Lines to Model Experimental Metastasis of High-Risk Neuroblastoma. Cancers, 15(19), 4693. https://doi.org/10.3390/cancers15194693