
Unraveling the Bone Tissue Microenvironment in Chronic Lymphocytic Leukemia

 ,
, 
 ,
,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Background on Bone Remodeling
3. Bone Tissue Erosion in Chronic Lymphocytic Leukemia Patients
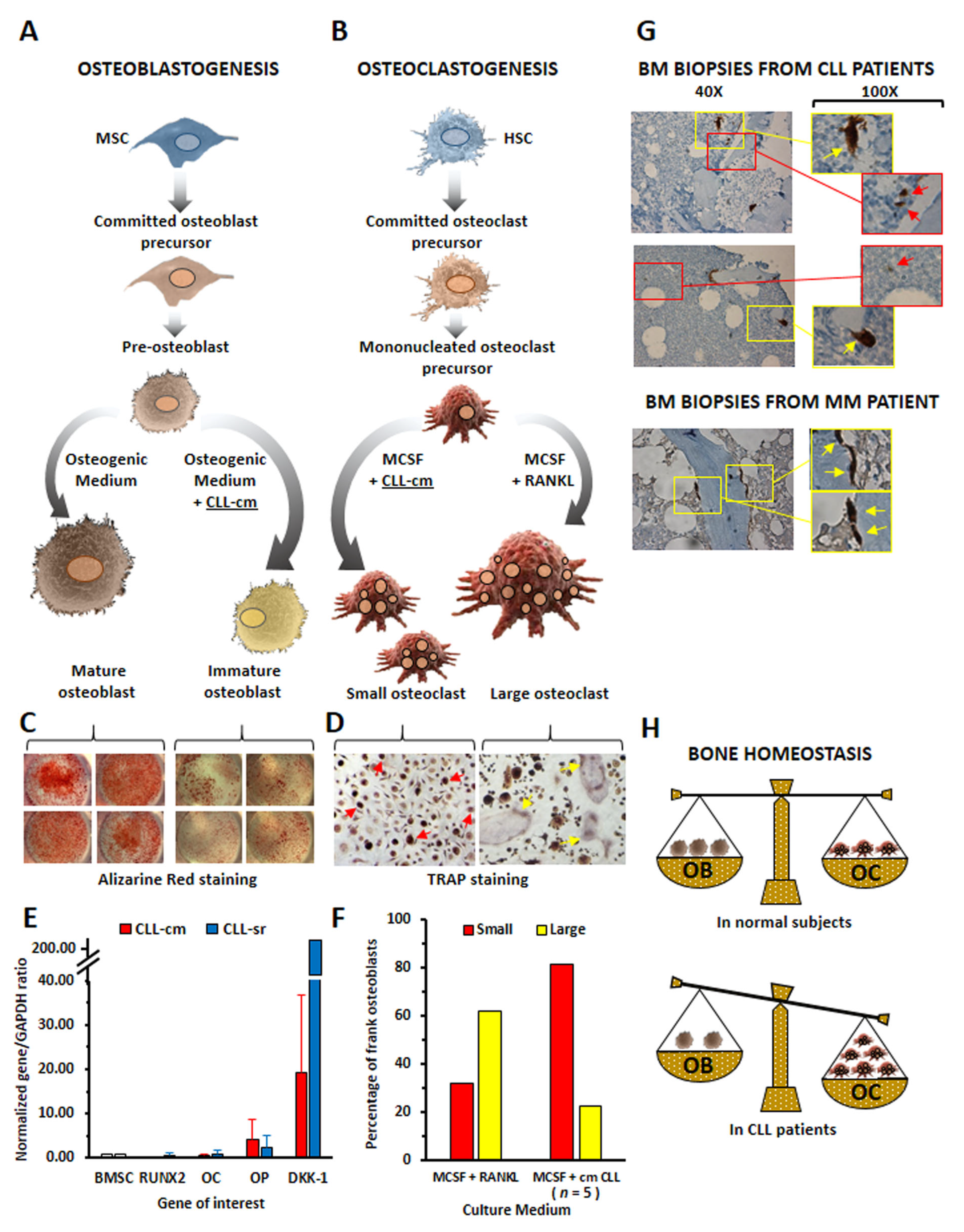
4. CLL Cells Affect Osteoblast and Osteoclast Differentiation
5. Cytokines Produced by Neoplastic B Cells Are Involved in Bone Tissue Derangement of CLL Patients
6. Chemokines: Can They Be Additional Players in CLL Bone Remodeling?
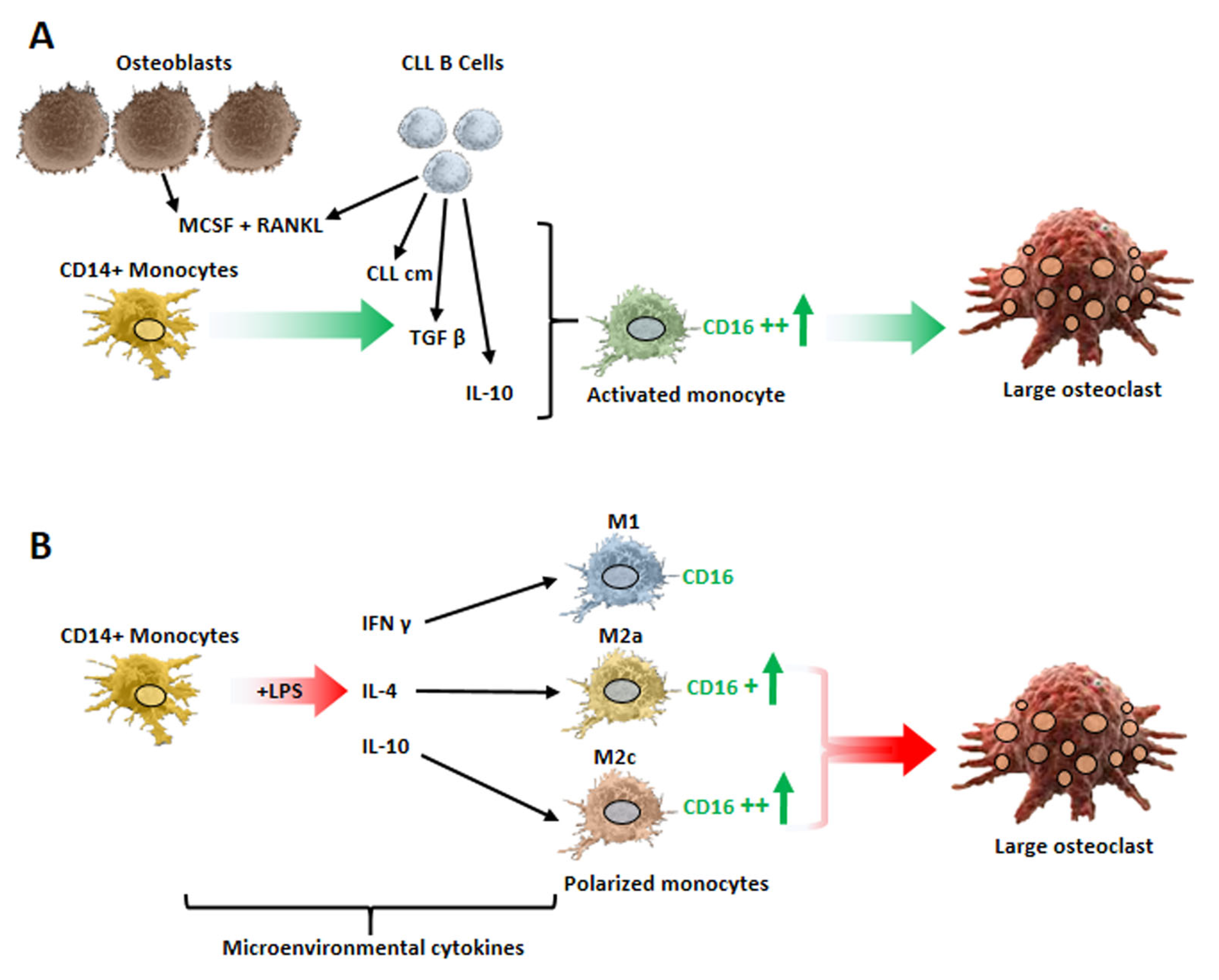
7. Monocyte Polarization by Leukemic B Cells and Bone Tissue Remodeling in CLL Patients
8. Disruption of the Endosteal Niche in CLL Patients: Cause and Effect of the CLL Clonal Expansion?
9. Impact of BCR Signaling Inhibitors on the CLL Endosteal Niche
10. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Primers 2017, 3, 16096. [Google Scholar] [CrossRef] [PubMed]
- Puente, X.S.; Jares, P.; Campo, E. Chronic lymphocytic leukemia and mantle cell lymphoma: Crossroads of genetic and microenvironment interactions. Blood 2018, 131, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- Muscarella, A.M.; Aguirre, S.; Hao, X.; Waldvogel, S.M.; Zhang, X.H. Exploiting bone niches: Progression of disseminated tumor cells to metastasis. J. Clin. Investig. 2021, 131, e143764. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Takegahara, N.; Kim, H.; Choi, Y. Updating osteoimmunology: Regulation of bone cells by innate and adaptive immunity. Nat. Rev. Rheumatol. 2018, 14, 146–156. [Google Scholar] [CrossRef]
- Marini, C.; Bruno, S.; Fiz, F.; Campi, C.; Piva, R.; Cutrona, G.; Matis, S.; Nieri, A.; Miglino, M.; Ibatici, A.; et al. Functional Activation of Osteoclast Commitment in Chronic Lymphocytic Leukaemia: A Possible Role for RANK/RANKL Pathway. Sci. Rep. 2017, 7, 14159. [Google Scholar] [CrossRef]
- Giannoni, P.; Marini, C.; Cutrona, G.; Matis, S.; Capra, M.C.; Puglisi, F.; Luzzi, P.; Pigozzi, S.; Gaggero, G.; Neri, A.; et al. Chronic lymphocytic leukemia cells impair osteoblastogenesis and promote osteoclastogenesis: Role of TNFalpha, IL-6 and IL-11 cytokines. Haematologica 2021, 106, 2598–2612. [Google Scholar] [CrossRef]
- Giannoni, P.; Marini, C.; Cutrona, G.; Todoerti, K.; Neri, A.; Ibatici, A.; Sambuceti, G.; Pigozzi, S.; Mora, M.; Ferrarini, M.; et al. A High Percentage of CD16+ Monocytes Correlates with the Extent of Bone Erosion in Chronic Lymphocytic Leukemia Patients: The Impact of Leukemic B Cells in Monocyte Differentiation and Osteoclast Maturation. Cancers 2022, 14, 5979. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Blair, H.C.; Larrouture, Q.C.; Li, Y.; Lin, H.; Beer-Stoltz, D.; Liu, L.; Tuan, R.S.; Robinson, L.J.; Schlesinger, P.H.; Nelson, D.J. Osteoblast Differentiation and Bone Matrix Formation In Vivo and In Vitro. Tissue Eng. Part B Rev. 2017, 23, 268–280. [Google Scholar] [CrossRef]
- Kim, J.S.; Choi, M.; Choi, J.Y.; Kim, J.Y.; Kim, J.Y.; Song, J.S.; Ivashkiv, L.B.; Lee, E.Y. Implication of the Association of Fibrinogen Citrullination and Osteoclastogenesis in Bone Destruction in Rheumatoid Arthritis. Cells 2020, 9, 2720. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, N.K.; Lee, S.Y. Current Understanding of RANK Signaling in Osteoclast Differentiation and Maturation. Mol. Cells 2017, 40, 706–713. [Google Scholar] [PubMed]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef]
- Li, J.; Sarosi, I.; Yan, X.Q.; Morony, S.; Capparelli, C.; Tan, H.L.; McCabe, S.; Elliott, R.; Scully, S.; Van, G.; et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 1566–1571. [Google Scholar] [CrossRef]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Xu, J.; Yu, L.; Liu, F.; Wan, L.; Deng, Z. The effect of cytokines on osteoblasts and osteoclasts in bone remodeling in osteoporosis: A review. Front. Immunol. 2023, 14, 1222129. [Google Scholar] [CrossRef]
- Zhou, P.; Zheng, T.; Zhao, B. Cytokine-mediated immunomodulation of osteoclastogenesis. Bone 2022, 164, 116540. [Google Scholar] [CrossRef]
- Brylka, L.J.; Schinke, T. Chemokines in Physiological and Pathological Bone Remodeling. Front. Immunol. 2019, 10, 2182. [Google Scholar] [CrossRef]
- Bacchiarri, F.; Gozzetti, A.; Mondanelli, N.; Lazzi, S.; Bocchia, M. A case of bone lesion in a patient with relapsed chronic lymphocytic leukemia and review of the literature. Clin. Case Rep. 2022, 10, e05379. [Google Scholar] [CrossRef]
- Fiz, F.; Marini, C.; Piva, R.; Miglino, M.; Massollo, M.; Bongioanni, F.; Morbelli, S.; Bottoni, G.; Campi, C.; Bacigalupo, A.; et al. Adult advanced chronic lymphocytic leukemia: Computational analysis of whole-body CT documents a bone structure alteration. Radiology 2014, 271, 805–813. [Google Scholar] [CrossRef]
- Chappard, D.; Rossi, J.F.; Bataillle, R.; Alexandre, C. Osteoclast cytomorphometry and scanning electron microscopy of bone eroded surfaces during leukemic disorders. Scanning Microsc. 1990, 4, 323–328. [Google Scholar]
- Rossi, J.F.; Chappard, D.; Marcelli, C.; Laplante, J.; Commes, T.; Baldet, P.; Janbon, C.; Jourdan, J.; Alexandre, C.; Bataille, R. Micro-osteoclast resorption as a characteristic feature of B-cell malignancies other than multiple myeloma. Br. J. Haematol. 1990, 76, 469–475. [Google Scholar] [CrossRef]
- Chappard, D.; Rossi, J.F.; Bataille, R.; Alexandre, C. Osteoclast cytomorphometry demonstrates an abnormal population in B cell malignancies but not in multiple myeloma. Calcif. Tissue Int. 1991, 48, 13–17. [Google Scholar] [CrossRef]
- Schmiedel, B.J.; Scheible, C.A.; Nuebling, T.; Kopp, H.G.; Wirths, S.; Azuma, M.; Schneider, P.; Jung, G.; Grosse-Hovest, L.; Salih, H.R. RANKL expression, function, and therapeutic targeting in multiple myeloma and chronic lymphocytic leukemia. Cancer Res. 2013, 73, 683–694. [Google Scholar] [CrossRef]
- Borge, M.; Delpino, M.V.; Podaza, E.; Stanganelli, C.; Palau-Nagore, V.; Roisman, A.; Slavutsky, I.; Palacios, M.F.; Ledesma, I.; Arra, A.; et al. Soluble RANKL production by leukemic cells in a case of chronic lymphocytic leukemia with bone destruction. Leuk. Lymphoma 2016, 57, 2468–2471. [Google Scholar] [CrossRef] [PubMed]
- Alankus, B.; Ecker, V.; Vahl, N.; Braun, M.; Weichert, W.; Macher-Goppinger, S.; Gehring, T.; Neumayer, T.; Zenz, T.; Buchner, M.; et al. Pathological RANK signaling in B cells drives autoimmunity and chronic lymphocytic leukemia. J. Exp. Med. 2021, 218, e20200517. [Google Scholar] [CrossRef] [PubMed]
- Long, C.L.; Humphrey, M.B. Osteoimmunology: The expanding role of immunoreceptors in osteoclasts and bone remodeling. Bonekey Rep. 2012, 1, 59. [Google Scholar] [CrossRef] [PubMed]
- Kotake, S.; Nanke, Y. Effect of TNFalpha on osteoblastogenesis from mesenchymal stem cells. Biochim. Biophys. Acta 2014, 1840, 1209–1213. [Google Scholar] [CrossRef]
- Sen, M.; Lauterbach, K.; El-Gabalawy, H.; Firestein, G.S.; Corr, M.; Carson, D.A. Expression and function of wingless and frizzled homologs in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2000, 97, 2791–2796. [Google Scholar] [CrossRef]
- Papadaki, H.A.; Kritikos, H.D.; Gemetzi, C.; Koutala, H.; Marsh, J.C.; Boumpas, D.T.; Eliopoulos, G.D. Bone marrow progenitor cell reserve and function and stromal cell function are defective in rheumatoid arthritis: Evidence for a tumor necrosis factor alpha-mediated effect. Blood 2002, 99, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, D.S.; Yun, H.; Kim, S.; Lee, N.; Kim, H.; Rho, J. Regulation of Osteoclast Differentiation by Cytokine Networks. Immune Netw. 2018, 18, e8. [Google Scholar] [CrossRef] [PubMed]
- Harmer, D.; Falank, C.; Reagan, M.R. Interleukin-6 Interweaves the Bone Marrow Microenvironment, Bone Loss, and Multiple Myeloma. Front. Endocrinol. 2018, 9, 788. [Google Scholar] [CrossRef] [PubMed]
- Sims, N.A. Influences of the IL-6 cytokine family on bone structure and function. Cytokine 2021, 146, 155655. [Google Scholar] [CrossRef] [PubMed]
- Foa, R.; Massaia, M.; Cardona, S.; Tos, A.G.; Bianchi, A.; Attisano, C.; Guarini, A.; di Celle, P.F.; Fierro, M.T. Production of tumor necrosis factor-alpha by B-cell chronic lymphocytic leukemia cells: A possible regulatory role of TNF in the progression of the disease. Blood 1990, 76, 393–400. [Google Scholar] [CrossRef]
- Ferrajoli, A.; Keating, M.J.; Manshouri, T.; Giles, F.J.; Dey, A.; Estrov, Z.; Koller, C.A.; Kurzrock, R.; Thomas, D.A.; Faderl, S.; et al. The clinical significance of tumor necrosis factor-alpha plasma level in patients having chronic lymphocytic leukemia. Blood 2002, 100, 1215–1219. [Google Scholar] [CrossRef]
- Burger, J.A. Chemokines and chemokine receptors in chronic lymphocytic leukemia (CLL): From understanding the basics towards therapeutic targeting. Semin. Cancer Biol. 2010, 20, 424–430. [Google Scholar] [CrossRef]
- Davids, M.S.; Burger, J.A. Cell Trafficking in Chronic Lymphocytic Leukemia. Open J. Hematol. 2012, 3, 3. [Google Scholar] [CrossRef]
- Schrottner, P.; Leick, M.; Burger, M. The role of chemokines in B cell chronic lymphocytic leukaemia: Pathophysiological aspects and clinical impact. Ann. Hematol. 2010, 89, 437–446. [Google Scholar] [CrossRef]
- van Attekum, M.H.; Eldering, E.; Kater, A.P. Chronic lymphocytic leukemia cells are active participants in microenvironmental cross-talk. Haematologica 2017, 102, 1469–1476. [Google Scholar] [CrossRef]
- Ghia, P.; Strola, G.; Granziero, L.; Geuna, M.; Guida, G.; Sallusto, F.; Ruffing, N.; Montagna, L.; Piccoli, P.; Chilosi, M.; et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur. J. Immunol. 2002, 32, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Scielzo, C.; Apollonio, B.; Scarfo, L.; Janus, A.; Muzio, M.; Ten Hacken, E.; Ghia, P.; Caligaris-Cappio, F. The functional in vitro response to CD40 ligation reflects a different clinical outcome in patients with chronic lymphocytic leukemia. Leukemia 2011, 25, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- De Cecco, L.; Capaia, M.; Zupo, S.; Cutrona, G.; Matis, S.; Brizzolara, A.; Orengo, A.M.; Croce, M.; Marchesi, E.; Ferrarini, M.; et al. Interleukin 21 Controls mRNA and MicroRNA Expression in CD40-Activated Chronic Lymphocytic Leukemia Cells. PLoS ONE 2015, 10, e0134706. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, A. Chemokines and Bone. Handb. Exp. Pharmacol. 2020, 262, 231–258. [Google Scholar] [PubMed]
- Liu, Z.; Liang, W.; Kang, D.; Chen, Q.; Ouyang, Z.; Yan, H.; Huang, B.; Jin, D.; Chen, Y.; Li, Q. Increased Osteoblastic Cxcl9 Contributes to the Uncoupled Bone Formation and Resorption in Postmenopausal Osteoporosis. Clin. Interv. Aging. 2020, 15, 1201–1212. [Google Scholar] [CrossRef]
- Phan, Q.T.; Chua, K.Y.; Jin, A.; Winkler, C.; Koh, W.P. CXCL9 Predicts the Risk of Osteoporotic Hip Fracture in a Prospective Cohort of Chinese Men-A Matched Case-Control Study. J. Bone Miner. Res. 2022, 37, 1843–1849. [Google Scholar] [CrossRef]
- Yan, X.J.; Dozmorov, I.; Li, W.; Yancopoulos, S.; Sison, C.; Centola, M.; Jain, P.; Allen, S.L.; Kolitz, J.E.; Rai, K.R.; et al. Identification of outcome-correlated cytokine clusters in chronic lymphocytic leukemia. Blood 2011, 118, 5201–5210. [Google Scholar] [CrossRef]
- Kashyap, M.K.; Kumar, D.; Jones, H.; Amaya-Chanaga, C.I.; Choi, M.Y.; Melo-Cardenas, J.; Ale-Ali, A.; Kuhne, M.R.; Sabbatini, P.; Cohen, L.J.; et al. Ulocuplumab (BMS-936564 / MDX1338): A fully human anti-CXCR4 antibody induces cell death in chronic lymphocytic leukemia mediated through a reactive oxygen species-dependent pathway. Oncotarget 2016, 7, 2809–2822. [Google Scholar] [CrossRef]
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Dell’Aquila, M.; Kipps, T.J. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell-derived factor-1. Blood 2000, 96, 2655–2663. [Google Scholar] [CrossRef]
- Tsukada, N.; Burger, J.A.; Zvaifler, N.J.; Kipps, T.J. Distinctive features of “nurselike” cells that differentiate in the context of chronic lymphocytic leukemia. Blood 2002, 99, 1030–1037. [Google Scholar] [CrossRef]
- Fiorcari, S.; Maffei, R.; Atene, C.G.; Potenza, L.; Luppi, M.; Marasca, R. Nurse-Like Cells and Chronic Lymphocytic Leukemia B Cells: A Mutualistic Crosstalk inside Tissue Microenvironments. Cells 2021, 10, 217. [Google Scholar] [CrossRef] [PubMed]
- Mesaros, O.; Jimbu, L.; Neaga, A.; Popescu, C.; Berceanu, I.; Tomuleasa, C.; Fetica, B.; Zdrenghea, M. Macrophage Polarization in Chronic Lymphocytic Leukemia: Nurse-Like Cells Are the Caretakers of Leukemic Cells. Biomedicines 2020, 8, 516. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, P.; Pietra, G.; Travaini, G.; Quarto, R.; Shyti, G.; Benelli, R.; Ottaggio, L.; Mingari, M.C.; Zupo, S.; Cutrona, G.; et al. Chronic Lymphocytic Leukemia Nurse-like cells express the hepatocyte growth factor receptor (c-MET) and indoleamine 2,3-dioxygenase and display features of immunosuppressive type 2 skewed macrophages. Haematologica 2014, 99, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Audrito, V.; Serra, S.; Brusa, D.; Mazzola, F.; Arruga, F.; Vaisitti, T.; Coscia, M.; Maffei, R.; Rossi, D.; Wang, T.; et al. Extracellular nicotinamide phosphoribosyltransferase (NAMPT) promotes M2 macrophage polarization in chronic lymphocytic leukemia. Blood 2015, 125, 111–123. [Google Scholar] [CrossRef]
- Bhattacharya, N.; Diener, S.; Idler, I.S.; Rauen, J.; Habe, S.; Busch, H.; Habermann, A.; Zenz, T.; Dohner, H.; Stilgenbauer, S.; et al. Nurse-like cells show deregulated expression of genes involved in immunocompetence. Br. J. Haematol. 2011, 154, 349–356. [Google Scholar] [CrossRef]
- Filip, A.A.; Cisel, B.; Koczkodaj, D.; Wasik-Szczepanek, E.; Piersiak, T.; Dmoszynska, A. Circulating microenvironment of CLL: Are nurse-like cells related to tumor-associated macrophages? Blood Cells Mol. Dis. 2013, 50, 263–270. [Google Scholar] [CrossRef]
- Lob, S.; Konigsrainer, A.; Rammensee, H.G.; Opelz, G.; Terness, P. Inhibitors of indoleamine-2,3-dioxygenase for cancer therapy: Can we see the wood for the trees? Nat. Rev. Cancer 2009, 9, 445–452. [Google Scholar] [CrossRef]
- Serra, S.; Vaisitti, T.; Audrito, V.; Bologna, C.; Buonincontri, R.; Chen, S.S.; Arruga, F.; Brusa, D.; Coscia, M.; Jaksic, O.; et al. Adenosine signaling mediates hypoxic responses in the chronic lymphocytic leukemia microenvironment. Blood Adv. 2016, 1, 47–61. [Google Scholar] [CrossRef]
- Jitschin, R.; Braun, M.; Buttner, M.; Dettmer-Wilde, K.; Bricks, J.; Berger, J.; Eckart, M.J.; Krause, S.W.; Oefner, P.J.; Le Blanc, K.; et al. CLL-cells induce IDOhi CD14+HLA-DRlo myeloid-derived suppressor cells that inhibit T-cell responses and promote TRegs. Blood 2014, 124, 750–760. [Google Scholar] [CrossRef]
- Zarobkiewicz, M.; Kowalska, W.; Chocholska, S.; Tomczak, W.; Szymanska, A.; Morawska, I.; Wojciechowska, A.; Bojarska-Junak, A. High M-MDSC Percentage as a Negative Prognostic Factor in Chronic Lymphocytic Leukaemia. Cancers 2020, 12, 2614. [Google Scholar] [CrossRef]
- Lindstrom, V.; Aittoniemi, J.; Jylhava, J.; Eklund, C.; Hurme, M.; Paavonen, T.; Oja, S.S.; Itala-Remes, M.; Sinisalo, M. Indoleamine 2,3-dioxygenase activity and expression in patients with chronic lymphocytic leukemia. Clin. Lymphoma Myeloma Leuk. 2012, 12, 363–365. [Google Scholar] [CrossRef]
- Maffei, R.; Bulgarelli, J.; Fiorcari, S.; Bertoncelli, L.; Martinelli, S.; Guarnotta, C.; Castelli, I.; Deaglio, S.; Debbia, G.; De Biasi, S.; et al. The monocytic population in chronic lymphocytic leukemia shows altered composition and deregulation of genes involved in phagocytosis and inflammation. Haematologica 2013, 98, 1115–1123. [Google Scholar] [CrossRef]
- Kowalska, W. Expression of CD163 and HLA-DR molecules on the monocytes in chronic lymphocytic leukemia patients. Folia Histochem. Cytobiol. 2020, 58, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Bolzoni, M.; Ronchetti, D.; Storti, P.; Donofrio, G.; Marchica, V.; Costa, F.; Agnelli, L.; Toscani, D.; Vescovini, R.; Todoerti, K.; et al. IL21R expressing CD14(+)CD16(+) monocytes expand in multiple myeloma patients leading to increased osteoclasts. Haematologica 2017, 102, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Koga, T.; Okamoto, K.; Sakaguchi, S.; Arai, K.; Yasuda, H.; Takai, T.; Kodama, T.; Morio, T.; Geha, R.S.; et al. Tyrosine kinases Btk and Tec regulate osteoclast differentiation by linking RANK and ITAM signals. Cell 2008, 132, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Park, O.J.; Kim, J.; Kwon, Y.; Yun, C.H.; Han, S.H. Modulation of macrophage subtypes by IRF5 determines osteoclastogenic potential. J. Cell Physiol. 2019, 234, 23033–23042. [Google Scholar] [CrossRef]
- Chiu, Y.G.; Shao, T.; Feng, C.; Mensah, K.A.; Thullen, M.; Schwarz, E.M.; Ritchlin, C.T. CD16 (FcRgammaIII) as a potential marker of osteoclast precursors in psoriatic arthritis. Arthritis Res. Ther. 2010, 12, R14. [Google Scholar] [CrossRef] [PubMed]
- Danilin, S.; Merkel, A.R.; Johnson, J.R.; Johnson, R.W.; Edwards, J.R.; Sterling, J.A. Myeloid-derived suppressor cells expand during breast cancer progression and promote tumor-induced bone destruction. Oncoimmunology 2012, 1, 1484–1494. [Google Scholar] [CrossRef]
- Kirkwood, K.L.; Zhang, L.; Thiyagarajan, R.; Seldeen, K.L.; Troen, B.R. Myeloid-Derived Suppressor Cells at the Intersection of Inflammaging and Bone Fragility. Immunol. Investig. 2018, 47, 844–854. [Google Scholar] [CrossRef]
- Sawant, A.; Deshane, J.; Jules, J.; Lee, C.M.; Harris, B.A.; Feng, X.; Ponnazhagan, S. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res. 2013, 73, 672–682. [Google Scholar] [CrossRef]
- Sawant, A.; Ponnazhagan, S. Myeloid-derived suppressor cells as osteoclast progenitors: A novel target for controlling osteolytic bone metastasis. Cancer Res. 2013, 73, 4606–4610. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Liang, M.; Yang, T.; Ji, J.; Jose Kumar Sreena, G.S.; Hou, X.; Cao, M.; Feng, Z. The Immunoregulatory Role of Myeloid-Derived Suppressor Cells in the Pathogenesis of Rheumatoid Arthritis. Front. Immunol. 2020, 11, 568362. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Zhang, J.; Lwin, S.T.; Edwards, J.R.; Edwards, C.M.; Mundy, G.R.; Yang, X. Osteoclasts in multiple myeloma are derived from Gr-1+CD11b+myeloid-derived suppressor cells. PLoS ONE 2012, 7, e48871. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro-Spinetti, E.; Taichman, R.S.; Balduino, A. The bone marrow endosteal niche: How far from the surface? J. Cell Biochem. 2015, 116, 6–11. [Google Scholar] [CrossRef]
- Audrito, V.; Vaisitti, T.; Serra, S.; Bologna, C.; Brusa, D.; Malavasi, F.; Deaglio, S. Targeting the microenvironment in chronic lymphocytic leukemia offers novel therapeutic options. Cancer Lett. 2013, 328, 27–35. [Google Scholar] [CrossRef]
- Li, J.; Lu, L.; Liu, Y.; Yu, X. Bone marrow adiposity during pathologic bone loss: Molecular mechanisms underlying the cellular events. J. Mol. Med. 2022, 100, 167–183. [Google Scholar] [CrossRef]
- Muruganandan, S.; Sinal, C.J. The impact of bone marrow adipocytes on osteoblast and osteoclast differentiation. IUBMB Life 2014, 66, 147–155. [Google Scholar] [CrossRef]
- van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef]
- Rosen, C.J.; Bouxsein, M.L. Mechanisms of disease: Is osteoporosis the obesity of bone? Nat. Clin. Pract. Rheumatol. 2006, 2, 35–43. [Google Scholar] [CrossRef]
- Liu, H.; He, J.; Koh, S.P.; Zhong, Y.; Liu, Z.; Wang, Z.; Zhang, Y.; Li, Z.; Tam, B.T.; Lin, P.; et al. Reprogrammed marrow adipocytes contribute to myeloma-induced bone disease. Sci. Transl. Med. 2019, 11, eaau9087. [Google Scholar] [CrossRef]
- Chen, S.S.; Chiorazzi, N. Functional consequences of inhibition of Bruton’s tyrosine kinase by ibrutinib in chronic lymphocytic leukemia. Hematol. Oncol. 2023, 41 (Suppl. S1), 119–128. [Google Scholar] [CrossRef] [PubMed]
- McCay, J.; Gribben, J.G. The role of BTK inhibitors on the tumor microenvironment in CLL. Leuk. Lymphoma 2022, 63, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Gokhale, S.; Jung, J.; Spirollari, E.; Tsai, J.; Arceo, J.; Wu, B.W.; Victor, E.; Xie, P. Multifaceted Immunomodulatory Effects of the BTK Inhibitors Ibrutinib and Acalabrutinib on Different Immune Cell Subsets—Beyond B Lymphocytes. Front. Cell Dev. Biol. 2021, 9, 727531. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, Y.; Yin, Q. Effects of ibrutinib on T-cell immunity in patients with chronic lymphocytic leukemia. Front. Immunol. 2022, 13, 962552. [Google Scholar] [CrossRef] [PubMed]
- Mhibik, M.; Wiestner, A.; Sun, C. Harnessing the Effects of BTKi on T Cells for Effective Immunotherapy against CLL. Int. J. Mol. Sci. 2019, 21, 68. [Google Scholar] [CrossRef]
- Kondo, K.; Shaim, H.; Thompson, P.A.; Burger, J.A.; Keating, M.; Estrov, Z.; Harris, D.; Kim, E.; Ferrajoli, A.; Daher, M.; et al. Ibrutinib modulates the immunosuppressive CLL microenvironment through STAT3-mediated suppression of regulatory B-cell function and inhibition of the PD-1/PD-L1 pathway. Leukemia 2018, 32, 960–970. [Google Scholar] [CrossRef]
- Shinohara, M.; Chang, B.Y.; Buggy, J.J.; Nagai, Y.; Kodama, T.; Asahara, H.; Takayanagi, H. The orally available Btk inhibitor ibrutinib (PCI-32765) protects against osteoclast-mediated bone loss. Bone 2014, 60, 8–15. [Google Scholar] [CrossRef]
- Shumilov, E.; Wulf, G.; Strobel, P.; Hasenkamp, J.; Hellige, N.; Bleckmann, A.; Haase, D.; Braulke, F.; Jung, W.; Schanz, J.; et al. Osteolytic lesions occur rarely in patients with B-CLL and may respond well to ibrutinib. Leuk. Lymphoma 2016, 57, 2476–2480. [Google Scholar] [CrossRef]
- Niemann, C.U.; Herman, S.E.; Maric, I.; Gomez-Rodriguez, J.; Biancotto, A.; Chang, B.Y.; Martyr, S.; Stetler-Stevenson, M.; Yuan, C.M.; Calvo, K.R.; et al. Disruption of in vivo Chronic Lymphocytic Leukemia Tumor-Microenvironment Interactions by Ibrutinib--Findings from an Investigator-Initiated Phase II Study. Clin. Cancer Res. 2016, 22, 1572–1582. [Google Scholar] [CrossRef]
- Fiorcari, S.; Maffei, R.; Audrito, V.; Martinelli, S.; Ten Hacken, E.; Zucchini, P.; Grisendi, G.; Potenza, L.; Luppi, M.; Burger, J.A.; et al. Ibrutinib modifies the function of monocyte/macrophage population in chronic lymphocytic leukemia. Oncotarget 2016, 7, 65968–65981. [Google Scholar] [CrossRef]
- Boissard, F.; Fournie, J.J.; Quillet-Mary, A.; Ysebaert, L.; Poupot, M. Nurse-like cells mediate ibrutinib resistance in chronic lymphocytic leukemia patients. Blood Cancer J. 2015, 5, e355. [Google Scholar] [CrossRef] [PubMed]
- Merchand-Reyes, G.; Santhanam, R.; Robledo-Avila, F.H.; Weigel, C.; Ruiz-Rosado, J.D.; Mo, X.; Partida-Sanchez, S.; Woyach, J.A.; Oakes, C.C.; Tridandapani, S.; et al. Disruption of Nurse-like Cell Differentiation as a Therapeutic Strategy for Chronic Lymphocytic Leukemia. J. Immunol. 2022, 209, 1212–1223. [Google Scholar] [CrossRef] [PubMed]
- Solman, I.G.; Blum, L.K.; Burger, J.A.; Kipps, T.J.; Dean, J.P.; James, D.F.; Mongan, A. Impact of long-term ibrutinib treatment on circulating immune cells in previously untreated chronic lymphocytic leukemia. Leuk. Res. 2021, 102, 106520. [Google Scholar] [CrossRef] [PubMed]
- Ariza, Y.; Murata, M.; Ueda, Y.; Yoshizawa, T. Bruton’s tyrosine kinase (Btk) inhibitor tirabrutinib suppresses osteoclastic bone resorption. Bone Rep. 2019, 10, 100201. [Google Scholar] [CrossRef]
- Pokhrel, N.K.; Kim, Y.G.; Kim, H.J.; Kim, H.J.; Lee, J.H.; Choi, S.Y.; Kwon, T.G.; Lee, H.J.; Kim, J.Y.; Lee, Y. A novel Bruton’s tyrosine kinase inhibitor, acalabrutinib, suppresses osteoclast differentiation and Porphyromonas gingivalis lipopolysaccharide-induced alveolar bone resorption. J. Periodontol. 2019, 90, 546–554. [Google Scholar] [CrossRef]
- Yeon, J.T.; Kim, K.J.; Son, Y.J.; Park, S.J.; Kim, S.H. Idelalisib inhibits osteoclast differentiation and pre-osteoclast migration by blocking the PI3Kdelta-Akt-c-Fos/NFATc1 signaling cascade. Arch. Pharm. Res. 2019, 42, 712–721. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannoni, P.; Marini, C.; Cutrona, G.; Sambuceti, G.M.; Fais, F.; de Totero, D. Unraveling the Bone Tissue Microenvironment in Chronic Lymphocytic Leukemia. Cancers 2023, 15, 5058. https://doi.org/10.3390/cancers15205058
Giannoni P, Marini C, Cutrona G, Sambuceti GM, Fais F, de Totero D. Unraveling the Bone Tissue Microenvironment in Chronic Lymphocytic Leukemia. Cancers. 2023; 15(20):5058. https://doi.org/10.3390/cancers15205058
Chicago/Turabian StyleGiannoni, Paolo, Cecilia Marini, Giovanna Cutrona, Gian Mario Sambuceti, Franco Fais, and Daniela de Totero. 2023. "Unraveling the Bone Tissue Microenvironment in Chronic Lymphocytic Leukemia" Cancers 15, no. 20: 5058. https://doi.org/10.3390/cancers15205058
APA StyleGiannoni, P., Marini, C., Cutrona, G., Sambuceti, G. M., Fais, F., & de Totero, D. (2023). Unraveling the Bone Tissue Microenvironment in Chronic Lymphocytic Leukemia. Cancers, 15(20), 5058. https://doi.org/10.3390/cancers15205058