Simple Summary
The MAP kinases pathway has shown a key role in the pathogenesis of colorectal cancer. The development of targeted therapy against BRAF-mutated tumors is changing the management of advanced disease. A proper understanding of the mechanisms of acquired resistance is essential to optimize the results of systemic treatment. We aim to review the current knowledge and potential fields of research regarding the use of BRAF inhibitors in metastatic colorectal tumors.
Abstract
Metastatic colorectal cancer (mCRC) with mutated BRAF exhibits distinct biological and molecular features that set it apart from other subtypes of CRC. Current standard treatment for these tumors involves a combination of chemotherapy (CT) and VEGF inhibitors. Recently, targeted therapy against BRAF and immunotherapy (IT) for cases with microsatellite instability (MSI) have been integrated into clinical practice. While targeted therapy has shown promising results, resistance to treatment eventually develops in a significant portion of responsive patients. This article aims to review the available literature on mechanisms of resistance to BRAF inhibitors (BRAFis) and potential therapeutic strategies to overcome them.
1. Introduction
CRC is currently one of the most common neoplasms, being the third most frequent tumor in men and the second in women [1]. Its incidence is rising in Western countries, with up to 70% of CRC being sporadic and associated with environmental factors, like tobacco, alcohol, diet, sedentary lifestyle, or obesity [2]. Globally, CRC accounts for 11% of all new cancer cases worldwide [3], with up to a quarter presenting distant metastases at diagnosis. Among the remaining three-quarters eligible for surgery, nearly half will develop metastases during the course of the disease [2]. Hence, this poses a significant healthcare challenge.
The BRAF proto-oncogene is located on chromosome 7 and consists of 18 exons [2]. Mutation in this gene is present in 7% of all tumors, with the majority being melanomas, where BRAF mutation is observed in up to 50% [4]. In mCRC, the BRAF mutation frequency hovers around 12% [5]. BRAF mutation in mCRC carries independent prognostic value and significant associations with clinical and biological disease characteristics [6]. This has led to a growing interest in genetic testing for BRAF mutations and the development of targeted therapies against them.
2. Molecular Biology
2.1. Carcinogenesis
The carcinogenesis of colorectal neoplasias starts with the accumulation of various genetic and epigenetic mutations that transform healthy epithelial tissue into benign neoplasms (adenomatous polyps), which later progress to dysplastic polyps and eventually invasive carcinomas. BRAF mutation is believed to be involved in the transformation of healthy epithelium into serrated adenomas (sessile or traditional), potentially representing an early event in CRC progression [7]. There are two main pathways through which adenomatous polyps transform into CRC: microsatellite instability (MSI) and chromosomal instability (CIN) [2]. Most CRCs develop through the latter pathway. One of the most frequently implicated genes is APC, which forms the beta-catenin destruction complex that activates the Wnt signaling pathway. Alteration of the APC gene (most commonly a 5q deletion) inhibits beta-catenin destruction, resulting in hyperactivation of the Wnt signaling pathway, leading to increased cellular proliferation and invasion. In terms of MSI tumors, they result from both the mismatch repair pathway (MMR) and the hypermethylated CpG island phenotype (CIMP) [2]. In the MMR pathway, mutations occur in genes encoding DNA repair proteins (MLH-1, MSH2, MSH-6, PMS-2), leading to progressive accumulation of unrepaired DNA abnormalities. These DNA molecules code for truncated proteins that serve as neoantigens, making these tumors more immunogenic than others. On the other hand, the CIMP pathway involves hypermethylation of the repair protein genes, specifically, CpG islands in the MMR enzyme promoter region. This extensive methylation inactivates repair proteins, resulting in epigenetic instability. Sporadic microsatellite instability is associated with a higher prevalence of BRAF mutations compared to tumors with microsatellite stability (MSS) or non-polyposis hereditary CRC [8]. In sporadic CRC, BRAF mutation is observed in 60% of tumors with MSI, whereas it is seen in only 5–10% of MSS tumors [1]. Approximately 20% of patients with mutated BRAF V600E concurrently exhibit microsatellite instability [9].
2.2. BRAF Mutation
BRAF is a serine/threonine kinase belonging to the RAF family, crucial in the Mitogen-Activated Protein Kinase (MAPK) signaling pathway, also known as the RAS/RAF/MEK/ERK cascade. This pathway controls cell growth and proliferation in response to various growth factors [5].
Different subtypes of RAF protein exist: A, B, and C, with subtype B (BRAF) being the most significant player in MEK and exhibiting more mutations. It comprises three domains: CR1 and CR2 located in the N-terminal region, where CR1 is the binding domain and CR2 is the regulatory domain. The third domain, CR3, is situated in the C-terminal region and is the catalytic domain [10]. Apart from its role in the MAPK pathway, BRAF is involved in other cellular processes, like migration (via RHO small GTPases), apoptosis (via BCL-2 regulation), and survival (via the HIPPO pathway) [7]. BRAF mutations are classified into three classes, class I being the most frequent mutation type (representing 95%) [2], occurring in codon 600, where valine is changed to glutamine (BRAF V600E). Although this mutation is present in other neoplastic diseases, CRC is unique in its description of glutamic acid substitution at codon 600, while other tumors more commonly feature lysine or methionine substitutions [11]. Class II BRAF mutations involve mutations in codons 601/597 and exhibit a prognosis similar to class I mutations. Finally, class III mutations (like BRAF D594G or G596N) act by amplifying RAS or CRAF. In class I and II mutations, the protein escapes the negative feedback that would normally inhibit its signaling under physiological conditions, leading to permanent activation. However, class III mutations require a second hit to be oncogenic, such as KRAS mutation, NRAS mutation, or EGFR receptor activation [12]. Furthermore, class III mutations impair the kinase (kinase-impairing mutation), while V600E mutations activate the kinase (kinase-activating mutation), possibly explaining the better prognosis of the former: Mutations like D594G or G596N exhibit an overall survival (OS) of 60.7 months, significantly longer than the 11.4 months seen in BRAF V600E mCRC or the 43 months in BRAF wild-type subtypes [13]. While class II mutations coexist with other pathway mutations in 10–20% of cases, class I BRAF mutation is almost mutually exclusive with other MAPK pathway mutations [14], with coexistence of RAF and RAS mutations extremely rare. In a report involving 2530 patients from three randomized trials (COIN, PICCOLO, and FOCUS), such coexistence was observed in only eight cases (0.3%) [15]. Furthermore, there is a subclassification of the different BRAF mutations, as mentioned earlier, which also implies prognostic differences:
- -
- BRAF V600E: In general terms, it confers a worse prognosis, with an overall survival between 9 and 19 months. In the CALGB 80405 first-line trial, patients with the BRAF V600E mutation were administered FOLFOX or FOLFIRI plus Bevacizumab or Cetuximab compared to those with BRAF wild type. An average overall survival of 13.5 months was observed compared to 30.6 months in the BRAF wild-type patients (p = 0.001) [16]. They exhibit a poorer response to chemotherapy, as well as a higher incidence of peritoneal metastases, which results in symptomatic issues significantly affecting the patient’s quality of life (abdominal pain, ascites, intestinal or urinary obstruction, etc.) [17]. Regarding pathological anatomy, they often present mucinous histology and are poorly differentiated. They are more commonly associated with a specific phenotype: women, advanced age, and tumors preferentially located in the right colon. As mentioned earlier, at the molecular level, it is frequent to observe KRAS wild-type tumors, and there is a higher coexistence with MSI. In a study involving 3063 patients, 35% of dMMR CCRm had a concurrent BRAF V600E mutation, while only 7% of pMMR tumors had this mutation (p < 0.001) [9].
- -
- BRAF non-V600E: These are more frequently observed in men, at younger ages than BRAF V600E mutations, and they are more commonly located in the left colon. They usually coexist with mutations in KRAS, and there is a lower association with MSI.
Another subclassification of tumors with BRAF mutations is based on genetic expression:
- -
- BM1: This is the most frequent (observed in up to 70%). It is defined by the activation of the KRAS/AKT pathway, dysregulation of mTOR/4EBP, and increased immune infiltration [18]. Generally, it confers a worse prognosis, although it exhibits a greater response to BRAF, MEK, and EGFR inhibitors compared to BMS-2 [19].
- -
- BM2: This represents the remaining 30%, and it is characterized by the dysregulation of cell cycle checkpoints [18].
A classification into four molecular subgroups (CMS 1–4) has also been developed, which has a significant association with the clinical and biological characteristics of the tumor [15]. It is worth noting that up to 60–70% of CRC tumors with mutated BRAF belong to the CMS1 subtype, also known as immune MSI, which is characterized by a high mutation rate, but a low prevalence of somatic copy number alterations (SCNAs). These tumors encompass those with MSI, which could explain the success of the combination of BRAF inhibitors and immunotherapy [20,21].
3. Mechanism of Action of BRAF Inhibitors (BRAFis)
There is a wide variety of BRAFis, all of which have a similar mechanism of action that ultimately inhibits downstream signaling of BRAF within the MAPK pathway. Some of the most commonly used ones are vemurafenib, dabrafenib, and encorafenib [22]. They selectively and competitively inhibit the mutated kinase, targeting the ATP-binding domain of the monomeric form of BRAF [23]. Most of them are also active against other forms of RAF, such as CRAF [1]. They are administered orally and are generally well tolerated. All of them can be safely administered in patients with a glomerular filtration rate (GFR) greater than 30 mL/min, and there is currently no safety data available for lower GFRs. Encorafenib has demonstrated a longer pharmacodynamic activity than other BRAF inhibitors. In a study, it was observed that despite the fact that the drug concentrations required to inhibit BRAF V600E were similar among the three drugs, the half-life of encorafenib was significantly longer (30 h) than that of dabrafenib (2 h) and vemurafenib (0.5 h) [24]. The most common adverse effects associated with its administration are palmoplantar erythroderma [24], reported in up to 67% of CCRm patients with BRAF V600E mutation treated with encorafenib monotherapy. The most common dose-limiting toxicity was peripheral neuropathy, manifesting as neuralgia (4.1%) [1]. Vemurafenib and dabrafenib have a different toxicity profile compared to encorafenib, as they are more frequently associated with digestive alterations (vomiting or diarrhea), rather than neurological events.
4. Current Treatment Landscape in BRAF-Mutated mCRC
4.1. BRAF-Mutated mCRC without MSI: First-Line Systemic Treatment
Current data on BRAF-mutated colon tumors have shown worse outcomes compared to BRAF wild-type tumors. In the FOCUS study with 711 patients, the median overall survival (OS) after standard treatment was about 12 months, compared to nearly 30 months in patients with BRAF wild-type tumors [25]. Despite not observing statistically significant differences in terms of PFS, only 33% of BRAF-mutated patients received second-line treatment, compared to 50% in BRAF wild-type cases [17]. In an attempt to overcome the poor response to standard chemotherapy, treatment with EGFR inhibitors (EGFRis) was explored as an alternative. However, a meta-analysis concluded that EGFR blockade does not enhance the efficacy of standard chemotherapy in BRAF-mutated mCRC [26,27]. Despite promising results initially obtained in the TRIBE study with FOLFOXIRI + Bevacizumab versus FOLFIRI + Bevacizumab in the BRAF V600E mutated subgroup (median OS of 19 and 10.7 months, respectively; median PFS of 7.5 and 5.5 months, respectively), subsequent studies showed that the triplet chemotherapy has not shown to be outright superior to doublet regimens, while increasing toxicity [28]. However, the FIRE 4.5 study, which compares FOLFOXIRI + Cetuximab and FOLFOXIRI + Bevacizumab as first-line treatment in CRC with BRAF mutated, recommends the use of the triplet + Bevacizumab as the preferred option for patients with ECOG 0-1 [29]. In the ESMO guidelines, this combination is also referenced as the first choice for fit patients [30]. The recent CAIRO [31] study shows positive results in favor of FOLFOXIRI + Bevacizumab vs. FOLFOX/FOLFIRI + Bevacizumab in right-sided CRC patients with initially unresectable hepatic metastases and/or RAS/BRAF V600E mutation, with a PFS of 10.6 vs. 9 months, ORR of 52.1% vs. 32%, and R0/1 of 51.4% vs. 37.4%, respectively. However, these results were achieved at the expense of increased toxicity, with a rate of G3–G4 events of 75% in patients who received the triplet compared to 58.5% in those who did not.
Therefore, the current standard of care for first-line treatment in non-MSI mCRC with BRAF mutations includes standard chemotherapy containing fluoropyrimidines (FOLFOX vs. FOLFIRI) combined with VEGF inhibitors (Bevacizumab) [32], knowing that FOLFOXIRI + Bevacizumab can be administered to selected patients.
4.2. BRAF-Mutated mCRC with MSI
As mentioned earlier, MSI tumors are more immunogenic than other tumor subtypes, as the truncated proteins resulting from errors in unrepaired DNA strands function as neoantigens that activate the immune system. The KEYNOTE-177 study demonstrated that pembrolizumab (anti-PD-1) in monotherapy was superior to standard chemotherapy in the first-line setting. Patients receiving immunotherapy had a progression-free survival (PFS) of 16.5 months compared to 8.2 months in the chemotherapy arm (HR: 0.60, p = 0.0002). The PFS benefit was also observed in subgroups carrying BRAF mutations (HR 0.48; 95% CI 0.27–0.86), while tumors with KRAS or NRAS mutations did not derive this benefit (HR 1.19, 95% CI 0.68–2.07) [33]. Therefore, having a BRAF mutation does not necessarily lead to a worse response to immunotherapy in MSI patients. Immunotherapy’s benefit in MSI tumors has been confirmed in other studies [34,35], making it the current first-line treatment for MSI-positive mCRC. Various studies have been designed to explore the combination of immunotherapy and targeted therapy as a therapeutic option for these tumors. In a phase II trial, dabrafenib, trametinib, and spartalizumab (anti-PD-1) were administered to a total of 21 patients with BRAF V600E mutations who had not received prior immunotherapy or BRAFi treatment. An objective response rate (ORR) of 35% and a disease control rate (DCR) of 75% were achieved [36].
4.3. BRAF-Mutated mCRC without MSI: Second-Line Systemic Treatment
Due to limited response to first-line treatment in BRAF-mutated mCRC, studies were initiated in the second-line setting to assess the efficacy of targeted treatment against this mutation: BRAF inhibitors (BRAFis). Unlike melanoma, BRAFi monotherapy showed limited effectiveness: a phase I study with vemurafenib (PLX4032) achieved a partial response (PR) of 5% and a PFS of 3.7 months [37]. Adding therapies like cetuximab and irinotecan to vemurafenib (known as the VIC regimen) in a phase IB study showed slight improvement, with a median PFS of 7.7 months [38], still far from the desired outcomes. The new standard of care for BRAF-mutated mCRC comes from the BEACON study, a phase III trial that randomized 665 pre-treated mCRC patients into three arms: triplet therapy of BRAFi, EGFRi, and MEKi (encorafenib + cetuximab + binimetinib) or the doublet (encorafenib + cetuximab) versus the control arm (investigator’s choice of irinotecan or FOLFIRI + cetuximab) [39]. The study showed clear clinical benefit in both targeted therapy arms compared to the control arm, in patients treated with cetuximab + encorafenib (PFS 4.3 vs. 1.5 months, HR 0.44; OS 9.3 vs. 5.9 months, HR 0.61) and in those treated with cetuximab + encorafenib + binimetinib (PFS 4.5 vs. 1.5 months, HR 0.42; OS 9.3 vs. 5.9 months, HR 0.6). Upon analyzing the results, it was concluded that the doublet had a similar overall efficacy to the triplet. Both regimens improved OS, ORR, and PFS with manageable toxicity (though grade 3 toxicity was slightly higher in the triplet and control arms than in the doublet arm) and similar treatment discontinuation rates. However, the study was not powered sufficiently to formally compare the doublet with the triplet. As a result, cetuximab + encorafenib has become the standard of care for pre-treated patients (not in the first-line setting). This treatment was FDA-approved as of April 2020 [5]. Ongoing studies aim to evaluate the combination of EGFRi, BRAFi, and MEKi in untreated tumors. The ANCHOR study is a phase II trial evaluating the use of encorafenib + binimetinib + cetuximab in the first-line setting. Positive results were obtained, with an ORR of 47.8% and a DCR of 85% (response rates of 50% and stable disease rates of 35%). However, the PFS was disappointing at 5.8 months [40]. Another study exploring different first-line BRAFi options is the BREAKWATER study (phase III), which randomizes 870 patients to receive encorafenib + cetuximab; chemotherapy ± bevacizumab; or chemotherapy + encorafenib + cetuximab [41]. Some preliminary results appear promising, as there is an observed ORR of around 67% and a PFS of approximately 10 months, with data on tolerable toxicity so far [42].
5. Mechanisms of Resistance to BRAFi and Therapeutic Options
Despite the promising results shown by BRAFi treatments in combination with other MAPK pathway inhibitors (EGFRi or MEKi) in BRAF-mutated metastatic colorectal cancer (mCRC), the emergence of resistance mechanisms to these drugs limits their clinical benefit. While the response rate after double or triple MAPK pathway blockade ranges from 20 to 40% [4], the observed responses are short-lived. This is why resistance mechanisms to BRAFi have been recently studied. They are primarily attributed to the molecular heterogeneity of the tumor. It is common for patients to concurrently present multiple resistance mechanisms [5]. Furthermore, if a cell line is resistant to a specific treatment (BRAFi, MEKi, EGFRi), it will also be resistant to other drugs sharing the same mechanism of action, not just that specific drug (class resistance) [4]. Most of the studies on BRAFi resistance have been conducted based on the prior knowledge in melanoma, although it is not easily extrapolated to colorectal cancer (CCR) due to their distinct origins and molecular biology. BRAFi treatment for melanomas, both in monotherapy and in combination with MEKi, shows a higher response rate compared to mCRC, where monotherapy with vemurafenib does not provide benefit [37]. One of the reasons explaining this difference in response rate is the embryological origin of both types of neoplasms. CCR arises from epithelial cells, which highly express EGFR. Inhibiting only a part of the MAPK pathway (BRAFi or MEKi in monotherapy) triggers a feedback loop that leads to EGFR hyperactivation and subsequent tumor growth [43,44,45]. This phenomenon does not occur in melanoma, which originates from neural crest cells where EGFR is not expressed, thus the feedback loop is not effective. Another hypothesis known as the “big bang” theory [46] suggests that the cellular populations forming the tumor are heterogeneous from the beginning of carcinogenesis. The tumor comprises subpopulations with different mutations (BRAF or KRAS) at its inception. Therefore, treating with BRAFi eliminates subpopulations with mutations at that level, potentially selecting for a KRAS-mutated population resistant to BRAFi. Understanding the MAPK pathway functioning and the physiological negative feedback suppressed by BRAFi monotherapy led to the concurrent administration of drugs inhibiting the pathway at various levels (doublets or triplets with EGFRi and MEKi). Although response rates initially increased, some tumors could escape this drug combination. The analysis of circulating DNA in blood samples from patients included in the BEACON trial revealed that the majority of resistance mechanisms were due to reactivation of the MAPK pathway, despite prior inhibition of BRAF, EGFR, and/or MEK [47]. To better comprehend the underlying resistance mechanisms, Oddo et al. [4] selected cell populations resistant to BRAFi or MEKi monotherapy, but sensitive to different drug combinations (BRAFi + MEKi, BRAFi + EGFRi, MEKi + EGFRi). These cells were exposed to various drugs to generate resistant descendants, aiming to study the molecular mechanisms by which resistance developed and potential therapeutic strategies to overcome it. The drugs included BRAFi (vemurafenib, encorafenib, dabrafenib), MEKi (selumetinib and trametinib), EGFRi (cetuximab), and the PI3K inhibitor apelisib.
5.1. Hyperactivation of Other Tyrosine-Kinase Receptors
In cell populations developing resistance to EGFRi, an activation of the MAPK pathway through alternative receptor tyrosine kinases (RTKs) like Her2 or MET was observed [48]. In fact, there are studies demonstrating the clinical benefit of combining BRAF inhibitors (BRAFis) with MET inhibitors (METis) to address this mechanism of resistance [49,50]. Similarly, in BRAFi-resistant melanomas, alternative mechanisms converging on pathway activation were noted, such as overexpression of other RTKs (PDGFR or IGFR-1) [51]. It is also hypothesized that heterogeneous genetic alterations could activate the MAPK pathway alternatively, despite its blockade at different levels.
5.2. Feedback Mechanisms and ERK Hyperphosphorylation
Acquired resistance generally results in a retrograde hyperactivation of the MAPK pathway. In the presence of a BRAF V600E mutation, the entire pathway downstream is activated, causing a negative feedback loop (upstream) where the final link (ERK) generates a signal that inhibits the pathway. Physiologically, this self-regulates the MAPK pathway, but mutated BRAF proteins escape conventional cellular regulation mechanisms. Administering BRAFi stops ERK activation, disrupting the negative feedback loop. Consequently, EGFR hyperactivation occurs, leading to increased ERK phosphorylation [45]. While there was no difference in the amount of MEK/ERK/AKT between parental and daughter cells, the latter exhibited increased ERK phosphorylation (and sometimes AKT phosphorylation) following combined therapies, while ERK in parental cells remained inhibited [4]. Therefore, increased phosphorylated ERK serves as a mechanism of acquired resistance to BRAFi and could be a potential therapeutic target. ERK inhibitors (ERKis) have been developed for this purpose. Ulixertinib has shown efficacy in tumors with BRAF (V600E and non-V600E) and NRAS mutations, with low toxicity [52]. Another ERKi that exhibited activity in BRAF V600E-mutated mCRC monotherapy is GDC-0994 [53]. Studies also combine ERKi with other targeted therapies. The phase Ib/II HERKULES-3 trial (NCT05039177) combines ERAS-007 (ERKi) with encorafenib and cetuximab in patients with the BRAF V600E mutation. Another ongoing study combines a different ERKi (LTT462) with dabrafenib and encorafenib. Concurrent administration of cyclin inhibitors (abemaciclib) with LY3214996, an ERKi, has also been tested (NCT02857270). Additionally, drugs targeting Src homology-2 domain-containing protein tyrosine phosphatase 2 (Shp2), a positive modulator of ERK, have been developed and could mediate acquired resistance. A phase Ib trial combines a Shp2 inhibitor (TNO155) with dabrafenib and trametinib or ERKi [5]. Although this is still preclinical activity, awaiting results, exploring ERK inhibition as a standard practice is worth considering. For now, the question of whether monotherapy or combination with BRAFi is more beneficial remains unresolved, but early results lean towards the combination (enhancing ERKi cytotoxicity) [4].
5.3. Structural Modifications of BRAF
The MAPK pathway cascade involves dimerization and subsequent activation of RAF. Despite the extensive clinical use of RAF inhibitors, both dabrafenib and vemurafenib or encorafenib solely inhibit monomeric RAF. An interesting strategy would be to inhibit the dimeric form once it has formed. Pan-RAF inhibitors are being developed for this purpose, such as ponatinib Hybrid Inhibitor 1 (PHI1) [54]. Other therapeutic strategies aim to prevent dimer formation with so-called BRAF paradox breakers. In a phase I/II trial evaluating the paradox breaker PLX8394, an objective response rate (ORR) of 22% was achieved in a population of 45 patients with BRAF mutations or fusions [55]. Aberrant splicing generates BRAF forms not sensitive to conventional BRAFi (like BRAF V600E DEx), or truncated forms that permanently activate MEK/ERK signaling. Both mechanisms could be involved in BRAFi resistance [51].
5.4. Amplifications and Acquired Mutations
When examining the DNA from resistant cells, an increase in the EGFR gene copy number was observed in those exposed to the BRAFi + MEKi or BRAFi + EGFRi combinations [4]. While EGFR protein overexpression has been described through immunohistochemistry in cases of primary BRAFi-resistant mutated BRAF CCR [45], gene amplification of EGFR has not been established in any study. Other gene amplifications observed in the DNA of resistant cells included KRAS and BRAF (in cell models subjected to the BRAFi + EGFRi and MEKi + EGFRi combinations, respectively). Amplification of KRAS G13D has also been in vitro in melanomas treated with MEKi [56]. These amplifications were determined by PCR and confirmed by FISH. They were found exclusively in the genetic material of resistant cells, absent in parental cells. HER2 or MET amplification was not observed in any case [4]. These findings support the theory that BRAFi resistance mechanisms involve increased activity of other components of the cascade, perpetually keeping the pathway active. Mutations acquired were identified through Sanger sequencing in resistant cell DNA: exons 2, 3, and 4 of KRAS; exons 2 and 3 of NRAS; exon 15 of BRAF; and exons 2 and 3 of MAP2K1 (gene encoding MEK) [4]. All cells retained the original BRAF V600E mutation, while the described acquired mutations were found only in daughter cells. The most frequently observed resistance mechanism was KRAS alteration, particularly in exons 2 and 4 (G12D, G13D, A146T/V) [4]. In some cases, different KRAS mutations coexisted within the same cell population, suggesting polyclonality. A point mutation was also identified in the EGFR ectodomain (G465R) as an acquired resistance mechanism, enabling ERK phosphorylation and increased cell growth even in the presence of vemurafenib + cetuximab. This mutation had been previously described in BRAF wild-type CCR resistant to cetuximab or panitumumab [57], but had not been observed in mutated BRAF tumors until then. These findings highlight the therapeutic utility of inhibiting the MAPK pathway at various vertical levels. This principle is supported by the high response rate observed in melanomas treated with the BRAFi and MEKi dual combination, although results are not fully extrapolatable to mCRC due to the molecular characteristics of each tumor. In tested resistant cells, EGFR overexpression constituted a resistance mechanism to therapeutic doublets (BRAFi + MEKo or + EGFRi), whereas administering a triplet with BRAFi + EGFRi + MEKi restored cell sensitivity to treatment [4]. In another recent study, cells developing resistance to the drug triplet maintained both the BRAF V600E mutation and KRAS mutation (G12D or G13D) [58], demonstrating that the coexistence of both mutations confers greater treatment resistance to the tumor. However, this condition is very rare, as previously mentioned [15]. Various mechanisms of acquired BRAFi resistance have been described in other tumors, mainly in melanoma, such as the loss of neurofibromin 1 (NF1). NF1 is a tumor suppressor that physiologically inhibits RAS. Its loss results in constant RAS activation of the MAPK pathway, despite targeted therapies. It has been described in 4% of mutated BRAF melanomas [59] and plays a significant role in developing BRAFi resistance.
5.5. Cell Cycle Dysregulation
Another mechanism involves altered cell cycle regulation. Cyclin D1 binds with CDK4 and CDK6, which phosphorylate retinoblastoma protein, initiating the cell cycle. Cells with hyperactivating cyclin D1 mutations have shown resistance to BRAFi. When this mutation is additionally associated with CDK4 mutations, resistance is further increased [60].
5.6. PI3K/AKT/mTOR Pathway Alterations
As reflected in Figure 1, the PI3K/AKT/mTOR pathway converges with the RAS/RAF/MEK/ERK pathway toward the nucleus. In CCR cell samples, PI3K is more activated compared to melanoma cells [61], leading to hypotheses of whether PI3K could be inhibited to bypass BRAFi resistance. Although controversial, current data seem to discount the significant role of the PI3K pathway in developing BRAFi resistance. In a phase II trial conducted with dabrafenib and trametinib, three out of five patients responding to treatment had concurrent PI3KCA mutations, with one achieving a complete response. All patients with PI3K mutations responded to treatment [62]. In the SWOG S1406 study comparing the VIC regimen (mentioned earlier) and standard chemotherapy with cetuximab, the presence of concurrent PI3K mutations increased progression-free survival (HR of 0.3 vs. 0.6 for PI3K wt) [37]. A comparison between encorafenib + cetuximab and the same doublet with the addition of a PI3K inhibitor (PI3Ki), alpelisib, showed few differences in outcomes: ORR of 29% and 18%, respectively; median PFS of 3.7 months or 4.2 months, respectively, with more toxicity in the alpelisib group [63]. Other hypotheses explored in melanoma regarding BRAFi resistance involve the PI3K/AKT pathway. In cell populations with overexpressed Hepatocyte growth factor (HGF) or its receptor c-MET, hyperactivation of the PI3K/AKT pathway is observed, subsequently activating the RAS/RAF/MEK/ERK pathway. In vitro, sensitivity to HGF or c-MET inhibition was observed, suggesting c-MET could be another therapeutic target [64]. Another therapeutic option explored is inhibiting the PI3K pathway at earlier stages, when it is still interconnected with the MAPK pathway.
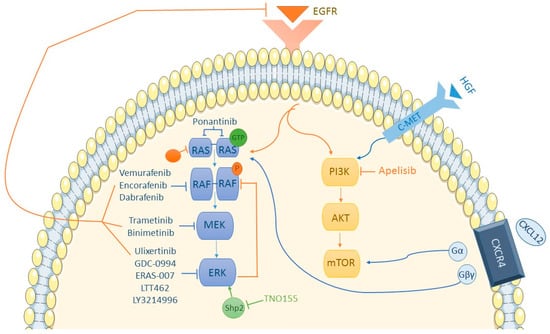
Figure 1.
MAPK and PI3K/AKT/mTOR pathway.
5.7. Other Potential Mechanisms
Chemokine receptor 4 (CXCR4) is overexpressed in many tumor cells, while its expression is diminished or absent in healthy tissues [65]. CXCR4′s presence in mCRC is associated with increased treatment resistance [66]. Its ligand, CXCL12 or stromal-cell-derived factor-1 (SDF-1), when binding to the receptor, divides it into an alpha subunit (activating the RAS/RAF pathway) and a beta/gamma subunit (activating the PI3K/AKT/mTOR pathway). Hence, crosstalk between both pathways makes CXCR4 a potential new target for BRAFi-resistant mutated BRAF tumors. Wnt/beta-catenin pathway involvement in acquired BRAFi resistance has also been observed. BRAF inhibition leads to positive feedback on this pathway [5], promoting cell proliferation, migration, and invasion. Additionally, R-Spondin (RSPO) fusions, which also activate the Wnt/beta-catenin pathway, are significantly associated with BRAF mutations, making the combination of BRAFi + Wnti an interesting therapeutic option. A phase I clinical trial for mCRC patients with the BRAF V600E mutation combines a Wnti (WNT974) with encorafenib and cetuximab [5]. It has been observed that SRC kinases are systematically activated in BRAF V600E CRCs and contribute to increasing tumor growth independently of ERK signaling, through beta-catenin (CTNNB1). The activation of these kinases is facilitated by prostaglandin E2. As we know, the enzyme COX-2 is essential for prostaglandin synthesis, so using a COX-2 inhibitor in combination with BRAF inhibitors (BRAFis) and/or EGFR inhibitors (EGFRis) is an interesting therapeutic strategy that has already shown longer-lasting suppression of tumor growth in patient-derived tumor xenograft models [67]. Furthermore, to enhance the antitumoral activity of BRAFi, VEGFi could be used [68,69], showing long-lasting tumor responses.
6. Prognosis and Predictive Biomarkers
As previously mentioned, patients with mutated BRAF CRC have a poor prognosis, despite receiving BRAFi. However, there is a subgroup of patients who exhibit a long response to these drugs and better survival. To attempt to define the mentioned prognostic heterogeneity, the BeCool study [70] developed a prognostic scale ranging from 0 to 16 points based on clinical and analytical criteria. It divided patients into low-risk (0–4), intermediate (5–8), and high-risk (9–16) categories, and significant differences were observed in terms of life expectancy in each subgroup (OS of 29.6, 15.5, and 6.6, respectively). It is an easy-to-use tool in clinical practice and could be useful for stratifying patients when enrolling them in future studies.
It is also important to mention the predictive role of the BRAF allele fraction (AF) in patients with BRAF V600E CRC who have received treatment with BRAFi + EGFRi ± MEKi. In the prospective cohort designed by Ros et al. [71], it is shown that patients with a high AF (≥2%) experience an increased occurrence of liver metastases and a worse PFS (HR 2.97, 95% CI 1.55–5.69) and OS (HR 3.28, 95% CI 1.58–6.81) compared to patients with a low AF (<2%). Therefore, determining the AF in blood samples can be useful in assessing tumor aggressiveness and identifying the subgroup of patients who may benefit from more intensive treatment.
In the aforementioned BEACON study [39], an attempt was made to establish a correlation between the clinical outcomes and molecular findings in tumor samples. It was concluded that in those BM1 or CMS4 subtypes, there is an increased inflammatory response, as well as an increased ORR in patients receiving the triplet (cetuximab + encorafenib + binimetinib) compared to the doublet (cetuximab + encorafenib): for CMS4, the ORR was 33.3% [95% CI: 21.7–46.7], and for BM1, it was 33.3% [95% CI: 21.4–47.1] in the triplet arm, compared to CMS4: 19.2% [95% CI: 9.6–32.5] and BM1: 14.9% [95% CI: 6.2–28.3] in the doublet arm [72]. Therefore, the molecular subtype of the tumor can serve as a prognostic marker when deciding which patients should receive triple therapy.
As mentioned previously, there is crosstalk between the MAPK pathway and the Wnt/Beta-catenin pathway that can modulate the antitumoral activity of targeted therapy. Recently, it has been observed that patients with MSS BRAF V600E CRC with mutations in RNF43 (a negative regulator of the Wnt pathway) exhibit a better antitumoral response to BRAFi and/or EGFRi than those with RNF43 wild type: they have a longer PFS (HR 0.30; 95% CI 0.12–0.75) and overall survival (OS) (HR 0.26; 95% CI 0.10–0.71). Therefore, RNF43 mutation could be used as a predictive biomarker for treatment response [73,74].
7. Conclusions
Mutated BRAF mCRC exhibits distinct biological behavior compared to other molecular subtypes of colorectal cancer. While the standard of care for years involved chemotherapy and anti-angiogenic agents, the development of targeted therapy has opened a new avenue for these patients. However, the emergence of BRAFi resistance presents a challenge in providing greater survival and quality of life for these patients. Therefore, continued research into resistance mechanisms and strategies to overcome them is imperative.
Author Contributions
P.G.: conceptualization, writing—original draft preparation; V.A.: writing—original draft preparation; M.S.R.: writing—original draft preparation; C.G.-M.: writing—review and editing; C.G.d.Q.: writing—review and editing; J.M.: writing—review and editing; J.C.C.: writing—review and editing; G.G.: writing—review and editing; I.O.: writing—review and editing; J.C.: writing—review and editing; Í.M.-D.: writing—review and editing; B.M.: writing—review and editing; B.d.F.: writing—review and editing; M.R.F.: supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available in this article.
Conflicts of Interest
M.R.F. declares the following conflicts of interest—Advisory: Servier, Amgen, Pierre Fabre, MSD; Speaker: Servier, Amgem, Merck, Pierre Fabre; Travels and other honoraria: Sanofi, Merck, Servier, Amgen. The other authors declare no conflicts of interest.
References
- Ros, J.; Baraibar, I.; Sardo, E.; Mulet, N.; Salvà, F.; Argilés, G.; Martini, G.; Ciardiello, D.; Cuadra, J.L.; Tabernero, J.; et al. BRAF, MEK and EGFR inhibition as treatment strategies in BRAF V600E metastatic colorectal cancer. Ther. Adv. Med. Oncol. 2021, 13, 175883592199297. [Google Scholar] [CrossRef]
- Molina-Cerrillo, J.; San Román, M.; Pozas, J.; Alonso-Gordoa, T.; Pozas, M.; Conde, E.; Rosas, M.; Grande, E.; García-Bermejo, M.L.; Carrato, A. BRAF Mutated Colorectal Cancer: New Treatment Approaches. Cancers 2020, 12, 1571. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Oddo, D.; Sennott, E.M.; Barault, L.; Valtorta, E.; Arena, S.; Cassingena, A.; Filiciotto, G.; Marzolla, G.; Elez, E.; Van Geel, R.M.J.M.; et al. Molecular Landscape of Acquired Resistance to Targeted Therapy Combinations in BRAF -Mutant Colorectal Cancer. Cancer Res. 2016, 76, 4504–4515. [Google Scholar] [CrossRef] [PubMed]
- Ciombor, K.K.; Strickler, J.H.; Bekaii-Saab, T.S.; Yaeger, R. BRAF -Mutated Advanced Colorectal Cancer: A Rapidly Changing Therapeutic Landscape. J. Clin. Oncol. 2022, 40, 2706–2715. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Barras, D. BRAF Mutation in Colorectal Cancer: An Update: Supplementary Issue: Biomarkers for Colon Cancer. Biomark Cancer 2015, 7s1, BIC.S25248. [Google Scholar] [CrossRef] [PubMed]
- Tran, B.; Kopetz, S.; Tie, J.; Gibbs, P.; Jiang, Z.Q.; Lieu, C.H.; Agarwal, A.; Maru, D.M.; Sieber, O.; Desai, J. Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer: Metastatic Pattern in BRAF Mutant CRC. Cancer 2011, 117, 4623–4632. [Google Scholar] [CrossRef]
- Venderbosch, S.; Nagtegaal, I.D.; Maughan, T.S.; Smith, C.G.; Cheadle, J.P.; Fisher, D.; Kaplan, R.; Quirke, P.; Seymour, M.T.; Richman, S.D.; et al. Mismatch Repair Status and BRAF Mutation Status in Metastatic Colorectal Cancer Patients: A Pooled Analysis of the CAIRO, CAIRO2, COIN, and FOCUS Studies. Clin. Cancer Res. 2014, 20, 5322–5330. [Google Scholar] [CrossRef]
- Kalady, M.F. Sessile Serrated Polyps: An Important Route to Colorectal Cancer. J. Natl. Compr. Cancer Netw. 2013, 11, 1585–1594. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell. 2018, 33, 125–136.e3. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Yaeger, R.; Rodrik-Outmezguine, V.S.; Tao, A.; Torres, N.M.; Chang, M.T.; Drosten, M.; Zhao, H.; Cecchi, F.; Hembrough, T.; et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated, R.A.S. Nature 2017, 548, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Caputo, F.; Santini, C.; Bardasi, C.; Cerma, K.; Casadei-Gardini, A.; Spallanzani, A.; Andrikou, K.; Cascinu, S.; Gelsomino, F. BRAF-Mutated Colorectal Cancer: Clinical and Molecular Insights. Int. J. Mol. Sci. 2019, 20, 5369. [Google Scholar] [CrossRef]
- Yaeger, R.; Kotani, D.; Mondaca, S.; Parikh, A.R.; Bando, H.; Van Seventer, E.E.; Taniguchi, H.; Zhao, H.; Thant, C.N.; De Stanchina, E.; et al. Response to Anti-EGFR Therapy in Patients with BRAF non-V600–Mutant Metastatic Colorectal Cancer. Clin. Cancer Res. 2019, 25, 7089–7097. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, J.F.; Fisher, D.; Smith, C.G.; Richman, S.D.; Elliott, F.; Brown, S.; Adams, R.; Maughan, T.; Quirke, P.; Cheadle, J.; et al. Investigating the poor outcomes ofBRAF-mutant advanced colorectal cancer: Analysis from 2530 patients in randomised clinical trials. Ann. Oncol. 2017, 28, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, F.; Ou, F.S.; Qu, X.; Zemla, T.J.; Niedzwiecki, D.; Tam, R.; Mahajan, S.; Goldberg, R.M.; Bertagnolli, M.M.; Blanke, C.D.; et al. Mutational Analysis of Patients with Colorectal Cancer in CALGB/SWOG 80405 Identifies New Roles of Microsatellite Instability and Tumor Mutational Burden for Patient Outcome. J. Clin. Oncol. 2019, 37, 1217–1227. [Google Scholar] [CrossRef]
- Ducreux, M.; Chamseddine, A.; Laurent-Puig, P.; Smolenschi, C.; Hollebecque, A.; Dartigues, P.; Samallin, E.; Boige, V.; Malka, D.; Gelli, M. Molecular targeted therapy of BRAF -mutant colorectal cancer. Ther. Adv. Med. Oncol. 2019, 11, 175883591985649. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef]
- Middleton, G.; Yang, Y.; Campbell, C.D.; André, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; et al. BRAF -Mutant Transcriptional Subtypes Predict Outcome of Combined BRAF, MEK, and EGFR Blockade with Dabrafenib, Trametinib, and Panitumumab in Patients with Colorectal Cancer. Clin. Cancer Res. 2020, 26, 2466–2476. [Google Scholar] [CrossRef]
- Morris, V.K.; Kennedy, E.B.; Baxter, N.N.; Benson, A.B.; Cercek, A.; Cho, M.; Ciombor, K.K.; Cremolini, C.; Davis, A.; Deming, D.A.; et al. Treatment of Metastatic Colorectal Cancer: ASCO Guideline. J. Clin. Oncol. 2023, 41, 678–700. [Google Scholar] [CrossRef]
- Tian, J.; Chen, J.H.; Chao, S.X.; Pelka, K.; Giannakis, M.; Hess, J.; Burke, K.; Jorgji, V.; Sindurakar, P.; Braverman, J.; et al. Combined PD-1, BRAF and MEK inhibition in BRAFV600E colorectal cancer: A phase 2 trial. Nat. Med. 2023, 29, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W. BRAF inhibitors: The current and the future. Curr. Opin. Pharmacol. 2015, 23, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Hertzman Johansson, C.; Egyhazi Brage, S. BRAF inhibitors in cancer therapy. Pharmacol. Ther. 2014, 142, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Delord, J.P.; Robert, C.; Nyakas, M.; McArthur, G.A.; Kudchakar, R.; Mahipal, A.; Yamada, Y.; Sullivan, R.; Arance, A.; Kefford, R.F.; et al. Phase I Dose-Escalation and -Expansion Study of the BRAF Inhibitor Encorafenib (LGX818) in Metastatic BRAF -Mutant Melanoma. Clin. Cancer Res. 2017, 23, 5339–5348. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.D.; Seymour, M.T.; Chambers, P.; Elliott, F.; Daly, C.L.; Meade, A.M.; Taylor, G.; Barrett, J.H.; Quirke, P. KRAS and BRAF Mutations in Advanced Colorectal Cancer Are Associated with Poor Prognosis but Do Not Preclude Benefit from Oxaliplatin or Irinotecan: Results from the MRC FOCUS Trial. J. Clin. Oncol. 2009, 27, 5931–5937. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Perrone, F.; Biondani, P.; Maggi, C.; Lampis, A.; Bertan, C.; Venturini, F.; Tondulli, L.; Ferrari, D.; Ricci, V.; et al. Single agent panitumumab in KRAS wild-type metastatic colorectal cancer patients following cetuximab-based regimens: Clinical outcome and biomarkers of efficacy. Cancer Biol. Ther. 2013, 14, 1098–1103. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; Di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonati, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef]
- Cremolini, C.; Antoniotti, C.; Stein, A.; Bendell, J.; Gruenberger, T.; Rossini, D.; Masi, G.; Ongaro, E.; Hurwitz, H.; Falcone, A.; et al. Individual Patient Data Meta-Analysis of FOLFOXIRI Plus Bevacizumab Versus Doublets Plus Bevacizumab as Initial Therapy of Unresectable Metastatic Colorectal Cancer. J. Clin. Oncol. 2020, 38, 3314–3324. [Google Scholar] [CrossRef]
- Stintzing, S.; Heinrich, K.; Tougeron, D.; Modest, D.P.; Schwaner, I.; Eucker, J.; Pihusch, R.; Stauch, M.; Kaiser, F.; Kahl, C.; et al. FOLFOXIRI Plus Cetuximab or Bevacizumab as First-Line Treatment of BRAFV600E-Mutant Metastatic Colorectal Cancer: The Randomized Phase II FIRE-4.5 (AIO KRK0116) Study. J. Clin. Oncol. 2023, 41, 4143–4153. [Google Scholar] [CrossRef]
- Cervantes, A.; Adam, R.; Roselló, S.; Arnold, D.; Normanno, N.; Taïeb, J.; Seligmann, J.; De Baere, T.; Osterlund, P.; Yoshino, T.; et al. Metastatic colorectal cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. 2023, 34, 10–32. [Google Scholar] [CrossRef]
- Punt, C.J.A.; Bond, M.J.G.; Bolhuis, K.; Loosveld, O.; Helgason, H.H. FOLFOXIRI + bevacizumab versus FOLFOX/FOLFIRI + bevacizumab in patients with initially unresectable colorectal liver metastases (CRLM) and right-sided and/or RAS/BRAFV600E-mutated primary tumor: Phase III CAIRO5 study of the Dutch Colorectal Cancer Group. J. Clin. Oncol. 2022, 40, LBA3506. [Google Scholar] [CrossRef]
- Masi, G.; Loupakis, F.; Salvatore, L.; Fornaro, L.; Cremolini, C.; Cupini, S.; Ciarlo, A.; Del Monte, F.; Cortesi, E.; Amoroso, D.; et al. Bevacizumab with FOLFOXIRI (irinotecan, oxaliplatin, fluorouracil, and folinate) as first-line treatment for metastatic colorectal cancer: A phase 2 trial. Lancet Oncol. 2010, 11, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): Final analysis of a randomised, open-label, phase 3 study. Lancet Oncol. 2022, 23, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability–High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Corcoran, R.; Giannakis, M.; Allen, J.; Chen, J.; Pelka, K.; Chao, S.; Meyerhardt, J.; Enzinger, A.; Enzinger, P.; McCleary, N.; et al. SO-26 Clinical efficacy of combined BRAF, MEK, and PD-1 inhibition in BRAFV600E colorectal cancer patients. Ann. Oncol. 2020, 31, S226–S227. [Google Scholar] [CrossRef]
- Kopetz, S.; Guthrie, K.A.; Morris, V.K.; Lenz, H.J.; Magliocco, A.M.; Maru, D.; Yan, Y.; Lanman, R.; Manyam, G.; Hong, D.S.; et al. Randomized Trial of Irinotecan and Cetuximab with or Without Vemurafenib in BRAF-Mutant Metastatic Colorectal Cancer (SWOG S1406). J. Clin. Oncol. 2021, 39, 285–294. [Google Scholar] [CrossRef]
- Hong, D.S.; Morris, V.K.; El Osta, B.; Sorokin, A.V.; Janku, F.; Fu, S.; Overman, M.J.; Piha-Paul, S.; Subbiah, V.; Kee, B.; et al. Phase IB Study of Vemurafenib in Combination with Irinotecan and Cetuximab in Patients with Metastatic Colorectal Cancer with BRAF V600E Mutation. Cancer Discov. 2016, 6, 1352–1365. [Google Scholar] [CrossRef]
- Tabernero, J.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib Plus Cetuximab as a New Standard of Care for Previously Treated BRAF V600E–Mutant Metastatic Colorectal Cancer: Updated Survival Results and Subgroup Analyses from the BEACON Study. J. Clin. Oncol. 2021, 39, 273–284. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Taieb, J.; Yaeger, R.; Yoshino, T.; Grothey, A.; Maiello, E.; Elez, E.; Dekervel, J.; Ross, P.; Ruiz-Casado, A.; et al. ANCHOR CRC: Results From a Single-Arm, Phase II Study of Encorafenib Plus Binimetinib and Cetuximab in Previously Untreated BRAFV600E-Mutant Metastatic Colorectal Cancer. J. Clin. Oncol. 2023, 41, 2628–2637. [Google Scholar] [CrossRef]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Ciardiello, F.; Desai, J.; Kim, T.W.; Maughan, T.; Van Cutsem, E.; Wasan, H.S.; Yoshino, T.; et al. BREAKWATER: Randomized phase 3 study of encorafenib (enco) + cetuximab (cetux) ± chemotherapy for first-line (1L) treatment (tx) of BRAF V600E-mutant (BRAF V600E) metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2021, 39, TPS3619. [Google Scholar] [CrossRef]
- Tabernero, J.; Yoshino, T.; Kim, T.W.; Yaeger, R.; Desai, J.; Wasan, H.S.; Van Cutsem, E.; Ciardiello, F.; Maughan, T.; Eng, C.; et al. LBA26 BREAKWATER safety lead-in (SLI): Encorafenib (E) + cetuximab (C) + chemotherapy (chemo) for BRAFV600E metastatic colorectal cancer (mCRC). Ann. Oncol. 2022, 33, S1392–S1393. [Google Scholar] [CrossRef]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; De Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [PubMed]
- Prahallad, A.; Sun, C.; Huang, S.; Di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; Della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-Mediated Reactivation of MAPK Signaling Contributes to Insensitivity of BRAF-Mutant Colorectal Cancers to RAF Inhibition with Vemurafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef]
- Kopetz, S.; Murphy, D.A.; Pu, J.; Yaeger, R.; Ciardiello, F.; Desai, J.; Van Cutsem, E.; Wasan, H.S.; Yoshino, T.; Alkuzweny, B.; et al. 316O Genomic mechanisms of acquired resistance of patients (pts) with BRAF V600E-mutant (mt) metastatic colorectal cancer (mCRC) treated in the BEACON study. Ann. Oncol. 2022, 33, S681–S682. [Google Scholar] [CrossRef]
- Misale, S.; Di Nicolantonio, F.; Sartore-Bianchi, A.; Siena, S.; Bardelli, A. Resistance to Anti-EGFR Therapy in Colorectal Cancer: From Heterogeneity to Convergent Evolution. Cancer Discov. 2014, 4, 1269–1280. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Oddo, D.; Gloghini, A.; Valtorta, E.; Berenato, R.; Barault, L.; Caporale, M.; Busico, A.; Morano, F.; Gualeni, A.V.; et al. MET-Driven Resistance to Dual EGFR and BRAF Blockade May Be Overcome by Switching from EGFR to MET Inhibition in BRAF -Mutated Colorectal Cancer. Cancer Discov. 2016, 6, 963–971. [Google Scholar] [CrossRef]
- Ros, J.; Elez, E. Overcoming acquired MET amplification after encorafenib-cetuximab in BRAF-V600E mutated colorectal cancer. Eur. J. Cancer 2022, 172, 326–328. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS–ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug. Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.J.; Infante, J.R.; Janku, F.; Wong, D.J.L.; Sosman, J.A.; Keedy, V.; Patel, M.R.; Shapiro, G.I.; Mier, J.W.; Tolcher, A.W.; et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov. 2018, 8, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Varga, A.; Soria, J.C.; Hollebecque, A.; LoRusso, P.; Bendell, J.; Huang, S.M.A.; Wagle, M.C.; Okrah, K.; Liu, L.; Murray, E.; et al. A First-in-Human Phase I Study to Evaluate the ERK1/2 Inhibitor GDC-0994 in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2020, 26, 1229–1236. [Google Scholar] [CrossRef] [PubMed]
- Cotto-Rios, X.M.; Agianian, B.; Gitego, N.; Zacharioudakis, E.; Giricz, O.; Wu, Y.; Zou, Y.; Verma, A.; Poulikakos, P.I.; Gavathiotis, E. Inhibitors of BRAF dimers using an allosteric site. Nat. Commun. 2020, 11, 4370. [Google Scholar] [CrossRef]
- Koumaki, K.; Kontogianni, G.; Kosmidou, V.; Pahitsa, F.; Kritsi, E.; Zervou, M.; Chatziioannou, A.; Souliotis, V.L.; Papadodima, O.; Pintzas, A. BRAF paradox breakers PLX8394, PLX7904 are more effective against BRAFV600Ε CRC cells compared with the BRAF inhibitor PLX4720 and shown by detailed pathway analysis. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166061. [Google Scholar] [CrossRef]
- Sale, M.J.; Balmanno, K.; Saxena, J.; Ozono, E.; Wojdyla, K.; McIntyre, R.E.; Gilley, R.; Woroniuk, A.; Howarth, K.D.; Hughes, G.; et al. MEK1/2 inhibitor withdrawal reverses acquired resistance driven by BRAFV600E amplification whereas KRASG13D amplification promotes EMT-chemoresistance. Nat. Commun. 2019, 10, 2030. [Google Scholar] [CrossRef]
- Arena, S.; Bellosillo, B.; Siravegna, G.; Martínez, A.; Cañadas, I.; Lazzari, L.; Ferruz, N.; Russo, M.; Misale, S.; González, I.; et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin. Cancer Res. 2015, 21, 2157–2166. [Google Scholar] [CrossRef]
- Ahronian, L.G.; Sennott, E.M.; Van Allen, E.M.; Wagle, N.; Kwak, E.L.; Faris, J.E.; Godfrey, J.T.; Nishimura, K.; Lynch, K.D.; Mermel, C.H.; et al. Clinical Acquired Resistance to RAF Inhibitor Combinations in BRAF -Mutant Colorectal Cancer through MAPK Pathway Alterations. Cancer Discov. 2015, 5, 358–367. [Google Scholar] [CrossRef]
- Mar, V.J.; Wong, S.Q.; Li, J.; Scolyer, R.A.; McLean, C.; Papenfuss, A.T.; Tothill, R.W.; Kakavand, H.; Mann, G.J.; Thompson, J.F.; et al. BRAF/NRAS Wild-Type Melanomas Have a High Mutation Load Correlating with Histologic and Molecular Signatures of UV Damage. Clin. Cancer Res. 2013, 19, 4589–4598. [Google Scholar] [CrossRef]
- Smalley, K.S.M.; Lioni, M.; Palma, M.D.; Xiao, M.; Desai, B.; Egyhazi, S.; Hansson, J.; Wu, H.; King, A.J.; Van Belle, P.; et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E–mutated melanomas. Mol. Cancer Ther. 2008, 7, 2876–2883. [Google Scholar] [CrossRef]
- Mao, M.; Tian, F.; Mariadason, J.M.; Tsao, C.C.; Lemos, R.; Dayyani, F.; Gopal, Y.N.V.; Jiang, Z.Q.; Wistuba, I.I.; Tang, X.M.; et al. Resistance to BRAF Inhibition in BRAF-Mutant Colon Cancer Can Be Overcome with PI3K Inhibition or Demethylating Agents. Clin. Cancer Res. 2013, 19, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, R.B.; André, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAF V600E-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef] [PubMed]
- Van Geel, R.M.J.M.; Tabernero, J.; Elez, E.; Bendell, J.C.; Spreafico, A.; Schuler, M.; Yoshino, T.; Delord, J.P.; Yamada, Y.; Lolkema, M.P.; et al. A Phase Ib Dose-Escalation Study of Encorafenib and Cetuximab with or without Alpelisib in Metastatic BRAF -Mutant Colorectal Cancer. Cancer Discov. 2017, 7, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef]
- Xu, C.; Zheng, L.; Li, D.; Chen, G.; Gu, J.; Chen, J. CXCR4 overexpression is correlated with poor prognosis in colorectal cancer. Life Sci. 2018, 208, 333–340. [Google Scholar] [CrossRef]
- Ottaiano, A.; Scala, S.; Normanno, N.; Botti, G.; Tatangelo, F.; Di Mauro, A.; Capozzi, M.; Facchini, S.; Tafuto, S.; Nasti, G. Prognostic and Predictive Role of CXC Chemokine Receptor 4 in Metastatic Colorectal Cancer Patients. Appl. Immunohistochem. Mol. Morphol. 2020, 28, 755–760. [Google Scholar] [CrossRef]
- Ruiz-Saenz, A.; Atreya, C.E.; Wang, C.; Pan, B.; Dreyer, C.A.; Brunen, D.; Prahallad, A.; Muñoz, D.P.; Ramms, D.J.; Burghi, V.; et al. A reversible SRC-relayed COX2 inflammatory program drives resistance to BRAF and EGFR inhibition in BRAFV600E colorectal tumors. Nat. Cancer 2023, 4, 240–256. [Google Scholar] [CrossRef]
- Bottos, A.; Martini, M.; Di Nicolantonio, F.; Comunanza, V.; Maione, F.; Minassi, A.; Appendino, G.; Bussolino, F.; Bardelli, A. Targeting Oncogenic Serine/Threonine-Protein Kinase BRAF in Cancer Cells Inhibits Angiogenesis and Abrogates Hypoxia. Proc. Natl. Acad. Sci. USA 2012, 109, E353–E359. Available online: https://pnas.org/doi/full/10.1073/pnas.1105026109 (accessed on 20 October 2023). [CrossRef]
- Comunanza, V.; Corà, D.; Orso, F.; Consonni, F.M.; Middonti, E.; Di Nicolantonio, F.; Buzdin, A.; Sica, A.; Medico, E.; Sangiolo, D.; et al. VEGF blockade enhances the antitumor effect of BRAFV600E inhibition. EMBO Mol. Med. 2017, 9, 219–237. [Google Scholar] [CrossRef]
- Loupakis, F.; Intini, R.; Cremolini, C.; Orlandi, A.; Sartore-Bianchi, A.; Pietrantonio, F.; Pella, N.; Spallanzani, A.; Dell’Aquila, E.; Scartozzi, M.; et al. A validated prognostic classifier for BRAF-mutated metastatic colorectal cancer: The ‘BRAF BeCool’ study. Eur. J. Cancer 2019, 118, 121–130. [Google Scholar] [CrossRef]
- Ros, J.; Matito, J.; Villacampa, G.; Comas, R.; Garcia, A.; Martini, G.; Baraibar, I.; Saoudi, N.; Salvà, F.; Martin, Á.; et al. Plasmatic BRAF-V600E allele fraction as a prognostic factor in metastatic colorectal cancer treated with BRAF combinatorial treatments. Ann. Oncol. 2023, 34, 543–552. [Google Scholar] [CrossRef]
- Kopetz, S.; Murphy, D.A.; Pu, J.; Ciardiello, F.; Desai, J.; Grothey, A.; Van Cutsem, E.; Wasan, H.S.; Yaeger, R.; Yoshino, T.; et al. Molecular correlates of clinical benefit in previously treated patients (pts) with BRAF V600E-mutant metastatic colorectal cancer (mCRC) from the BEACON study. J. Clin. Oncol. 2021, 39, 3513. [Google Scholar] [CrossRef]
- Elez, E.; Ros, J.; Fernández, J.; Villacampa, G.; Moreno-Cárdenas, A.B.; Arenillas, C.; Bernatowicz, K.; Comas, R.; Li, S.; Kodack, D.P.; et al. RNF43 mutations predict response to anti-BRAF/EGFR combinatory therapies in BRAFV600E metastatic colorectal cancer. Nat. Med. 2022, 28, 2162–2170. [Google Scholar] [CrossRef] [PubMed]
- Quintanilha, J.C.F.; Graf, R.P.; Oxnard, G.R. BRAF V600E and RNF43 Co-mutations Predict Patient Outcomes with Targeted Therapies in Real-World Cases of Colorectal Cancer. Oncologist 2023, 28, e171–e174. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).