
Rhabdomyosarcoma: Current Therapy, Challenges, and Future Approaches to Treatment Strategies

 ,
,  , ,
, ,  ,
,  , , ,
, , ,  , , ,
, , ,  , , ,
, , ,  , ,
, ,  add
Show full author list
add
Show full author list
Abstract
Simple Summary
Abstract
1. Introduction
2. RMS Subtypes

| Embryonal | Alveolar | Pleomorphic | Spindle Cell/Sclerosing ++ | |
|---|---|---|---|---|
| Prevalence | 2.6% (most common) [2] * | 1.0% (common) [2] * | Rare [2,20] | Rare [2,20] |
| Age | Bimodal distribution: peak incidence ages 0–4 > 14–18 [2,22] | Late childhood/adolescents [3,23] | 40–70 yrs of age, peak during 6th decade of life [27] | Children [4,28] |
| Gender predominance | Male [4] | None | Male [3] | NA |
| Subtypes | Spindle cell and Botryoid subtypes | NA + | Classic, round cell, and spindle cell subtypes | NA |
| Primary tumor location | Head/neck, superior nasal quadrants, eye socket, bladder, and prostate [23] | Trunk and extremities, inferior orbit [23] | Lower extremities [3,4] | Head/neck region, paratesticular region [4,23] |
| Genetics | 80% have loss of heterozygosity at 11p15 (IGF-2 gene) [3]; associated with familial cancer syndromes, e.g., LFS, NF1 | 60% are t(2:13)(q35:114): PAX3-FOXO1 positive [3]; 20% are t(1;13)(p36;q14): PAX7-FOXO1 positive [1]; 20% are FN; resemble ERMS characteristics/prognosis [3] | NA | NA |
| Histology | Immature rhabdomyoblast, less dense stromal rich background vs. ARMS, lacks alveolar pattern [24] | Densely packed, small, round cells lining septations that resemble fetal alveoli [4] | Differentiated from high-grade soft tissue sarcomas by the presence of skeletal muscle proteins on immunohistochemistry [3,4] | NA |
3. RMS Classification
4. RMS Epidemiology
5. RMS Treatment
5.1. Temsirolimus
5.2. Vincristine
| Treatment | Clinical Trial Phase | Reference |
|---|---|---|
| Ifosfamide/vinorelbine | III | [84] |
| Ifosfamide/doxorubicin | III | [85] |
| Vincristine, dactinomycin, and cyclophosphamide or vincristine, dactinomycin, and cyclophosphamide/vincristine and irinotecan | III | [86] |
| Trabectedin | II | [87] |
| Irinotecan or vincristine and irinotecan | II | [13] |
| Vincristine, doxorubicin, and cyclophosphamide/ifosfamide and etoposide | II | [88] |
| Vincristine, irinotecan, and temozolomide | N/A | [89] |
| Vincristine and irinotecan + vincristine, doxorubicin, and cyclophosphamide/ ifosfamide and etoposide + temozolomide | II | [71] |
| Temozolomide + irinotecan | Preclinical (mouse models) | [90] |
5.3. Doxorubicin
5.4. Actinomycin D (Dactinomycin)
5.5. Cyclophosphamide
5.6. Ifosfamide
5.7. Melphalan
5.8. Etoposide
5.9. Irinotecan
5.10. Volasertib
6. Apoptosis—General Considerations
6.1. Avoidance of Apoptosis by RMS Cells
6.2. Antineoplastic Agents Targeting the Apoptosis Pathway in RMS
7. Autophagy Process
Targeting Autophagy to Increase the Effectiveness of Chemotherapy in Rhabdomyosarcoma
8. General Concepts of Unfolded Protein Response and Its Link to RMS
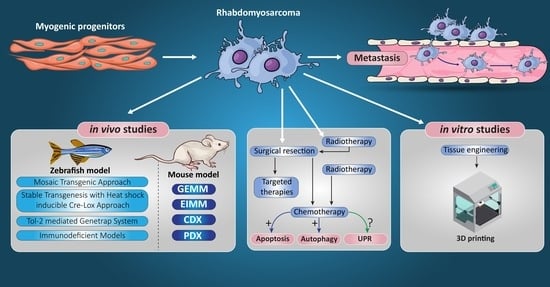
9. RMS In Vivo Models
9.1. RMS In Vivo Mouse Models
9.1.1. Genetically Engineered Mouse Models (GEMMs)
9.1.2. Environmental-Induced Mouse Models (EIMMs)
9.1.3. Cell-Line-Derived Xenograft Mouse Models (CDXs)
9.1.4. Patient-Derived Xenograft Mouse Models (PDXs)
9.2. Zebrafish Models to Study RMS
9.2.1. Mosaic Transgenic Approach
9.2.2. Stable Transgenesis with Heat-Shock-Inducible Cre-LoxP Approach
9.2.3. Tol-2-Mediated Gene Trap System
9.2.4. Immunodeficient/Compromised Zebrafish Models to Study RMS
10. Tissue Engineering Basics
Application of 3D Printing in Muscles and Rhabdomyosarcoma Tissue Engineering and Treatment
11. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 17-DMAG | 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin |
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| 3-MA | 3-Methyladenine |
| 4D | Four-dimensional |
| 5-FU | 5-fluorouracil |
| AD | Actinomycin D |
| ADP | Adenosine diphosphate |
| ADR | Adriamycin |
| AF | Anti-Fas death receptor antibody |
| AIF | Apoptotic-inducing factor |
| AIM | ATG8-interacting motif |
| ALK | Anaplastic lymphoma kinase |
| AM | Additive manufacturing |
| AMP | Adenosine monophosphate |
| AMPK | AMP-activated protein kinase |
| ARMS | Alveolar rhabdomyosarcoma |
| ATF4 | Transcription factor 4 |
| ATF6 | Activating transcription factor 6 |
| ATG | Autophagy related genes |
| ATP | Adenosine triphosphate |
| BAG3 | Bcl-2-associated athanogene 3 |
| BD | Benzenediazonium sulphate |
| cdh15 | Cadherin 15 |
| CDK | Cyclin-dependent kinase |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CDXs | Cell-line derived xenograft mouse models |
| CM | Carbonaceous material |
| CMA | Chaperone-mediated autophagy |
| CMP | Chaperone-mediated autophagy |
| COG-STS | Children’s oncology group soft tissue sarcoma |
| CPT | Camptothecin |
| CPX | Ciclopirox olamine |
| CQ | Chloroquine |
| CSC | Cancer stem cells |
| CT | Computed tomography |
| CYP450 | Cytochrome P450 |
| DAPI | 4′,6-diamidino-2-phenylindole |
| DMA | Dynamic mechanical analysis |
| DNA | Deoxyribonucleic acid |
| Dox | Doxorubicin |
| ECM | Extracellular matrix |
| EIMMs | Environmentally induced mouse models |
| EMT | Epithelial to mesenchymal transition |
| ER | Endoplasmic reticulum |
| ERAD | ER associated degradation machinery |
| ERK1/2 | Extracellular signal-regulated kinase 1/2 |
| ERMS | Embryonal rhabdomyosarcoma |
| FACS | Fluorescence-activated cell sorting |
| FADD | Fas-associated protein with death domain |
| FAPs | Fibro-adipogenic progenitors |
| FBS | Fetal bovine serum |
| FCS | Fetal calf serum |
| FDA | Food and drug administration |
| FDG PET scan | Fluorodeoxyglucose (FDG)-positron emission tomography (PET) |
| FFF | Fused filament fabrication |
| FFS | Failure free survival |
| FG + | PAX3-FOXO1 fusion genes positive |
| FGFR4 | Fibroblast growth factor receptor 4 |
| FN | Fusion-negative |
| FOXO1 | Forkhead box protein O1 |
| FP | Fusion-positive |
| GBM | Glioblastoma multiforme |
| GDP | Guanosine diphosphate |
| GelMA | Gelatin-methacryloyl |
| GEMMs | Genetically engineered mouse models |
| GFP | Green fluorescent protein |
| GHPA | Gelatin-hydroxyphenyl propionic acid |
| GO | Graphene oxide |
| Grp78 | ER chaperone glucose regulated protein 78 |
| GSK3 | Glycogen synthase kinase 3 |
| GSTP1 | Glutathione S-transferase P1 |
| GTP | Guanosine-5′-triphosphate |
| HandE | Hematoxylin and eosin |
| HAS | Human serum albumin |
| HDAC6 | Histone deacetylase 6 |
| HDACIs | Histone deacetylases inhibitors |
| HDACs | Histone deacetylases |
| Hgf | Hepatocyte growth factor |
| Hh | Hedgehog |
| HMG-CoA | β-Hydroxy β-methylglutaryl-CoA |
| hRD | Human rhabdomyosarcoma |
| HSMM | Human skeletal muscle myoblast |
| HSR | Heat shock response |
| ICE | Carboplatin, Epirubicin, and Etoposide |
| ICE | Carboplatin, Epirubicin, and Etoposide |
| ID | Ifosfamide/doxorubicin |
| IE | Ifosfamide/etoposide |
| IGF1R | Insulin growth factor 1 receptor |
| IGF2 | Insulin growth factor 2 |
| IgG1 | Immunoglobulin G1 |
| il2rga | Interlukin-2 receptor gamma a |
| IL-6 | Interleukin-6 |
| IMRT | Intensity modulated radiation therapy |
| IRE | Inositol requiring enzyme |
| IRE1α | Inositol requiring enzyme 1α |
| IRS | Intergroup rhabdomyosarcoma study |
| IVA | Ifosfamide, vincristine, and actinomycin D |
| JAK/STAT | Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathway |
| KEAP1 | Kelch like-ECH-associated protein 1 |
| KRAS | The gene Kirsten rat sarcoma viral oncogene homolog |
| LAMP2A | Lysosomal chaperone-mediated autophagy receptor |
| LC3 | Light chain 3 |
| LFS | Li-Fraumeni syndrome |
| LIR | LC3-interacting region |
| MAPK | Mitogen-activated protein kinase |
| MDM2 | Murine double minute 2 |
| MDR | Multidrug resistance |
| MEV | Mevalonate |
| miRs | Muscle-specific microRNAs |
| MMP9 | The matrix metalloproteinase 9 |
| MRI | Magnetic resonance imaging |
| mRNA | Messenger RNA |
| mTOR | Mammalian target of rapamycin |
| MTX | Methotrexate |
| myf5 | Myogenic factor 5 |
| MyoD1 | Myogenic differentiation 1 |
| NF1 | Neurofibromatosis type I |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| NSCLCs | Non-small-cell lung cancers |
| OZO-H | 4-phenyl-1,3,2-oxathiazolylium-5-oleate |
| PARP | Poly (ADP-ribose) polymerase |
| PAS | Pre-autophagosomal structure |
| PBT | Proton beam therapy |
| PCL | Poly(ɛ-caprolactone) |
| PDGF-β | Platelet-derived growth factor β |
| PDGFR | Platelet-derived growth factor receptor |
| PDXs | Patient-derived xenograft mouse models |
| PERK | Protein kinase R like endoplasmic reticulum kinase |
| P-gp | P-glycoprotein |
| PI3K | Phosphoinositide 3-kinase |
| PI3P | Phosphatidylinositol 3-phosphate |
| PLK1 | Polo-like kinase-1 |
| PLKs | Polo-like kinases |
| PNET | Primitive neuroectodermal tumors |
| PQC | Protein quality control system |
| prkdc | Protein kinase DNA-activated catalytic polypeptide |
| PRMS | Pleomorphic rhabdomyosarcoma |
| PVA | Poly vinyl alcohol |
| RAC1 | Ras-related C3 botulinum toxin substrate 1 |
| rag2 | Recombination activating 2 |
| RIDD | Regulated IRE1 dependent decay |
| RMS | Rhabdomyosarcoma |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| ROS1 | c-ros oncogene 1 |
| RTCB | RNA 2′,3′-cyclic phosphate and 5′-OH ligase |
| RTKs | Receptor tyrosine kinases |
| S6K1 | Ribosomal protein S6 kinase 1 |
| SAHA | Suberoylanilide hydroxamic acid |
| SAM | Syngeneic allograft model |
| SAR | Structure–activity relationship |
| SDH | Silibinin di-hemisuccinate |
| shATG7 | shRNA sequence against ATG7 |
| SHH | Sonic hedgehog |
| SIOP | International Society of Pediatric Oncology |
| siRNA | Short interfering RNA or silencing RNA |
| SIRT | The mammalian Sirtuin |
| SLA | Stereolithography |
| Smac | Second mitochondria-derived activator of caspase |
| ST80 | The cytoplasmic histone deacetylase 6 inhibitor ST80 |
| tBID | Truncated BID |
| TGF | Transforming growth factor |
| TMZ | Temozolomide |
| TNBC | Triple negative breast cancer |
| TNFR | Tumor necrosis factor receptor |
| TPCs | Tumor propagating cells |
| TRAILR | TNF-related apoptosis-inducing ligand receptor |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| Tv6 | Tenovin-6 |
| ULK1/2 | Unc-51 Like autophagy activating kinase 1/2 |
| UPR | The unfolded protein response |
| UPS | The ubiquitin-proteasome system |
| UTR | Untranslated region |
| UV | Ultraviolet |
| VAC | Vincristine, actinomycin D, and cyclophosphamide |
| VAI | Vincristine and dactinomycin, ifosfamide |
| V-ATPase | Vacuolar H+ ATPase |
| VEGF | Vascular endothelial growth factor |
| VI | Vincristine and irinotecan |
| VIE | Vincristine, ifosfamide, and etoposide |
| VIT | Vincristine, irinotecan, and temozolomide |
| VM | Vincristine/melphalan |
| VML | Volumetric muscle loss |
| Vps | Vacuolar protein sorting |
| VTC | Vincristine, topotecan, and cyclophosphamide |
| WHO | The World Health Organization |
| XBP1 | X-box-binding protein 1 |
| XIAP | X-chromosome linked IAP protein |
References
- Shern, J.F.; Yohe, M.E.; Khan, J. Pediatric rhabdomyosarcoma. Crit. Rev. Oncog. 2015, 20, 227–243. [Google Scholar] [CrossRef]
- Ognjanovic, S.; Linabery, A.M.; Charbonneau, B.; Ross, J.A. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975–2005. Cancer 2009, 115, 4218–4226. [Google Scholar] [CrossRef]
- Ruiz-Mesa, C.; Goldberg, J.M.; Coronado Munoz, A.J.; Dumont, S.N.; Trent, J.C. Rhabdomyosarcoma in adults: New perspectives on therapy. Curr. Treat. Options Oncol. 2015, 16, 27. [Google Scholar] [CrossRef]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Primers 2019, 5, 1. [Google Scholar] [CrossRef]
- Crist, W.M.; Anderson, J.R.; Meza, J.L.; Fryer, C.; Raney, R.B.; Ruymann, F.B.; Breneman, J.; Qualman, S.J.; Wiener, E.; Wharam, M.; et al. Intergroup rhabdomyosarcoma study-IV: Results for patients with nonmetastatic disease. J. Clin. Oncol. 2001, 19, 3091–3102. [Google Scholar] [CrossRef]
- Arndt, C.A.; Stoner, J.A.; Hawkins, D.S.; Rodeberg, D.A.; Hayes-Jordan, A.A.; Paidas, C.N.; Parham, D.M.; Teot, L.A.; Wharam, M.D.; Breneman, J.C.; et al. Vincristine, actinomycin, and cyclophosphamide compared with vincristine, actinomycin, and cyclophosphamide alternating with vincristine, topotecan, and cyclophosphamide for intermediate-risk rhabdomyosarcoma: Children’s oncology group study D9803. J. Clin. Oncol. 2009, 27, 5182–5188. [Google Scholar] [CrossRef]
- Stevens, M.C.; Rey, A.; Bouvet, N.; Ellershaw, C.; Flamant, F.; Habrand, J.L.; Marsden, H.B.; Martelli, H.; Sanchez de Toledo, J.; Spicer, R.D.; et al. Treatment of nonmetastatic rhabdomyosarcoma in childhood and adolescence: Third study of the International Society of Paediatric Oncology--SIOP Malignant Mesenchymal Tumor 89. J. Clin. Oncol. 2005, 23, 2618–2628. [Google Scholar] [CrossRef]
- Oberlin, O.; Rey, A.; Sanchez de Toledo, J.; Martelli, H.; Jenney, M.E.; Scopinaro, M.; Bergeron, C.; Merks, J.H.; Bouvet, N.; Ellershaw, C.; et al. Randomized comparison of intensified six-drug versus standard three-drug chemotherapy for high-risk nonmetastatic rhabdomyosarcoma and other chemotherapy-sensitive childhood soft tissue sarcomas: Long-term results from the International Society of Pediatric Oncology MMT95 study. J. Clin. Oncol. 2012, 30, 2457–2465. [Google Scholar] [CrossRef]
- Ferrari, A.; Casanova, M.; Collini, P.; Meazza, C.; Luksch, R.; Massimino, M.; Cefalo, G.; Terenziani, M.; Spreafico, F.; Catania, S.; et al. Adult-type soft tissue sarcomas in pediatric-age patients: Experience at the Istituto Nazionale Tumori in Milan. J. Clin. Oncol. 2005, 23, 4021–4030. [Google Scholar] [CrossRef]
- Blakely, M.L.; Andrassy, R.J.; Raney, R.B.; Anderson, J.R.; Wiener, E.S.; Rodeberg, D.A.; Paidas, C.N.; Lobe, T.E.; Crist, W.M.; Intergroup Rhabdomyosarcoma Studies, I.t.I.V. Prognostic factors and surgical treatment guidelines for children with rhabdomyosarcoma of the perineum or anus: A report of Intergroup Rhabdomyosarcoma Studies I through IV, 1972 through 1997. J. Pediatr. Surg. 2003, 38, 347–353. [Google Scholar] [CrossRef]
- Crist, W.M.; Garnsey, L.; Beltangady, M.S.; Gehan, E.; Ruymann, F.; Webber, B.; Hays, D.M.; Wharam, M.; Maurer, H.M. Prognosis in children with rhabdomyosarcoma: A report of the intergroup rhabdomyosarcoma studies I and II. Intergroup Rhabdomyosarcoma Committee. J. Clin. Oncol. 1990, 8, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Pappo, A.S.; Anderson, J.R.; Crist, W.M.; Wharam, M.D.; Breitfeld, P.P.; Hawkins, D.; Raney, R.B.; Womer, R.B.; Parham, D.M.; Qualman, S.J.; et al. Survival after relapse in children and adolescents with rhabdomyosarcoma: A report from the Intergroup Rhabdomyosarcoma Study Group. J. Clin. Oncol. 1999, 17, 3487–3493. [Google Scholar] [CrossRef]
- Pappo, A.S.; Lyden, E.; Breitfeld, P.; Donaldson, S.S.; Wiener, E.; Parham, D.; Crews, K.R.; Houghton, P.; Meyer, W.H.; Children’s Oncology, G. Two consecutive phase II window trials of irinotecan alone or in combination with vincristine for the treatment of metastatic rhabdomyosarcoma: The Children’s Oncology Group. J. Clin. Oncol. 2007, 25, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, D.S.; Spunt, S.L.; Skapek, S.X.; Committee, C.O.G.S.T.S. Children’s Oncology Group’s 2013 blueprint for research: Soft tissue sarcomas. Pediatr. Blood Cancer 2013, 60, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Crist, W.; Gehan, E.A.; Ragab, A.H.; Dickman, P.S.; Donaldson, S.S.; Fryer, C.; Hammond, D.; Hays, D.M.; Herrmann, J.; Heyn, R.; et al. The third intergroup rhabdomyosarcoma study. J. Clin. Oncol. 1995, 13, 610–630. [Google Scholar] [CrossRef]
- Sultan, I.; Qaddoumi, I.; Yaser, S.; Rodriguez-Galindo, C.; Ferrari, A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: An analysis of 2,600 patients. J. Clin. Oncol. 2009, 27, 3391–3397. [Google Scholar] [CrossRef]
- Ferrari, A.; Dileo, P.; Casanova, M.; Bertulli, R.; Meazza, C.; Gandola, L.; Navarria, P.; Collini, P.; Gronchi, A.; Olmi, P.; et al. Rhabdomyosarcoma in adults. A retrospective analysis of 171 patients treated at a single institution. Cancer 2003, 98, 571–580. [Google Scholar] [CrossRef]
- Noujaim, J.; Thway, K.; Jones, R.L.; Miah, A.; Khabra, K.; Langer, R.; Kasper, B.; Judson, I.; Benson, C.; Kollar, A. Adult pleomorphic rhabdomyosarcoma: A multicentre retrospective study. Anticancer Res. 2015, 35, 6213–6217. [Google Scholar]
- Raney, R.B.; Maurer, H.M.; Anderson, J.R.; Andrassy, R.J.; Donaldson, S.S.; Qualman, S.J.; Wharam, M.D.; Wiener, E.S.; Crist, W.M. The Intergroup Rhabdomyosarcoma Study Group (IRSG): Major lessons from the IRS-I through IRS-IV studies as background for the current IRS-V treatment protocols. Sarcoma 2001, 5, 9–15. [Google Scholar] [CrossRef]
- Kallen, M.E.; Hornick, J.L. The 2020 WHO Classification: What’s new in soft tissue tumor pathology? Am. J. Surg. Pathol. 2022, 45, e1–e23. [Google Scholar] [CrossRef]
- Pappo, A.S.; Shapiro, D.N.; Crist, W.M.; Maurer, H.M. Biology and therapy of pediatric rhabdomyosarcoma. J. Clin. Oncol. 1995, 13, 2123–2139. [Google Scholar] [CrossRef]
- Kelly, K.M.; Womer, R.B.; Sorensen, P.H.; Xiong, Q.B.; Barr, F.G. Common and variant gene fusions predict distinct clinical phenotypes in rhabdomyosarcoma. J. Clin. Oncol. 1997, 15, 1831–1836. [Google Scholar] [CrossRef]
- Dasgupta, R.; Rodeberg, D.A. Update on rhabdomyosarcoma. Semin. Pediatr. Surg. 2012, 21, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Rudzinski, E.R.; Teot, L.A.; Anderson, J.R.; Moore, J.; Bridge, J.A.; Barr, F.G.; Gastier-Foster, J.M.; Skapek, S.X.; Hawkins, D.S.; Parham, D.M. Dense pattern of embryonal rhabdomyosarcoma, a lesion easily confused with alveolar rhabdomyosarcoma: A report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Am. J. Clin. Pathol. 2013, 140, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Malempati, S.; Hawkins, D.S. Rhabdomyosarcoma: Review of the Children’s Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatr. Blood Cancer 2012, 59, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Williamson, D.; Chisholm, J.; Wirapati, P.; Pierron, G.; Petel, F.; Concordet, J.P.; Thway, K.; Oberlin, O.; Pritchard-Jones, K.; et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol. 2012, 30, 1670–1677. [Google Scholar] [CrossRef]
- Furlong, M.A.; Mentzel, T.; Fanburg-Smith, J.C. Pleomorphic rhabdomyosarcoma in adults: A clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Mod. Pathol. 2001, 14, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Rudzinski, E.R.; Anderson, J.R.; Hawkins, D.S.; Skapek, S.X.; Parham, D.M.; Teot, L.A. The World Health Organization classification of skeletal muscle tumors in pediatric rhabdomyosarcoma: A report from the Children’s Oncology Group. Arch. Pathol. Lab. Med. 2015, 139, 1281–1287. [Google Scholar] [CrossRef]
- Enterline, H.T.; Horn, R.C., Jr. Alveolar rhabdomyosarcoma; a distinctive tumor type. Am. J. Clin. Pathol. 1958, 29, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Patton, R.B.; Horn, R.C., Jr. Rhabdomyosarcoma: Clinical and pathological features and comparison with human fetal and embryonal skeletal muscle. Surgery 1962, 52, 572–584. [Google Scholar]
- Toro, J.R.; Travis, L.B.; Wu, H.J.; Zhu, K.; Fletcher, C.D.; Devesa, S.S. Incidence patterns of soft tissue sarcomas, regardless of primary site, in the surveillance, epidemiology and end results program, 1978-2001: An analysis of 26,758 cases. Int. J. Cancer 2006, 119, 2922–2930. [Google Scholar] [CrossRef]
- Merlino, G.; Helman, L.J. Rhabdomyosarcoma-working out the pathways. Oncogene 1999, 18, 5340–5348. [Google Scholar] [CrossRef]
- Miller, R.W.; Young, J.L., Jr.; Novakovic, B. Childhood cancer. Cancer 1995, 75 (Suppl. S1), 395–405. [Google Scholar] [CrossRef] [PubMed]
- Stiller, C.A.; Parkin, D.M. International variations in the incidence of childhood soft-tissue sarcomas. Paediatr. Perinat. Epidemiol. 1994, 8, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Lychou, S.E.; Gustafsson, G.G.; Ljungman, G.E. Higher rates of metastatic disease may explain the declining trend in Swedish paediatric rhabdomyosarcoma survival rates. Acta Paediatr. 2016, 105, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A.; Kassira, N.; Cheung, M.C.; Koniaris, L.G.; Neville, H.L.; Sola, J.E. Rhabdomyosarcoma in children: A SEER population based study. J. Surg. Res. 2011, 170, e243–e251. [Google Scholar] [CrossRef]
- Grufferman, S.; Ruymann, F.; Ognjanovic, S.; Erhardt, E.B.; Maurer, H.M. Prenatal X-ray exposure and rhabdomyosarcoma in children: A report from the children’s oncology group. Cancer Epidemiol. Biomark. Prev. 2009, 18, 1271–1276. [Google Scholar] [CrossRef]
- Little, D.J.; Ballo, M.T.; Zagars, G.K.; Pisters, P.W.; Patel, S.R.; El-Naggar, A.K.; Garden, A.S.; Benjamin, R.S. Adult rhabdomyosarcoma: Outcome following multimodality treatment. Cancer 2002, 95, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Maurer, H.M.; Beltangady, M.; Gehan, E.A.; Crist, W.; Hammond, D.; Hays, D.M.; Heyn, R.; Lawrence, W.; Newton, W.; Ortega, J.; et al. The Intergroup Rhabdomyosarcoma Study-I. A final report. Cancer 1988, 61, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Maurer, H.M.; Gehan, E.A.; Beltangady, M.; Crist, W.; Dickman, P.S.; Donaldson, S.S.; Fryer, C.; Hammond, D.; Hays, D.M.; Herrmann, J.; et al. The Intergroup Rhabdomyosarcoma Study-II. Cancer 1993, 71, 1904–1922. [Google Scholar] [CrossRef]
- Borinstein, S.C.; Steppan, D.; Hayashi, M.; Loeb, D.M.; Isakoff, M.S.; Binitie, O.; Brohl, A.S.; Bridge, J.A.; Stavas, M.; Shinohara, E.T.; et al. Consensus and controversies regarding the treatment of rhabdomyosarcoma. Pediatric. Blood Cancer 2018, 65, e26809. [Google Scholar] [CrossRef]
- Gallego, S.; Bernabeu, D.; Garrido-Pontnou, M.; Guillen, G.; Hindi, N.; Juan-Ribelles, A.; Márquez, C.; Mata, C.; Orcajo, J.; Ramírez, G.; et al. GEIS-SEHOP clinical practice guidelines for the treatment of rhabdomyosarcoma. Clin. Transl. Oncol. 2021, 23, 2460–2473. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.; Rodeberg, D.; Breitfeld, P.P.; Raney, R.B.; Ullrich, F.; Donaldson, S. Does bladder preservation (as a surgical principle) lead to retaining bladder function in bladder/prostate rhabdomyosarcoma? Results from intergroup rhabdomyosarcoma study iv. J. Urol. 2004, 171, 2396–2403. [Google Scholar] [CrossRef]
- Chargari, C.; Haie-Meder, C.; Guérin, F.; Minard-Colin, V.; de Lambert, G.; Mazeron, R.; Escande, A.; Marsolat, F.; Dumas, I.; Deutsch, E.; et al. Brachytherapy Combined With Surgery for Conservative Treatment of Children With Bladder Neck and/or Prostate Rhabdomyosarcoma. Int. J. Radiat. Oncol. Biol. Phys. 2017, 98, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Rogers, T.N.; Seitz, G.; Fuchs, J.; Martelli, H.; Dasgupta, R.; Routh, J.C.; Hawkins, D.S.; Koscielniak, E.; Bisogno, G.; Rodeberg, D.A. Surgical management of paratesticular rhabdomyosarcoma: A consensus opinion from the Children’s Oncology Group, European paediatric Soft tissue sarcoma Study Group, and the Cooperative Weichteilsarkom Studiengruppe. Pediatr. Blood Cancer 2021, 68, e28938. [Google Scholar] [CrossRef]
- Debie, P.; Hernot, S. Emerging fluorescent molecular tracers to guide intra-operative surgical decision-making. Front. Pharmacol. 2019, 10, 510. [Google Scholar] [CrossRef]
- Rijs, Z.; Jeremiasse, B.; Shifai, N.; Gelderblom, H.; Sier, C.F.; Vahrmeijer, A.L.; van Leeuwen, F.W.; van der Steeg, A.F.; van de Sande, M.A. Introducing fluorescence-guided surgery for pediatric Ewing, osteo-, and rhabdomyosarcomas: A literature review. Biomedicines 2021, 9, 1388. [Google Scholar] [CrossRef]
- Hayes-Jordan, A.; Doherty, D.K.; West, S.D.; Raney, R.B.; Blakely, M.L.; Cox, C.S., Jr.; Andrassy, R.J.; Lally, K.P. Outcome after surgical resection of recurrent rhabdomyosarcoma. J. Pediatr. Surg. 2006, 41, 633–638. [Google Scholar] [CrossRef]
- Hensle, T.W.; Chang, D.T. Reconstructive surgery for children with pelvic rhabdomyosarcoma. Urol. Clin. N. Am. 2000, 27, 489–502. [Google Scholar] [CrossRef]
- Macedo, A., Jr.; Damázio, E.; Bacelar, H.; Rondon, A.; Ottoni, S.; Liguori, R.; Garrone, G.; Leslie, B.; Ortiz, V. Ten years’ minimum follow-up with the ileal continent catheterizable reservoir: A test of time. J. Pediatr. Urol. 2013, 9, 272–277. [Google Scholar] [CrossRef]
- Wang, J.; Chai, S.; Wang, R.; Zheng, G.; Zhang, K.; Huo, B.; Huo, X.; Jiang, Y.; Ji, Z.; Jiang, P.; et al. Expert consensus on computed tomography-assisted three-dimensional-printed coplanar template guidance for interstitial permanent radioactive. J. Cancer Res. Ther. 2019, 15, 1430–1434. [Google Scholar] [CrossRef] [PubMed]
- Réguerre, Y.; Martelli, H.; Rey, A.; Rogers, T.; Gaze, M.; Ben Arush, M.W.; Devalck, C.; Oberlin, O.; Stevens, M.; Orbach, D. Local therapy is critical in localised pelvic rhabdomyosarcoma: Experience of the International Society of Pediatric Oncology Malignant Mesenchymal Tumor (SIOP-MMT) committee. Eur. J. Cancer 2012, 48, 2020–2027. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.L.; Elze, M.C.; Casanova, M.; Geoerger, B.; Gaze, M.N.; Minard-Colin, V.; McHugh, K.; van Rijn, R.R.; Kelsey, A.; Martelli, H.; et al. The Impact of Radiation Therapy in Children and Adolescents With Metastatic Rhabdomyosarcoma. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, 968–978. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.C.; Venkatramani, R.; Okcu, M.F.; Nuchtern, J.G.; Vasudevan, S.A.; Mahajan, A.; Rainusso, N.C.; Allen-Rhoades, W.; Chintagumpala, M.; Paulino, A.C. Local therapy to distant metastatic sites in stage IV rhabdomyosarcoma. Pediatr. Blood Cancer 2018, 65, e26859. [Google Scholar] [CrossRef]
- Schoot, R.A.; Saeed, P.; Freling, N.J.; Blank, L.E.; Pieters, B.R.; van der Grient, J.N.; Strackee, S.D.; Bras, J.; Caron, H.N.; Merks, J.H.; et al. Local Resection and Brachytherapy for Primary Orbital Rhabdomyosarcoma: Outcome and Failure Pattern Analysis. Ophthalmic. Plast. Reconstr. Surg. 2016, 32, 354–360. [Google Scholar] [CrossRef]
- Ge, X.; Ma, J.; Dai, H.; Ren, L.; Li, Q.; Shi, J. Clinical research on the treatment effects of radioactive (125)I seeds interstitial brachytherapy on children with primary orbital rhabdomyosarcoma. Med. Oncol. 2014, 31, 27. [Google Scholar] [CrossRef]
- Bramwell, V.H. Management of advanced adult soft tissue sarcoma. Sarcoma 2003, 7, 43–55. [Google Scholar] [CrossRef]
- Heyn, R.M.; Holland, R.; Newton, W.A., Jr.; Tefft, M.; Breslow, N.; Hartmann, J.R. The role of combined chemotherapy in the treatment of rhabdomyosarcoma in children. Cancer 1974, 34, 2128–2142. [Google Scholar] [CrossRef]
- Spunt, S.L.; Smith, L.M.; Ruymann, F.B.; Qualman, S.J.; Donaldson, S.S.; Rodeberg, D.A.; Anderson, J.R.; Crist, W.M.; Link, M.P. Cyclophosphamide dose intensification during induction therapy for intermediate-risk pediatric rhabdomyosarcoma is feasible but does not improve outcome: A report from the soft tissue sarcoma committee of the children’s oncology group. Clin. Cancer Res. 2004, 10, 6072–6079. [Google Scholar] [CrossRef]
- Mascarenhas, L.; Chi, Y.Y.; Hingorani, P.; Anderson, J.R.; Lyden, E.R.; Rodeberg, D.A.; Indelicato, D.J.; Kao, S.C.; Dasgupta, R.; Spunt, S.L.; et al. Randomized phase II trial of bevacizumab or temsirolimus in combination with chemotherapy for first relapse rhabdomyosarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 2866–2874. [Google Scholar] [CrossRef]
- Mandell, L.; Ghavimi, F.; Peretz, T.; LaQuaglia, M.; Exelby, P. Radiocurability of microscopic disease in childhood rhabdomyosarcoma with radiation doses less than 4,000 cGy. J. Clin. Oncol. 1990, 8, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Oberlin, O.; Rey, A.; Lyden, E.; Bisogno, G.; Stevens, M.C.; Meyer, W.H.; Carli, M.; Anderson, J.R. Prognostic factors in metastatic rhabdomyosarcomas: Results of a pooled analysis from United States and European Cooperative Groups. J. Clin. Oncol. 2008, 26, 2384–2389. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, G.; Ferrari, A.; Prete, A.; Messina, C.; Basso, E.; Cecchetto, G.; Indolfi, P.; Scarzello, G.; D’Angelo, P.; De Sio, L.; et al. Sequential high-dose chemotherapy for children with metastatic rhabdomyosarcoma. Eur. J. Cancer 2009, 45, 3035–3041. [Google Scholar] [CrossRef]
- Sleijfer, S.; Ray-Coquard, I.; Papai, Z.; Le Cesne, A.; Scurr, M.; Schöffski, P.; Collin, F.; Pandite, L.; Marreaud, S.; De Brauwer, A. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: A phase II study from the European Organisation for Research and Treatment of Cancer–Soft Tissue and Bone Sarcoma Group (EORTC study 62043). J. Clin. Oncol. 2009, 27, 3126–3132. [Google Scholar] [PubMed]
- Van Der Graaf, W.T.; Blay, J.-Y.; Chawla, S.P.; Kim, D.-W.; Bui-Nguyen, B.; Casali, P.G.; Schöffski, P.; Aglietta, M.; Staddon, A.P.; Beppu, Y. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): A randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012, 379, 1879–1886. [Google Scholar] [CrossRef]
- Kim, A.; Widemann, B.C.; Krailo, M.; Jayaprakash, N.; Fox, E.; Weigel, B.; Blaney, S.M. Phase 2 trial of sorafenib in children and young adults with refractory solid tumors: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2015, 62, 1562–1566. [Google Scholar] [CrossRef]
- Santoro, A.; Comandone, A.; Basso, U.; Soto Parra, H.; De Sanctis, R.; Stroppa, E.; Marcon, I.; Giordano, L.; Lutman, F.; Boglione, A. Phase II prospective study with sorafenib in advanced soft tissue sarcomas after anthracycline-based therapy. Ann. Oncol. 2013, 24, 1093–1098. [Google Scholar] [CrossRef]
- Schöffski, P.; Wozniak, A.; Leahy, M.G.; Aamdal, S.; Rutkowski, P.; Bauer, S.; Richter, S.; Grünwald, V.; Debiec-Rychter, M.; Sciot, R. The tyrosine kinase inhibitor crizotinib does not have clinically meaningful activity in heavily pre-treated patients with advanced alveolar rhabdomyosarcoma with FOXO rearrangement: European Organisation for Research and Treatment of Cancer phase 2 trial 90101 ‘CREATE’. Eur. J. Cancer 2018, 94, 156–167. [Google Scholar]
- Geoerger, B.; Kieran, M.W.; Grupp, S.; Perek, D.; Clancy, J.; Krygowski, M.; Ananthakrishnan, R.; Boni, J.P.; Berkenblit, A.; Spunt, S.L. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur. J. Cancer 2012, 48, 253–262. [Google Scholar] [CrossRef]
- Weigel, B.; Malempati, S.; Reid, J.M.; Voss, S.D.; Cho, S.Y.; Chen, H.X.; Krailo, M.; Villaluna, D.; Adamson, P.C.; Blaney, S.M. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 452–456. [Google Scholar] [CrossRef]
- Malempati, S.; Weigel, B.J.; Chi, Y.Y.; Tian, J.; Anderson, J.R.; Parham, D.M.; Teot, L.A.; Rodeberg, D.A.; Yock, T.I.; Shulkin, B.L. The addition of cixutumumab or temozolomide to intensive multiagent chemotherapy is feasible but does not improve outcome for patients with metastatic rhabdomyosarcoma: A report from the Children’s Oncology Group. Cancer 2019, 125, 290–297. [Google Scholar] [CrossRef]
- Vardanyan, R. Chapter 10—Classes of Piperidine-Based Drugs. In Piperidine-Based Drug Discovery; Vardanyan, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 299–332. [Google Scholar] [CrossRef]
- Cai, P.; Tsao, R.; Ruppen, M.E. In vitro metabolic study of temsirolimus: Preparation, isolation, and identification of the metabolites. Drug Metabo. Dispos. 2007, 35, 1554–1563. [Google Scholar] [CrossRef]
- Proud, C.G. mTOR and its downstream targets. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: Waltham, MA, USA, 2013; pp. 194–199. [Google Scholar] [CrossRef]
- Moudi, M.; Go, R.; Yien, C.Y.; Nazre, M. Vinca alkaloids. Int. J. Prev. Med. 2013, 4, 1231–1235. [Google Scholar]
- Pellegrini, F.; Budman, D.R. Review: Tubulin function, action of antitubulin drugs, and new drug development. Cancer Investig. 2005, 23, 264–273. [Google Scholar] [CrossRef]
- Kerckhove, N.; Collin, A.; Condé, S.; Chaleteix, C.; Pezet, D.; Balayssac, D. Long-term effects, pathophysiological mechanisms, and risk factors of chemotherapy-induced peripheral neuropathies: A comprehensive literature review. Front. Pharmacol. 2017, 8, 86. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, J.; Xiao, M.; Li, W.; Miller, D.D. An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm. Res. 2012, 29, 2943–2971. [Google Scholar] [CrossRef]
- Bates, D.; Eastman, A. Microtubule destabilising agents: Far more than just antimitotic anticancer drugs. Br. J. Clin. Pharmacol. 2017, 83, 255–268. [Google Scholar] [CrossRef]
- Mittal, B.; Tulsyan, S.; Kumar, S.; Mittal, R.D.; Agarwal, G. Chapter Four—Cytochrome P450 in cancer susceptibility and treatment. Adv. Clin. Chem. 2015, 71, 77–139. [Google Scholar]
- Nabors, L.B.; Surboeck, B.; Grisold, W. Chapter 14—Complications from pharmacotherapy. In Handbook of Clinical Neurology; Berger, M.S., Weller, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 134, pp. 235–250. [Google Scholar]
- Agrawal, K. Vincristine. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–4. [Google Scholar] [CrossRef]
- Martino, E.; Casamassima, G.; Castiglione, S.; Cellupica, E.; Pantalone, S.; Papagni, F.; Rui, M.; Siciliano, A.M.; Collina, S. Vinca alkaloids and analogues as anti-cancer agents: Looking back, peering ahead. Bioorganic Med. Chem. Lett. 2018, 28, 2816–2826. [Google Scholar] [CrossRef]
- Bisogno, G.; De Salvo, G.L.; Bergeron, C.; Melcón, S.G.; Merks, J.H.; Kelsey, A.; Martelli, H.; Minard-Colin, V.; Orbach, D.; Glosli, H. Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 1566–1575. [Google Scholar] [CrossRef]
- Bisogno, G.; Jenney, M.; Bergeron, C.; Melcón, S.G.; Ferrari, A.; Oberlin, O.; Carli, M.; Stevens, M.; Kelsey, A.; De Paoli, A. Addition of dose-intensified doxorubicin to standard chemotherapy for rhabdomyosarcoma (EpSSG RMS 2005): A multicentre, open-label, randomised controlled, phase 3 trial. Lancet Oncol. 2018, 19, 1061–1071. [Google Scholar] [CrossRef]
- Hawkins, D.S.; Chi, Y.-Y.; Anderson, J.R.; Tian, J.; Arndt, C.A.; Bomgaars, L.; Donaldson, S.S.; Hayes-Jordan, A.; Mascarenhas, L.; McCarville, M.B. Addition of vincristine and irinotecan to vincristine, dactinomycin, and cyclophosphamide does not improve outcome for intermediate-risk rhabdomyosarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2018, 36, 2770–2777. [Google Scholar] [CrossRef]
- Baruchel, S.; Pappo, A.; Krailo, M.; Baker, K.S.; Wu, B.; Villaluna, D.; Lee-Scott, M.; Adamson, P.C.; Blaney, S.M. A phase 2 trial of trabectedin in children with recurrent rhabdomyosarcoma, Ewing sarcoma and non-rhabdomyosarcoma soft tissue sarcomas: A report from the Children’s Oncology Group. Eur. J. Cancer 2012, 48, 579–585. [Google Scholar] [CrossRef]
- Weigel, B.J.; Lyden, E.; Anderson, J.R.; Meyer, W.H.; Parham, D.M.; Rodeberg, D.A.; Michalski, J.M.; Hawkins, D.S.; Arndt, C.A. Intensive multiagent therapy, including dose-compressed cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, irinotecan, and radiation, in patients with high-risk rhabdomyosarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2016, 34, 117. [Google Scholar] [CrossRef]
- Setty, B.A.; Stanek, J.R.; Mascarenhas, L.; Miller, A.; Bagatell, R.; Okcu, F.; Nicholls, L.; Lysecki, D.; Gupta, A.A. VIncristine, irinotecan, and temozolomide in children and adolescents with relapsed rhabdomyosarcoma. Pediatr. Blood Cancer 2018, 65, e26728. [Google Scholar] [CrossRef]
- Igarashi, K.; Kawaguchi, K.; Kiyuna, T.; Murakami, T.; Miwa, S.; Nelson, S.D.; Dry, S.M.; Li, Y.; Singh, A.S.; Kimura, H. Temozolomide combined with irinotecan caused regression in an adult pleomorphic rhabdomyosarcoma patient-derived orthotopic xenograft (PDOX) nude-mouse model. Oncotarget 2017, 8, 75874. [Google Scholar] [CrossRef]
- Agrawal, K. Doxorubicin. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–5. [Google Scholar] [CrossRef]
- Hena, S.; Znad, H. Chapter Six—Membrane bioreactor for pharmaceuticals and personal care products removal from wastewater. In Comprehensive Analytical Chemistry; Chormey, D.S., Bakırdere, S., Turan, N.B., Engin, G.Ö., Eds.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 81, pp. 201–256. [Google Scholar]
- Rubin, E.H.; Hait, W.N. Drugs that target DNA topoisomerases. In Holland-Frei Cancer Medicine 8, 8th ed.; Hong, W.K., Best, R.C., Hait, W.N., Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Holland, J.F., Frei, E., Eds.; PMPH USA, Ltd.: Shelton, CT, USA, 2010; pp. 645–653. [Google Scholar]
- Prakash, V.; Timasheff, S.N. Mechanism of interaction of vinca alkaloids with tubulin: Catharanthine and vindoline. Biochemistry 1991, 30, 873–880. [Google Scholar] [CrossRef]
- Martins-Teixeira, M.B.; Carvalho, I. Antitumour anthracyclines: Progress and perspectives. ChemMedChem 2020, 15, 933–948. [Google Scholar] [CrossRef]
- Kwok, K.K.; Vincent, E.C.; Gibson, J.N. 36—Antineoplastic Drugs. In Pharmacology and Therapeutics for Dentistry, 7th ed.; Dowd, F.J., Johnson, B.S., Mariotti, A.J., Eds.; Mosby: Maryland Heights, MO, USA, 2017; pp. 530–562. [Google Scholar] [CrossRef]
- Avendaño, C.; Menéndez, J.C. Chapter 4—Anticancer Drugs Acting via Radical Species, Photosensitizers and Photodynamic Therapy of Cancer. In Medicinal Chemistry of Anticancer Drugs; Avendaño, C., Menéndez, J.C., Eds.; Elsevier: Amsterdam, The Netherlands, 2008; pp. 93–138. [Google Scholar] [CrossRef]
- Golomb, L.; Volarevic, S.; Oren, M. p53 and ribosome biogenesis stress: The essentials. FEBS Lett. 2014, 588, 2571–2579. [Google Scholar] [CrossRef]
- Ladds, M.; Laín, S. Small molecule activators of the p53 response. J. Mol. Cell Biol. 2019, 11, 245–254. [Google Scholar] [CrossRef]
- Chen, C.S.; Ho, D.R.; Chen, F.Y.; Chen, C.R.; Ke, Y.D.; Su, J.G. AKT mediates actinomycin D-induced p53 expression. Oncotarget 2014, 5, 693–703. [Google Scholar] [CrossRef]
- van Leeuwen, I.M.; Higgins, M.; Campbell, J.; Brown, C.J.; McCarthy, A.R.; Pirrie, L.; Westwood, N.J.; Laín, S. Mechanism-specific signatures for small-molecule p53 activators. Cell Cycle 2011, 10, 1590–1598. [Google Scholar] [CrossRef]
- Veal, G.J.; Cole, M.; Errington, J.; Parry, A.; Hale, J.; Pearson, A.D.J.; Howe, K.; Chisholm, J.C.; Beane, C.; Brennan, B.; et al. Pharmacokinetics of dactinomycin in a pediatric patient population: A United Kingdom Children’s Cancer Study Group. Clin. Cancer Res. 2005, 11, 5893–5899. [Google Scholar] [CrossRef]
- Melguizo, C.; Prados, J.; Fernández, J.E.; Vélez, C.; Alvarez, L.; Aránega, A. Actinomycin D causes multidrug resistance and differentiation in a human rhabdomyosarcoma cell line. Cell Mol. Biol. 1994, 40, 137–145. [Google Scholar]
- Rider, B.J. Cyclophosphamide. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–5. [Google Scholar] [CrossRef]
- Ralhan, R.; Kaur, J. Alkylating agents and cancer therapy. Expert. Opin. Ther. Pat. 2007, 17, 1061–1075. [Google Scholar] [CrossRef]
- Konstantinov, S.M.; Berger, M.R. Alkylating agents. In Encyclopedia of Molecular Pharmacology; Offermanns, S., Rosenthal, W., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 53–57. [Google Scholar] [CrossRef]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef]
- Giraud, B.; Hebert, G.; Deroussent, A.; Veal, G.J.; Vassal, G.; Paci, A. Oxazaphosphorines: New therapeutic strategies for an old class of drugs. Expert Opin. Drug Metab. Toxicol. 2010, 6, 919–938. [Google Scholar] [CrossRef]
- Shin, Y.J.; Kim, J.Y.; Moon, J.W.; You, R.M.; Park, J.Y.; Nam, J.H. Fatal Ifosfamide-induced metabolic encephalopathy in patients with recurrent epithelial ovarian cancer: Report of two cases. Cancer Res. Treat. 2011, 43, 260–263. [Google Scholar] [CrossRef]
- Kataria, P.S.; Kendre, P.P.; Patel, A.A. Ifosfamide-induced encephalopathy precipitated by aprepitant: A rarely manifested side effect of drug interaction. J. Pharmacol. Pharmacother. 2017, 8, 38–40. [Google Scholar] [CrossRef]
- Thirumaran, R.; Prendergast, G.C.; Gilman, P.B. Chapter 7—Cytotoxic Chemotherapy in Clinical Treatment of Cancer. In Cancer Immunother; Prendergast, G.C., Jaffee, E.M., Eds.; Academic Press: Burlington, VT, USA, 2007; pp. 101–116. [Google Scholar] [CrossRef]
- Dechant, K.L.; Brogden, R.N.; Pilkington, T.; Faulds, D. Ifosfamide/mesna. A review of its antineoplastic activity, pharmacokinetic properties and therapeutic efficacy in cancer. Drugs 1991, 42, 428–467. [Google Scholar] [CrossRef]
- Kerbusch, T.; de Kraker, J.; Keizer, H.J.; van Putten, J.W.; Groen, H.J.; Jansen, R.L.; Schellens, J.H.; Beijnen, J.H. Clinical pharmacokinetics and pharmacodynamics of ifosfamide and its metabolites. Clin. Pharmacokinet. 2001, 40, 41–62. [Google Scholar] [CrossRef]
- Falco, P.; Bringhen, S.; Avonto, I.; Gay, F.; Morabito, F.; Boccadoro, M.; Palumbo, A. Melphalan and its role in the management of patients with multiple myeloma. Expert. Rev. Anticancer Ther. 2007, 7, 945–957. [Google Scholar] [CrossRef]
- Osborne, M.R.; Lawley, P.D. Alkylation of DNA by melphalan with special reference to adenine derivatives and adenine-guanine cross-linking. Chem. Biol. Interact. 1993, 89, 49–60. [Google Scholar] [CrossRef]
- Sirohi, B.; Cunningham, D.; Powles, R.; Murphy, F.; Arkenau, T.; Norman, A.; Oates, J.; Wotherspoon, A.; Horwich, A. Long-term outcome of autologous stem-cell transplantation in relapsed or refractory Hodgkin’s lymphoma. Ann. Oncol. 2008, 19, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, E.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anticancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, A.; Zanetta, F.; Biamonti, G. Molecular mechanisms of etoposide. EXCLI J. 2015, 14, 95. [Google Scholar]
- Wagner, L.M. Fifteen years of irinotecan therapy for pediatric sarcoma: Where to next? Clin. Sarcoma Res. 2015, 5, 20. [Google Scholar] [CrossRef]
- Adams, D.J.; Wahl, M.L.; Flowers, J.L.; Sen, B.; Colvin, M.; Dewhirst, M.W.; Manikumar, G.; Wani, M.C. Camptothecin analogs with enhanced activity against human breast cancer cells. II. Impact of the tumor pH gradient. Cancer Chemother. Pharmacol. 2006, 57, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Zunino, F.; Dallavalleb, S.; Laccabuea, D.; Berettaa, G.; Merlinib, L.; Pratesi, G. Current status and perspectives in the development of camptothecins. Curr. Pharm. Des. 2002, 8, 2505–2520. [Google Scholar] [CrossRef]
- de Man, F.M.; Goey, A.K.L.; van Schaik, R.H.N.; Mathijssen, R.H.J.; Bins, S. Individualization of irinotecan treatment: A review of pharmacokinetics, pharmacodynamics, and pharmacogenetics. Clin. Pharmacokinet. 2018, 57, 1229–1254. [Google Scholar] [CrossRef]
- Xu, Y.; Villalona-Calero, M.A. Irinotecan: Mechanisms of tumor resistance and novel strategies for modulating its activity. Ann. Oncol. 2002, 13, 1841–1851. [Google Scholar] [CrossRef]
- Bailly, C. Irinotecan: 25 years of cancer treatment. Pharmacol. Res. 2019, 148, 104398. [Google Scholar] [CrossRef] [PubMed]
- Gjertsen, B.T.; Schöffski, P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia 2015, 29, 11–19. [Google Scholar] [CrossRef]
- Gutteridge, R.E.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 inhibitors in cancer therapy: From laboratory to clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Su, W.C.; Yen, C.J.; Hsu, C.H.; Su, W.P.; Yeh, K.H.; Lu, Y.S.; Cheng, A.L.; Huang, D.C.; Fritsch, H.; et al. A phase I study of two dosing schedules of volasertib (BI 6727), an intravenous polo-like kinase inhibitor, in patients with advanced solid malignancies. Br. J. Cancer 2014, 110, 2434–2440. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, D.; Impagnatiello, M.A.; Blaukopf, C.; Sommer, C.; Gerlich, D.W.; Roth, M.; Tontsch-Grunt, U.; Wernitznig, A.; Savarese, F.; Hofmann, M.H.; et al. Efficacy and mechanism of action of volasertib, a potent and selective inhibitor of Polo-like kinases, in preclinical models of acute myeloid leukemia. J. Pharmacol. Exp. Ther. 2015, 352, 579–589. [Google Scholar] [CrossRef]
- Janning, M.; Fiedler, W. Volasertib for the treatment of acute myeloid leukemia: A review of preclinical and clinical development. Future Oncol. 2014, 10, 1157–1165. [Google Scholar] [CrossRef]
- Gatz, S.A.; Aladowicz, E.; Casanova, M.; Chisholm, J.C.; Kearns, P.R.; Fulda, S.; Geoerger, B.; Schäfer, B.W.; Shipley, J.M. A Perspective on Polo-Like Kinase-1 Inhibition for the Treatment of Rhabdomyosarcomas. Front. Oncol. 2019, 9, 1271. [Google Scholar] [CrossRef]
- Macedo, A., Jr.; Ferreira, P.V.; Barroso, U., Jr.; Demarchi, G.T.; Garrone, G.; Liguori, R.; Caran, E.; Ortiz, V. Sexual function in teenagers after multimodal treatment of pelvic rhabdomyosarcoma: A preliminary report. J. Pediatr. Urol. 2010, 6, 605–608. [Google Scholar] [CrossRef]
- Gupta, A.A.; Chi, Y.Y.; Anderson, J.R.; Lyden, E.; Weigel, B.; Arndt, C.; Meyer, W.H.; Rosenberg, A.; Hawkins, D.S. Patterns of chemotherapy-induced toxicities and outcome in children and adolescents with metastatic rhabdomyosarcoma: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2017, 64, e26479. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Siri, M.; Dastghaib, S.; Zamani, M.; Rahmani-Kukia, N.; Geraylow, K.R.; Fakher, S.; Keshvarzi, F.; Mehrbod, P.; Ahmadi, M.; Mokarram, P.; et al. Autophagy, unfolded protein response, and neuropilin-1 cross-talk in SARS-CoV-2 infection: What can be learned from other coronaviruses. Int. J. Mol. Sci. 2021, 22, 5992. [Google Scholar] [CrossRef]
- Meier, P.; Finch, A.; Evan, G. Apoptosis in development. Nature 2000, 407, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef] [PubMed]
- Behrouj, H.; Seghatoleslam, A.; Mokarram, P.; Ghavami, S. Effect of casein kinase 1alpha inhibition on autophagy flux and the AKT/phospho-beta-catenin (S552) axis in HCT116, a RAS-mutated colorectal cancer cell line. Can. J. Physiol. Pharmacol. 2021, 99, 284–293. [Google Scholar] [CrossRef]
- Häcker, G. The morphology of apoptosis. Cell Tissue Res. 2000, 301, 5–17. [Google Scholar] [CrossRef]
- Hashemi, M.; Aftabi, S.; Moazeni-Roodi, A.; Sarani, H.; Wiechec, E.; Ghavami, S. Association of CASP8 polymorphisms and cancer susceptibility: A meta-analysis. Eur. J. Pharmacol. 2020, 881, 173201. [Google Scholar] [CrossRef]
- Goldar, S.; Khaniani, M.S.; Derakhshan, S.M.; Baradaran, B. Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac. J. Cancer Prevent. 2015, 16, 2129–2144. [Google Scholar] [CrossRef]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.R.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef]
- Ghavami, S.; Eshraghi, M.; Kadkhoda, K.; Mutawe, M.M.; Maddika, S.; Bay, G.H.; Wesselborg, S.; Halayko, A.J.; Klonisch, T.; Los, M. Role of BNIP3 in TNF-induced cell death--TNF upregulates BNIP3 expression. Biochim. Biophys. Acta 2009, 1793, 546–560. [Google Scholar] [CrossRef]
- Fulda, S.; Debatin, K.-M. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene 2006, 25, 4798–4811. [Google Scholar] [CrossRef]
- Sheikholeslami, K.; Ali Sher, A.; Lockman, S.; Kroft, D.; Ganjibakhsh, M.; Nejati-Koshki, K.; Shojaei, S.; Ghavami, S.; Rastegar, M. Simvastatin induces apoptosis in medulloblastoma brain tumor cells via mevalonate cascade prenylation substrates. Cancers 2019, 11, 994. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Cunnington, R.H.; Yeganeh, B.; Davies, J.J.; Rattan, S.G.; Bathe, K.; Kavosh, M.; Los, M.J.; Freed, D.H.; Klonisch, T.; et al. Autophagy regulates trans fatty acid-mediated apoptosis in primary cardiac myofibroblasts. Biochim. Biophys. Acta 2012, 1823, 2274–2286. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ghavami, S.; Sharma, P.; Yeganeh, B.; Ojo, O.O.; Jha, A.; Mutawe, M.M.; Kashani, H.H.; Los, M.J.; Klonisch, T.; Unruh, H.; et al. Airway mesenchymal cell death by mevalonate cascade inhibition: Integration of autophagy, unfolded protein response and apoptosis focusing on Bcl2 family proteins. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2014, 1843, 1259–1271. [Google Scholar] [CrossRef]
- Hashemi, M.; Karami, S.; Sarabandi, S.; Moazeni-Roodi, A.; Malecki, A.; Ghavami, S.; Wiechec, E. Association between PD-1 and PD-L1 polymorphisms and the risk of cancer: A meta-analysis of case-control studies. Cancers 2019, 11, 1150. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, M.; Moazeni-Roodi, A.; Ghavami, S. Association between CASP3 polymorphisms and overall cancer risk: A meta-analysis of case-control studies. J. Cell Biochem. 2018, 120, 7199–7210. [Google Scholar] [CrossRef]
- Li, H.; Zhu, H.; Xu, C.-J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Makin, G.; Dive, C. Apoptosis and cancer chemotherapy. Trends Cell Biol. 2001, 11, S22–S26. [Google Scholar] [CrossRef]
- Fernald, K.; Kurokawa, M. Evading apoptosis in cancer. Trends Cell Biol. 2013, 23, 620–633. [Google Scholar] [CrossRef]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Cao, L.; Yu, Y.; Bilke, S.; Walker, R.L.; Mayeenuddin, L.H.; Azorsa, D.O.; Yang, F.; Pineda, M.; Helman, L.J.; Meltzer, P.S. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 2010, 70, 6497–6508. [Google Scholar] [CrossRef]
- Taylor, J.G.T.; Cheuk, A.T.; Tsang, P.S.; Chung, J.-Y.; Song, Y.K.; Desai, K.; Yu, Y.; Chen, Q.-R.; Shah, K.; Youngblood, V.; et al. Identification of FGFR4-activating mutations in human rhabdomyosarcomas that promote metastasis in xenotransplanted models. J. Clin. Investig. 2009, 119, 3395–3407. [Google Scholar] [CrossRef] [PubMed]
- Packham, G.; White, E.L.; Eischen, C.M.; Yang, H.; Parganas, E.; Ihle, J.N.; Grillot, D.A.; Zambetti, G.P.; Nuñez, G.; Cleveland, J.L. Selective regulation of Bcl-XL by a Jak kinase-dependent pathway is bypassed in murine hematopoietic malignancies. Genes Dev. 1998, 12, 2475–2487. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Bertrand, F.E.; Ludwig, D.E.; Bäsecke, J.; Libra, M.; Stivala, F.; Milella, M.; Tafuri, A.; et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia 2008, 22, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Wesche, J.; Haglund, K.; Haugsten, E.M. Fibroblast growth factors and their receptors in cancer. Biochem. J. 2011, 437, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Taulli, R.; Scuoppo, C.; Bersani, F.; Accornero, P.; Forni, P.E.; Miretti, S.; Grinza, A.; Allegra, P.; Schmitt-Ney, M.; Crepaldi, T. Validation of met as a therapeutic target in alveolar and embryonal rhabdomyosarcoma. Cancer Res. 2006, 66, 4742–4749. [Google Scholar] [CrossRef]
- Wachtel, M.; Rakic, J.; Okoniewski, M.; Bode, P.; Niggli, F.; Schäfer, B.W. FGFR4 signaling couples to Bim and not Bmf to discriminate subsets of alveolar rhabdomyosarcoma cells. Int. J. Cancer 2014, 135, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Crose, L.E.S.; Etheridge, K.T.; Chen, C.; Belyea, B.; Talbot, L.J.; Bentley, R.C.; Linardic, C.M. FGFR4 blockade exerts distinct antitumorigenic effects in human embryonal versus alveolar rhabdomyosarcoma. Clin. Cancer Res. 2012, 18, 3780–3790. [Google Scholar] [CrossRef]
- Ehnman, M.; Missiaglia, E.; Folestad, E.; Selfe, J.; Strell, C.; Thway, K.; Brodin, B.; Pietras, K.; Shipley, J.; Östman, A.; et al. Distinct effects of ligand-induced PDGFRα and PDGFRβ signaling in the human rhabdomyosarcoma tumor cell and stroma cell compartments. Cancer Res. 2013, 73, 2139–2149. [Google Scholar] [CrossRef]
- Shukla, N.; Ameur, N.; Yilmaz, I.; Nafa, K.; Lau, C.-Y.; Marchetti, A.; Borsu, L.; Barr, F.G.; Ladanyi, M. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clin. Cancer Res. 2012, 18, 748–757. [Google Scholar] [CrossRef]
- Stewart, E.; McEvoy, J.; Wang, H.; Chen, X.; Honnell, V.; Ocarz, M.; Gordon, B.; Dapper, J.; Blankenship, K.; Yang, Y.; et al. Identification of Therapeutic Targets in Rhabdomyosarcoma through Integrated Genomic, Epigenomic, and Proteomic Analyses. Cancer Cell. 2018, 34, 411–426.e419. [Google Scholar] [CrossRef] [PubMed]
- Davicioni, E.; Graf Finckenstein, F.; Shahbazian, V.; Buckley, J.D.; Triche, T.J.; Anderson, M.J. Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 2006, 66, 6936. [Google Scholar] [CrossRef] [PubMed]
- Romualdi, C.; De Pittà, C.; Tombolan, L.; Bortoluzzi, S.; Sartori, F.; Rosolen, A.; Lanfranchi, G. Defining the gene expression signature of rhabdomyosarcoma by meta-analysis. BMC Genom. 2006, 7, 287. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, J.; Simpson, L.; Takahashi, M.; Miliaresis, C.; Myers, M.P.; Tonks, N.; Parsons, R. The PTEN/MMAC1 tumor suppressor induces cell death that is rescued by the AKT/protein kinase B oncogene. Cancer Res. 1998, 58, 5667–5672. [Google Scholar] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvesen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Keller, C.; Arenkiel, B.R.; Coffin, C.M.; El-Bardeesy, N.; DePinho, R.A.; Capecchi, M.R. Alveolar rhabdomyosarcomas in conditional Pax3: Fkhr mice: Cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004, 18, 2614–2626. [Google Scholar] [CrossRef]
- Li, Z.; Yu, X.; Shen, J.; Liu, Y.; Chan, M.T.V.; Wu, W.K.K. Micro RNA dysregulation in rhabdomyosarcoma: A new player enters the game. Cell Prolif 2015, 48, 511–516. [Google Scholar] [CrossRef]
- Huang, H.-J.; Liu, J.; Hua, H.; Li, S.-E.; Zhao, J.; Yue, S.; Yu, T.-T.; Jin, Y.-C.; Cheng, S.Y. MiR-214 and N-ras regulatory loop suppresses rhabdomyosarcoma cell growth and xenograft tumorigenesis. Oncotarget 2014, 5, 2161. [Google Scholar] [CrossRef]
- Diao, Y.; Guo, X.; Jiang, L.; Wang, G.; Zhang, C.; Wan, J.; Jin, Y.; Wu, Z. miR-203, a tumor suppressor frequently down-regulated by promoter hypermethylation in rhabdomyosarcoma. J. Biol. Chem. 2014, 289, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Megiorni, F.; Cialfi, S.; McDowell, H.P.; Felsani, A.; Camero, S.; Guffanti, A.; Pizer, B.; Clerico, A.; De Grazia, A.; Pizzuti, A.; et al. Deep Sequencing the microRNA profile in rhabdomyosarcoma reveals down-regulation of miR-378 family members. BMC Cancer 2014, 14, 880. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Y.; Liu, J.; Wang, X.; Yan, Q. Cyclophosphamide-induced apoptosis in A431 cells is inhibited by fucosyltransferase IV. J. Cell. Biochem. 2011, 112, 1376–1383. [Google Scholar] [CrossRef]
- Becker, R.; Ritter, A.; Eichhorn, U.; Lips, J.; Bertram, B.; Wiessler, M.; Zdzienicka, M.Z.; Kaina, B. Induction of DNA breaks and apoptosis in crosslink-hypersensitive V79 cells by the cytostatic drug β-D-glucosyl-ifosfamide mustard. Br. J. Cancer 2002, 86, 130–135. [Google Scholar] [CrossRef]
- Park, B.; Je, Y.T.; Chun, K.H. AKT is translocated to the mitochondria during etoposide-induced apoptosis of HeLa cells. Mol. Med. Rep. 2015, 12, 7577–7581. [Google Scholar] [CrossRef] [PubMed]
- Jeong, C.-H.; Chun, K.-S.; Kundu, J.; Park, B. Phosphorylation of Smac by Akt promotes the caspase-3 activation during etoposide-induced apoptosis in HeLa cells. Mol. Carcinog. 2015, 54, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-F.; Chen, C.-L.; Chang, W.-T.; Jan, M.-S.; Hsu, L.-J.; Wu, R.-H.; Fang, Y.-T.; Tang, M.-J.; Chang, W.-C.; Lin, Y.-S. Bcl-2 rescues ceramide- and etoposide-induced mitochondrial apoptosis through blockage of caspase-2 activation. J. Biol. Chem. 2005, 280, 23758–23765. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.-J.; Liu, S.; Liu, Y.; Zheng, D. Actinomycin D enhances TRAIL-induced caspase-dependent and -independent apoptosis in SH-SY5Y neuroblastoma cells. Neurosci. Res. 2007, 59, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-J.; Chiou, J.-T.; Lee, Y.-C.; Huang, C.-H.; Shi, Y.-J.; Chang, L.-S. SIRT3, PP2A and TTP protein stability in the presence of TNF-α on vincristine-induced apoptosis of leukaemia cells. J. Cell. Mol. Med. 2020, 24, 2552–2565. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-F.; Lin, J.-D.; Yeh, C.-N.; Huang, Y.-T.; Chou, T.-C.; Wong, R.J. Targeting PLKs as a therapeutic approach to well-differentiated thyroid cancer. Endocr. Relat. Cancer 2019, 26, 727–738. [Google Scholar] [CrossRef]
- Gomez-Bougie, P.; Oliver, L.; Le Gouill, S.; Bataille, R.; Amiot, M. Melphalan-induced apoptosis in multiple myeloma cells is associated with a cleavage of Mcl-1 and Bim and a decrease in the Mcl-1/Bim complex. Oncogene 2005, 24, 8076–8079. [Google Scholar] [CrossRef]
- Moghadam, A.R.; da Silva Rosa, S.C.; Samiei, E.; Alizadeh, J.; Field, J.; Kawalec, P.; Thliveris, J.; Akbari, M.; Ghavami, S.; Gordon, J.W. Autophagy modulates temozolomide-induced cell death in alveolar Rhabdomyosarcoma cells. Cell Death Discov. 2018, 4, 52. [Google Scholar] [CrossRef]
- Takeba, Y.; Sekine, S.; Kumai, T.; Matsumoto, N.; Nakaya, S.; Tsuzuki, Y.; Yanagida, Y.; Nakano, H.; Asakura, T.; Ohtsubo, T.; et al. Irinotecan-induced apoptosis is inhibited by increased P-glycoprotein expression and decreased p53 in human hepatocellular carcinoma cells. Biol. Pharm. Bull. 2007, 30, 1400–1406. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef]
- Hosoi, H.; Dilling, M.B.; Shikata, T.; Liu, L.N.; Shu, L.; Ashmun, R.A.; Germain, G.S.; Abraham, R.T.; Houghton, P.J. Rapamycin causes poorly reversible inhibition of mTOR and induces p53-independent apoptosis in human rhabdomyosarcoma cells. Cancer Res. 1999, 59, 886. [Google Scholar] [PubMed]
- Sehgal, S.N. Sirolimus: Its discovery, biological properties, and mechanism of action. Transplant. Proc. 2003, 35, S7–S14. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Andersen, J.S.; Mann, M.; Terada, N.; Korsmeyer, S.J. p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. USA 2001, 98, 9666–9670. [Google Scholar] [CrossRef] [PubMed]
- Kaylani, S.Z.; Xu, J.; Srivastava, R.K.; Kopelovich, L.; Pressey, J.G.; Athar, M. Rapamycin targeting mTOR and hedgehog signaling pathways blocks human rhabdomyosarcoma growth in xenograft murine model. Biochem. Biophys. Res. Commun. 2013, 435, 557–561. [Google Scholar] [CrossRef]
- Trucco, M.M.; Meyer, C.F.; Thornton, K.A.; Shah, P.; Chen, A.R.; Wilky, B.A.; Carrera-Haro, M.A.; Boyer, L.C.; Ferreira, M.F.; Shafique, U.; et al. A phase II study of temsirolimus and liposomal doxorubicin for patients with recurrent and refractory bone and soft tissue sarcomas. Clin. Sarcoma Res. 2018, 8, 21. [Google Scholar] [CrossRef]
- Miyoshi, K.; Kohashi, K.; Fushimi, F.; Yamamoto, H.; Kishimoto, J.; Taguchi, T.; Iwamoto, Y.; Oda, Y. Close correlation between CXCR4 and VEGF expression and frequent CXCR7 expression in rhabdomyosarcoma. Hum. Pathol. 2014, 45, 1900–1909. [Google Scholar] [CrossRef]
- McKian, K.P.; Haluska, P. Cixutumumab. Expert Opin. Investig. Drugs 2009, 18, 1025–1033. [Google Scholar] [CrossRef]
- Attias-Geva, Z.; Bentov, I.; Ludwig, D.L.; Fishman, A.; Bruchim, I.; Werner, H. Insulin-like growth factor-I receptor (IGF-IR) targeting with monoclonal antibody cixutumumab (IMC-A12) inhibits IGF-I action in endometrial cancer cells. Eur. J. Cancer 2011, 47, 1717–1726. [Google Scholar] [CrossRef]
- Chugh, R.; Griffith, K.A.; Davis, E.J.; Thomas, D.G.; Zavala, J.D.; Metko, G.; Brockstein, B.; Undevia, S.D.; Stadler, W.M.; Schuetze, S.M. Doxorubicin plus the IGF-1R antibody cixutumumab in soft tissue sarcoma: A phase I study using the TITE-CRM model. Ann. Oncol. 2015, 26, 1459–1464. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.K.; Tap, W.D.; Qin, L.-X.; Livingston, M.B.; Undevia, S.D.; Chmielowski, B.; Agulnik, M.; Schuetze, S.M.; Reed, D.R.; Okuno, S.H.; et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: A multicentre, open-label, phase 2 trial. Lancet Oncol. 2013, 14, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Asmane, I.; Watkin, E.; Alberti, L.; Duc, A.; Marec-Berard, P.; Ray-Coquard, I.; Cassier, P.; Decouvelaere, A.-V.; Ranchère, D.; Kurtz, J.-E.; et al. Insulin-like growth factor type 1 receptor (IGF-1R) exclusive nuclear staining: A predictive biomarker for IGF-1R monoclonal antibody (Ab) therapy in sarcomas. Eur. J. Cancer 2012, 48, 3027–3035. [Google Scholar] [CrossRef] [PubMed]
- Forde, P.M.; Rudin, C.M. Crizotinib in the treatment of non-small-cell lung cancer. Expert Opin. Pharmacother. 2012, 13, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Prabhash, K.; Noronha, V.; Joshi, A.; Desai, S. Crizotinib: A comprehensive review. South Asian J. Cancer 2013, 2, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhang, X.; Wu, Z.; Xu, X.; Guo, M.; Zhai, X.; Zuo, D.; Wu, Y. The novel ALK inhibitor ZX-29 induces apoptosis through inhibiting ALK and inducing ROS-mediated endoplasmic reticulum stress in Karpas299 cells. J. Biochem. Mol. Toxicol. 2021, 35, e22666. [Google Scholar] [CrossRef]
- Frentzel, J.; Sorrentino, D.; Giuriato, S. Targeting autophagy in ALK-associated cancers. Cancers 2017, 9, 161. [Google Scholar] [CrossRef]
- van Erp, A.E.M.; Hillebrandt-Roeffen, M.H.S.; van Houdt, L.; Fleuren, E.D.G.; van der Graaf, W.T.A.; Versleijen-Jonkers, Y.M.H. Targeting anaplastic lymphoma kinase (ALK) in rhabdomyosarcoma (RMS) with the second-generation ALK inhibitor ceritinib. Target. Oncol. 2017, 12, 815–826. [Google Scholar] [CrossRef]
- Wierdl, M.; Tsurkan, L.; Chi, L.; Hatfield, M.J.; Tollemar, V.; Bradley, C.; Chen, X.; Qu, C.; Potter, P.M. Targeting ALK in pediatric RMS does not induce antitumor activity in vivo. Cancer Chemother. Pharmacol. 2018, 82, 251–263. [Google Scholar] [CrossRef]
- Dolgikh, N.; Fulda, S. Rhabdomyosarcoma cells are susceptible to cell death by LDK378 alone or in combination with sorafenib independently of anaplastic lymphoma kinase status. Anticancer Drugs 2017, 28, 1118–1125. [Google Scholar] [CrossRef]
- Ferguson, M.; Hingorani, P.; Gupta, A.A. Emerging Molecular-Targeted Therapies in Early-Phase Clinical Trials and Preclinical Models. Am. Soc. Clin. Oncol. Educ. Book 2013, 33, 420–424. [Google Scholar] [CrossRef]
- Riedel, R.F.; Ballman, K.V.; Lu, Y.; Attia, S.; Loggers, E.T.; Ganjoo, K.N.; Livingston, M.B.; Chow, W.; Wright, J.; Ward, J.H.; et al. A randomized, double-blind, placebo-controlled, phase II study of regorafenib versus placebo in advanced/metastatic, treatment-rfractory liposarcoma: Results from the SARC024 study. Oncologist 2020, 25, e1655–e1662. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Sakai, R.; Rinehart, K.; Wang, A. Molecular and crystal structures of ecteinascidins: Potent antitumor compounds from the Caribbean tunicate Ecteinascidia turbinata. J. Biomol. Struct. Dyn. 1993, 10, 793–818. [Google Scholar]
- Banerjee, P.; Zhang, R.; Ivan, C.; Galletti, G.; Clise-Dwyer, K.; Barbaglio, F.; Scarfò, L.; Aracil, M.; Klein, C.; Wierda, W.; et al. Trabectedin reveals a strategy of immunomodulation in chronic lymphocytic leukemia. Cancer Immunol. Res. 2019, 7, 2036–2051. [Google Scholar] [CrossRef]
- Zhang, T.; Cooper, S.; Brockdorff, N. The interplay of histone modifications—writers that read. EMBO Rep. 2015, 16, 1467–1481. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC inhibitors in cancer development and therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef]
- Enßle, J.C.; Boedicker, C.; Wanior, M.; Vogler, M.; Knapp, S.; Fulda, S. Co-targeting of BET proteins and HDACs as a novel approach to trigger apoptosis in rhabdomyosarcoma cells. Cancer Lett. 2018, 428, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Laszig, S.; Boedicker, C.; Weiser, T.; Knapp, S.; Fulda, S. The novel dual BET/HDAC inhibitor TW09 mediates cell death by mitochondrial apoptosis in rhabdomyosarcoma cells. Cancer Lett. 2020, 486, 46–57. [Google Scholar] [CrossRef]
- Tomoyasu, C.; Kikuchi, K.; Kaneda, D.; Yagyu, S.; Miyachi, M.; Tsuchiya, K.; Iehara, T.; Sakai, T.; Hosoi, H. OBP-801, a novel histone deacetylase inhibitor, induces M-phase arrest and apoptosis in rhabdomyosarcoma cells. Oncol. Rep. 2019, 41, 643–649. [Google Scholar] [CrossRef]
- Heinicke, U.; Fulda, S. Chemosensitization of rhabdomyosarcoma cells by the histone deacetylase inhibitor SAHA. Cancer Lett. 2014, 351, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Bharathy, N.; Berlow, N.E.; Wang, E.; Abraham, J.; Settelmeyer, T.P.; Hooper, J.E.; Svalina, M.N.; Bajwa, Z.; Goros, M.W.; Hernandez, B.S.; et al. Preclinical rationale for entinostat in embryonal rhabdomyosarcoma. Skelet. Muscle 2019, 9, 12. [Google Scholar] [CrossRef]
- de Haan, R.; van Werkhoven, E.; van den Heuvel, M.M.; Peulen, H.M.U.; Sonke, G.S.; Elkhuizen, P.; van den Brekel, M.W.M.; Tesselaar, M.E.T.; Vens, C.; Schellens, J.H.M.; et al. Study protocols of three parallel phase 1 trials combining radical radiotherapy with the PARP inhibitor olaparib. BMC Cancer 2019, 19, 901. [Google Scholar] [CrossRef]
- Lesueur, P.; Lequesne, J.; Grellard, J.-M.; Dugué, A.; Coquan, E.; Brachet, P.-E.; Geffrelot, J.; Kao, W.; Emery, E.; Berro, D.H.; et al. Phase I/IIa study of concomitant radiotherapy with olaparib and temozolomide in unresectable or partially resectable glioblastoma: OLA-TMZ-RTE-01 trial protocol. BMC Cancer 2019, 19, 198. [Google Scholar] [CrossRef] [PubMed]
- Camero, S.; Ceccarelli, S.; De Felice, F.; Marampon, F.; Mannarino, O.; Camicia, L.; Vescarelli, E.; Pontecorvi, P.; Pizer, B.; Shukla, R.; et al. PARP inhibitors affect growth, survival and radiation susceptibility of human alveolar and embryonal rhabdomyosarcoma cell lines. J. Cancer Res. Clin. Oncol. 2019, 145, 137–152. [Google Scholar] [CrossRef]
- Werner, M.; Sacher, J.; Hohenegger, M. Mutual amplification of apoptosis by statin-induced mitochondrial stress and doxorubicin toxicity in human rhabdomyosarcoma cells. Br. J. Pharmacol. 2004, 143, 715–724. [Google Scholar] [CrossRef]
- Ahmadi, M.; Amiri, S.; Pecic, S.; Machaj, F.; Rosik, J.; Łos, M.J.; Alizadeh, J.; Mahdian, R.; da Silva Rosa, S.C.; Schaafsma, D.; et al. Pleiotropic effects of statins: A focus on cancer. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165968. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, J.; Shojaei, S.; da Silva Rosa, S.; Moghadam, A.R.; Zeki, A.A.; Hashemi, M.; Los, M.J.; Gordon, J.W.; Ghavami, S. Detection of small GTPase prenylation and GTP binding using membrane fractionation and GTPase-linked immunosorbent assay. J. Visu. Exp. 2018, 141, e57646. [Google Scholar]
- Yeganeh, B.; Wiechec, E.; Ande, S.R.; Sharma, P.; Moghadam, A.R.; Post, M.; Freed, D.H.; Hashemi, M.; Shojaei, S.; Zeki, A.A.; et al. Targeting the mevalonate cascade as a new therapeutic approach in heart disease, cancer and pulmonary disease. Pharmacol. Ther. 2014, 143, 87–110. [Google Scholar] [CrossRef] [PubMed]
- Koohestanimobarhan, S.; Salami, S.; Imeni, V.; Mohammadi, Z.; Bayat, O. Lipophilic statins antagonistically alter the major epithelial-to-mesenchymal transition signaling pathways in breast cancer stem–like cells via inhibition of the mevalonate pathway. J. Cell. Biochem. 2019, 120, 2515–2531. [Google Scholar] [CrossRef] [PubMed]
- De Duve, C.; Wattiaux, R. Functions of lysosomes. Ann. Rew. Physiol. 1966, 28, 435–492. [Google Scholar] [CrossRef] [PubMed]
- Musiwaro, P.; Smith, M.; Manifava, M.; Walker, S.A.; Ktistakis, N.T. Characteristics and requirements of basal autophagy in HEK 293 cells. Autophagy 2013, 9, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef]
- Habibzadeh, P.; Dastsooz, H.; Eshraghi, M.; Los, M.J.; Klionsky, D.J.; Ghavami, S. Autophagy: The potential link between SARS-CoV-2 and cancer. Cancers 2021, 13, 5721. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Siri, M.; Behrouj, H.; Dastghaib, S.; Zamani, M.; Likus, W.; Rezaie, S.; Hudecki, J.; Khazayel, S.; Los, M.J.; Mokarram, P.; et al. Casein Kinase-1-Alpha Inhibitor (D4476) sensitizes microsatellite instable colorectal cancer cells to 5-Fluorouracil via authophagy fux inhibition. Arch. Immunol. Ther. Exp. 2021, 69, 26. [Google Scholar] [CrossRef]
- Hinton, M.; Eltayeb, E.; Ghavami, S.; Dakshinamurti, S. Effect of pulsatile stretch on unfolded protein response in a new model of the pulmonary hypertensive vascular wall. Biochem. Biophys. Rep. 2021, 27, 101080. [Google Scholar] [CrossRef]
- Wang, B.; Abraham, N.; Gao, G.; Yang, Q. Dysregulation of autophagy and mitochondrial function in Parkinson’s disease. Transl. Neurodegener. 2016, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef]
- Eshraghi, M.; Adlimoghaddam, A.; Mahmoodzadeh, A.; Sharifzad, F.; Yasavoli-Sharahi, H.; Lorzadeh, S.; Albensi, B.C.; Ghavami, S. Alzheimer’s disease pathogenesis: Role of autophagy and mitophagy focusing in microglia. Int. J. Mol. Sci. 2021, 22, 3330. [Google Scholar] [CrossRef] [PubMed]
- Lorzadeh, S.; Kohan, L.; Ghavami, S.; Azarpira, N. Autophagy and the Wnt signaling pathway: A focus on Wnt/beta-catenin signaling. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118926. [Google Scholar] [CrossRef] [PubMed]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Oh, J.E.; Lee, H.K. Autophagy in innate recognition of pathogens and adaptive immunity. Yonsei Med. J. 2012, 53, 241–247. [Google Scholar] [CrossRef]
- Hombach-Klonisch, S.; Natarajan, S.; Thanasupawat, T.; Medapati, M.; Pathak, A.; Ghavami, S.; Klonisch, T. Mechanisms of therapeutic resistance in cancer (stem) cells with emphasis on thyroid cancer cells. Front. Endocrinol. 2014, 5, 37. [Google Scholar] [CrossRef]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.; Sher, A.A.; Coombs, K.M. The roles of apoptosis, autophagy and unfolded protein response in arbovirus, influenza virus, and HIV infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef]
- Ghavami, S.; Mutawe, M.M.; Schaafsma, D.; Yeganeh, B.; Unruh, H.; Klonisch, T.; Halayko, A.J. Geranylgeranyl transferase 1 modulates autophagy and apoptosis in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302, L420–L428. [Google Scholar] [CrossRef]
- Alizadeh, J.; Kochan, M.M.; Stewart, V.D.; Drewnik, D.A.; Hannila, S.S.; Ghavami, S. Inhibition of autophagy flux promotes secretion of chondroitin sulfate proteoglycans in primary rat astrocytes. Mol. Neurobiol. 2021, 58, 6077–6091. [Google Scholar] [CrossRef]
- Zeki, A.A.; Yeganeh, B.; Kenyon, N.J.; Post, M.; Ghavami, S. Autophagy in airway diseases: A new frontier in human asthma? Allergy 2016, 71, 5–14. [Google Scholar] [CrossRef]
- Lamb, C.A.; Yoshimori, T.; Tooze, S.A. The autophagosome: Origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013, 14, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; He, P.; Huang, Y.; Li, Y.F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.S.; Vats, S.; Chia, A.Y.; Tan, T.Z.; Deng, S.; Ong, M.S.; Arfuso, F.; Yap, C.T.; Goh, B.C.; Sethi, G.; et al. Dual role of autophagy in hallmarks of cancer. Oncogene 2018, 37, 1142–1158. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhao, L.; Kuang, M.; Zhang, B.; Liang, Z.; Yi, T.; Wei, Y.; Zhao, X. Autophagy in tumorigenesis and cancer therapy: Dr. Jekyll or Mr. Hyde? Cancer Lett. 2012, 323, 115–127. [Google Scholar] [CrossRef]
- Lim, S.M.; Hanif, E.A.M.; Chin, S.-F. Is targeting autophagy mechanism in cancer a good approach? The possible double-edge sword effect. Cell Biosci. 2021, 11, 56. [Google Scholar] [CrossRef]
- Chi, K.-H.; Wang, Y.-S.; Huang, Y.-C.; Chiang, H.-C.; Chi, M.-S.; Chi, C.-H.; Wang, H.-E.; Kao, S.-J. Simultaneous activation and inhibition of autophagy sensitizes cancer cells to chemotherapy. Oncotarget 2016, 7, 58075. [Google Scholar] [CrossRef]
- Rezaei Moghadam, A. Targeting Rhabdomyosarcoma with Temozolomide: How Autophagy Regulates TMZ-Induced Apoptosis in Rhabdomyosarcoma Cells; University of Manitoba: Winnipeg, MB, Canada, 2018. [Google Scholar]
- Alizadeh, J.; Zeki, A.A.; Mirzaei, N.; Tewary, S.; Moghadam, A.R.; Glogowska, A.; Nagakannan, P.; Eftekharpour, E.; Wiechec, E.; Gordon, J.W. Mevalonate cascade inhibition by simvastatin induces the intrinsic apoptosis pathway via depletion of isoprenoids in tumor cells. Sci. Rep. 2017, 7, 44841. [Google Scholar] [CrossRef]
- Likus, W.; Siemianowicz, K.; Bieńk, K.; Pakuła, M.; Pathak, H.; Dutta, C.; Wang, Q.; Shojaei, S.; Assaraf, Y.G.; Ghavami, S. Could drugs inhibiting the mevalonate pathway also target cancer stem cells? Drug Resist. Updates 2016, 25, 13–25. [Google Scholar] [CrossRef]
- Wang, C.-Y.; Liu, P.-Y.; Liao, J.K. Pleiotropic effects of statin therapy: Molecular mechanisms and clinical results. Trends Mol. Med. 2008, 14, 37–44. [Google Scholar] [CrossRef]
- Allen, S.C.; Mamotte, C.D. Pleiotropic and adverse effects of statins—Do epigenetics play a role? J. Pharmacol. Exp. Ther. 2017, 362, 319–326. [Google Scholar] [CrossRef]
- Labos, C.; Brophy, J.M.; Smith, G.D.; Sniderman, A.D.; Thanassoulis, G. Evaluation of the pleiotropic effects of statins: A reanalysis of the randomized trial evidence using Egger regression—Brief report. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 262–265. [Google Scholar] [CrossRef]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2013, 368, 576–577. [Google Scholar] [CrossRef] [PubMed]
- Gaist, D.; Hallas, J.; Friis, S.; Hansen, S.; Sørensen, H.T. Statin use and survival following glioblastoma multiforme. Cancer Epidemiol. 2014, 38, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Karamouzis, M.V.; Papavassiliou, A.G. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 2007, 6, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Matzno, S.; Yasuda, S.; Juman, S.; Yamamoto, Y.; Nagareya-Ishida, N.; Nakabayashi, T.; Matsuyama, K.; Tazuya-Murayama, K. Statin-induced apoptosis linked with membrane farnesylated Ras small G protein depletion, rather than geranylated Rho protein. J. Pharm. Pharmacol. 2005, 57, 1475–1484. [Google Scholar] [CrossRef]
- Tsubaki, M.; Fujiwara, D.; Takeda, T.; Kino, T.; Tomonari, Y.; Itoh, T.; Imano, M.; Satou, T.; Sakaguchi, K.; Nishida, S. The sensitivity of head and neck carcinoma cells to statins is related to the expression of their Ras expression status, and statin-induced apoptosis is mediated via suppression of the Ras/ERK and Ras/mTOR pathways. Clin. Exp. Pharmacol. Physiol. 2017, 44, 222–234. [Google Scholar] [CrossRef]
- Martirosyan, A.; Clendening, J.W.; Goard, C.A.; Penn, L.Z. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: Potential therapeutic relevance. BMC Cancer 2010, 10, 103. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Longo, J.; van Leeuwen, J.E.; Mullen, P.J.; Ba-Alawi, W.; Haibe-Kains, B.; Penn, L.Z. Statin-induced cancer cell death can be mechanistically uncoupled from prenylation of RAS family proteins. Cancer Res. 2018, 78, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Su, Z.; DeWitt, J.P.; Xie, L.; Chen, Y.; Li, X.; Han, L.; Li, D.; Xia, J.; Zhang, Y. Fluvastatin prevents lung adenocarcinoma bone metastasis by triggering autophagy. EBioMedicine 2017, 19, 49–59. [Google Scholar] [CrossRef]
- Peng, X.; Li, W.; Yuan, L.; Mehta, R.G.; Kopelovich, L.; McCormick, D.L. Inhibition of proliferation and induction of autophagy by atorvastatin in PC3 prostate cancer cells correlate with downregulation of Bcl2 and upregulation of miR-182 and p21. PLoS ONE 2013, 8, e70442. [Google Scholar] [CrossRef]
- Arndt, C.A. Role of doxorubicin in rhabdomyosarcoma: Is the answer knowable? Am. Soc. Clin. Oncol. Educ. Book 2012, 32, 621–623. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A.; Finley, K.D.; Mentzer, R.M. Cardioprotection requires taking out the trash. Basic Res. Cardiol. 2009, 104, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Takemura, G.; Kanamori, H.; Takeyama, T.; Watanabe, T.; Morishita, K.; Ogino, A.; Tsujimoto, A.; Goto, K.; Maruyama, R. Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovasc. Res. 2012, 96, 456–465. [Google Scholar] [CrossRef]
- Sishi, B.J.; Loos, B.; van Rooyen, J.; Engelbrecht, A.-M. Autophagy upregulation promotes survival and attenuates doxorubicin-induced cardiotoxicity. Biochem. Pharmacol. 2013, 85, 124–134. [Google Scholar] [CrossRef]
- Smuder, A.J.; Kavazis, A.N.; Min, K.; Powers, S.K. Doxorubicin-induced markers of myocardial autophagic signaling in sedentary and exercise trained animals. J. Appl. Physiol. 2013, 115, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.T.; Li, Z.L.; He, Z.X.; Qiu, J.X.; Zhou, S.F. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef]
- Li, Z.; Li, H.; Liu, B.; Luo, J.; Qin, X.; Gong, M.; Shi, B.; Wei, Y. Inhibition of miR-25 attenuates doxorubicin-induced apoptosis, reactive oxygen species production and DNA damage by targeting PTEN. Int. J. Med. Sci. 2020, 17, 1415. [Google Scholar] [CrossRef]
- Chen, M.B.; Shen, W.X.; Yang, Y.; Wu, X.Y.; Gu, J.H.; Lu, P.H. Activation of AMP-activated protein kinase is involved in vincristine-induced cell apoptosis in B16 melanoma cell. J. Cell Physiol. 2011, 226, 1915–1925. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef]
- Rubinstein, A.D.; Kimchi, A. Life in the balance–a mechanistic view of the crosstalk between autophagy and apoptosis. J. Cell Sci. 2012, 125, 5259–5268. [Google Scholar] [CrossRef]
- Kothari, A.; Hittelman, W.N.; Chambers, T.C. Cell cycle–dependent mechanisms underlie vincristine-induced death of primary acute lymphoblastic leukemia cells. Cancer Res. 2016, 76, 3553–3561. [Google Scholar] [CrossRef] [PubMed]
- Raney, R.B.; Walterhouse, D.O.; Meza, J.L.; Andrassy, R.J.; Breneman, J.C.; Crist, W.M.; Maurer, H.M.; Meyer, W.H.; Parham, D.M.; Anderson, J.R. Results of the Intergroup Rhabdomyosarcoma Study Group D9602 protocol, using vincristine and dactinomycin with or without cyclophosphamide and radiation therapy, for newly diagnosed patients with low-risk embryonal rhabdomyosarcoma: A report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. J. Clin. Oncol. 2011, 29, 1312. [Google Scholar] [PubMed]
- Walterhouse, D.O.; Pappo, A.S.; Meza, J.L.; Breneman, J.C.; Hayes-Jordan, A.; Parham, D.M.; Cripe, T.P.; Anderson, J.R.; Meyer, W.H.; Hawkins, D.S. Reduction of cyclophosphamide dose for patients with subset 2 low-risk rhabdomyosarcoma is associated with an increased risk of recurrence: A report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Cancer 2017, 123, 2368–2375. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.Y.C.; Philip Kuebler, J.; Tormey, D.C.; Anderson, S.; Pandya, K.J.; Borden, E.C.; Davis, T.E.; Trump, D.L. Phase ii evaluation of a combination of mitomycin C, vincristine, and cisplatin in advanced non-small cell lung cancer. Cancer 1986, 57, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Steiner, R.; Stewart, J.F.; Cantwell, B.M.; Minton, M.J.; Knight, R.K.; Rubens, R.D. Adriamycin alone or combined with vincristine in the treatment of advanced breast cancer. Eur. J. Cancer Clin. Oncol. 1983, 19, 1553–1557. [Google Scholar] [CrossRef]
- Haim, N.; Epelbaum, R.; Ben-Shahar, M.; Yarnitsky, D.; Simri, W.; Robinson, E. Full dose vincristine (without 2-mg dose limit) in the treatment of lymphomas. Cancer 1994, 73, 2515–2519. [Google Scholar] [CrossRef]
- Sarris, A.; Hagemeister, F.; Romaguera, J.; Rodriguez, M.; McLaughlin, P.; Tsimberidou, A.; Medeiros, L.; Samuels, B.; Pate, O.; Oholendt, M. Liposomal vincristine in relapsed non-Hodgkin’s lymphomas: Early results of an ongoing phase II trial. Ann. Oncol. 2000, 11, 69–72. [Google Scholar] [CrossRef]
- Rosenstock, J.G.; Evans, A.E.; Schut, L. Response to vincristine of recurrent brain tumors in children. J. Neurosurg. 1976, 45, 135–140. [Google Scholar] [CrossRef]
- Levin, V.; Edwards, M.; Wright, D.; Seager, M.; Schimberg, T.P.; Townsend, J.; Wilson, C.B. Modified procarbazine, CCNU, and vincristine (PCV 3) combination chemotherapy in the treatment of malignant brain tumors. Cancer Treat. Rep. 1980, 64, 237–244. [Google Scholar]
- Villikka, K.; Kivistö, K.T.; Mäenpää, H.; Joensuu, H.; Neuvonen, P.J. Cytochrome P450-inducing antiepileptics increase the clearance of vincristine in patients with brain tumors. Clin. Pharmacol. Ther. 1999, 66, 589–593. [Google Scholar]
- Wang, Y.; Xia, C.; Lv, Y.; Li, C.; Mei, Q.; Li, H.; Wang, H.; Li, S. Crosstalk influence between P38MAPK and autophagy on mitochondria-mediated apoptosis induced by anti-fas antibody/actinomycin D in human hepatoma bel-7402 cells. Molecules 2017, 22, 1705. [Google Scholar] [CrossRef]
- Dong, X.; Yang, Y.; Zhou, Y.; Bi, X.; Zhao, N.; Zhang, Z.; Li, L.; Hang, Q.; Zhang, R.; Chen, D. Glutathione S-transferases P1 protects breast cancer cell from adriamycin-induced cell death through promoting autophagy. Cell Death Differ. 2019, 26, 2086–2099. [Google Scholar] [CrossRef]
- Seitz, G.; Bonin, M.; Fuchs, J.; Poths, S.; Ruck, P.; Warmann, S.W.; Armeanu-Ebinger, S. Inhibition of glutathione-S-transferase as a treatment strategy for multidrug resistance in childhood rhabdomyosarcoma. Int. J. Oncol. 2010, 36, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Tan, S.-H.; Nicolas, V.; Bauvy, C.; Yang, N.-D.; Zhang, J.; Xue, Y.; Codogno, P.; Shen, H.-M. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013, 23, 508–523. [Google Scholar] [CrossRef] [PubMed]
- Salerno, M.; Avnet, S.; Bonuccelli, G.; Hosogi, S.; Granchi, D.; Baldini, N. Impairment of lysosomal activity as a therapeutic modality targeting cancer stem cells of embryonal rhabdomyosarcoma cell line RD. PLoS ONE 2014, 9, e110340. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Shen, T.; Shang, C.; Luo, Y.; Liu, L.; Yan, J.; Li, Y.; Huang, S. Ciclopirox induces autophagy through reactive oxygen species-mediated activation of JNK signaling pathway. Oncotarget 2014, 5, 10140. [Google Scholar] [CrossRef][Green Version]
- Peron, M.; Bonvini, P.; Rosolen, A. Effect of inhibition of the ubiquitin-proteasome system and Hsp90 on growth and survival of rhabdomyosarcoma cells in vitro. BMC Cancer 2012, 12, 233. [Google Scholar] [CrossRef]
- Rapino, F.; Jung, M.; Fulda, S. BAG3 induction is required to mitigate proteotoxicity via selective autophagy following inhibition of constitutive protein degradation pathways. Oncogene 2014, 33, 1713–1724. [Google Scholar] [CrossRef] [PubMed]
- Sadeghabadi, Z.A.; Nourbakhsh, M.; Pasalar, P.; Emamgholipour, S.; Golestani, A.; Larijani, B.; Razzaghy-Azar, M. Reduced gene expression of sirtuins and active AMPK levels in children and adolescents with obesity and insulin resistance. Obes. Res. Pract. 2018, 12, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Jing, E.; Gesta, S.; Kahn, C.R. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007, 6, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Bonham, E.M.; Hannon, B.E.; Amick, T.R.; Baylin, S.B.; O’Hagan, H.M. Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. J. Mol. Cell Biol. 2016, 8, 244–254. [Google Scholar] [CrossRef]
- Wu, S.; Jiang, J.; Liu, J.; Wang, X.; Gan, Y.; Tang, Y. Meta-analysis of SIRT1 expression as a prognostic marker for overall survival in gastrointestinal cancer. Oncotarget 2017, 8, 62589. [Google Scholar] [CrossRef] [PubMed]
- Grbesa, I.; Pajares, M.J.; Martinez-Terroba, E.; Agorreta, J.; Mikecin, A.-M.; Larrayoz, M.; Idoate, M.A.; Gall-Troselj, K.; Pio, R.; Montuenga, L.M. Expression of sirtuin 1 and 2 is associated with poor prognosis in non-small cell lung cancer patients. PLoS ONE 2015, 10, e0124670. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Su, L.; Chen, W.Y. The emerging and diverse roles of sirtuins in cancer: A clinical perspective. OncoTargets Ther. 2013, 6, 1399. [Google Scholar]
- Ma, L.; Maruwge, W.; Strambi, A.; D’arcy, P.; Pellegrini, P.; Kis, L.; De Milito, A.; Lain, S.; Brodin, B. SIRT1 and SIRT2 inhibition impairs pediatric soft tissue sarcoma growth. Cell Death Dis. 2014, 5, e1483. [Google Scholar] [CrossRef]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef]
- Jones, P.; George, A. The ABC transporter structure and mechanism: Perspectives on recent research. Cell. Mol. Life Sci. CMLS 2004, 61, 682–699. [Google Scholar] [CrossRef]
- Borst, P.; Evers, R.; Kool, M.; Wijnholds, J. A family of drug transporters: The multidrug resistance-associated proteins. J. Natl. Cancer Inst. 2000, 92, 1295–1302. [Google Scholar] [CrossRef]
- Hui, R.C.; Francis, R.E.; Guest, S.K.; Costa, J.R.; Gomes, A.R.; Myatt, S.S.; Brosens, J.J.; Lam, E.W. Doxorubicin activates FOXO3a to induce the expression of multidrug resistance gene ABCB1 (MDR1) in K562 leukemic cells. Mol. Cancer Ther. 2008, 7, 670–678. [Google Scholar] [CrossRef]
- Hill, C.R.; Cole, M.; Errington, J.; Malik, G.; Boddy, A.V.; Veal, G.J. Characterisation of the clinical pharmacokinetics of actinomycin D and the influence of ABCB1 pharmacogenetic variation on actinomycin D disposition in children with cancer. Clin. Pharmacokinet. 2014, 53, 741–751. [Google Scholar] [CrossRef]
- Hill, C.R.; Jamieson, D.; Thomas, H.D.; Brown, C.D.; Boddy, A.V.; Veal, G.J. Characterisation of the roles of ABCB1, ABCC1, ABCC2 and ABCG2 in the transport and pharmacokinetics of actinomycin D in vitro and in vivo. Biochem. Pharmacol. 2013, 85, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Brakora, K.A.; Seiden, M.V. Inhibition of ABCB1 (MDR1) and ABCB4 (MDR3) expression by small interfering RNA and reversal of paclitaxel resistance in human ovarian cancer cells. Mol. Cancer Ther. 2004, 3, 833–838. [Google Scholar] [CrossRef]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.-L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) induction defines a common resistance mechanism in paclitaxel-and olaparib-resistant ovarian cancer cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [PubMed]
- El-Khoury, V.; Breuzard, G.; Fourré, N.; Dufer, J. The histone deacetylase inhibitor trichostatin A downregulates human MDR1 (ABCB1) gene expression by a transcription-dependent mechanism in a drug-resistant small cell lung carcinoma cell line model. Br. J. Cancer 2007, 97, 562–573. [Google Scholar] [CrossRef]
- Limtrakul, P.; Chearwae, W.; Shukla, S.; Phisalphong, C.; Ambudkar, S.V. Modulation of function of three ABC drug transporters, P-glycoprotein (ABCB1), mitoxantrone resistance protein (ABCG2) and multidrug resistance protein 1 (ABCC1) by tetrahydrocurcumin, a major metabolite of curcumin. Mol. Cell. Biochem. 2007, 296, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Nieth, C.; Lage, H. Induction of the ABC-Transporters Mdr1/P-gp (Abcb1), Mrp1 (Abcc1), and Bcrp (Abcg2) during establishment of multidrug resistance following exposure to mitoxantrone. J. Chemother. 2005, 17, 215–223. [Google Scholar] [CrossRef]
- Michaelis, M.; Rothweiler, F.; Wurglics, M.; Aniceto, N.; Dittrich, M.; Zettl, H.; Wiese, M.; Wass, M.; Ghafourian, T.; Schubert-Zsilavecz, M. Substrate-specific effects of pirinixic acid derivatives on ABCB1-mediated drug transport. Oncotarget 2016, 7, 11664. [Google Scholar] [CrossRef]
- Chiu, L.-Y.; Ko, J.-L.; Lee, Y.-J.; Yang, T.-Y.; Tee, Y.-T.; Sheu, G.-T. L-type calcium channel blockers reverse docetaxel and vincristine-induced multidrug resistance independent of ABCB1 expression in human lung cancer cell lines. Toxicol. Lett. 2010, 192, 408–418. [Google Scholar] [CrossRef]
- Pitchakarn, P.; Ohnuma, S.; Pintha, K.; Pompimon, W.; Ambudkar, S.V.; Limtrakul, P. Kuguacin J isolated from Momordica charantia leaves inhibits P-glycoprotein (ABCB1)-mediated multidrug resistance. J. Nutr. Biochem. 2012, 23, 76–84. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, M.; Liu, L.-X. The overexpression of multidrug resistance-associated proteins and gankyrin contribute to arsenic trioxide resistance in liver and gastric cancer cells. Oncol. Rep. 2009, 22, 73–80. [Google Scholar] [PubMed]
- Zhou, T.; Niu, W.; Yuan, Z.; Guo, S.; Song, Y.; Di, C.; Xu, X.; Tan, X.; Yang, L. ABCA1 is coordinated with ABCB1 in the arsenic-resistance of human cells. Appl. Biochem. Biotechnol. 2019, 187, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, M.; Prince, H.M. Romidepsin for Relapsed and Refractory Cutaneous T-Cell Lymphoma. Clin. Med. Insights Dermatol. 2012, 5, 21. [Google Scholar] [CrossRef]
- Grant, C.; Rahman, F.; Piekarz, R.; Peer, C.; Frye, R.; Robey, R.W.; Gardner, E.R.; Figg, W.D.; Bates, S.E. Romidepsin: A new therapy for cutaneous T-cell lymphoma and a potential therapy for solid tumors. Expert Rev. Anticancer Ther. 2010, 10, 997–1008. [Google Scholar] [CrossRef]
- Tufan, A.; Babaoglu, M.O.; Akdogan, A.; Yasar, U.; Calguneri, M.; Kalyoncu, U.; Karadag, O.; Hayran, M.; Ertenli, A.I.; Bozkurt, A. Association of drug transporter gene ABCB1 (MDR1) 3435C to T polymorphism with colchicine response in familial Mediterranean fever. J. Rheumatol. 2007, 34, 1540–1544. [Google Scholar]
- Ozen, F.; Silan, C.; Uludag, A.; Candan, F.; Silan, F.; Ozdemir, S.; Atik, S.; Ozdemir, O. Association between ABCB1 (MDR1) gene 3435 C> T polymorphism and colchicine unresponsiveness of FMF patients. Ren. Fail. 2011, 33, 899–903. [Google Scholar] [CrossRef]
- Onaciu, A.; Munteanu, R.; Munteanu, V.C.; Gulei, D.; Raduly, L.; Feder, R.I.; Pirlog, R.; Atanasov, A.G.; Korban, S.S.; Irimie, A.; et al. Spontaneous and induced animal models for cancer research. Diagnostics 2020, 10, 660. [Google Scholar] [CrossRef]
- Emmink, B.L.; Van Houdt, W.J.; Vries, R.G.; Hoogwater, F.J.; Govaert, K.M.; Verheem, A.; Nijkamp, M.W.; Steller, E.J.; Jimenez, C.R.; Clevers, H. Differentiated human colorectal cancer cells protect tumor-initiating cells from irinotecan. Gastroenterology 2011, 141, 269–278. [Google Scholar] [CrossRef]
- Han, J.Y.; Lim, H.S.; Yoo, Y.K.; Shin, E.S.; Park, Y.H.; Lee, S.Y.; Lee, J.E.; Lee, D.H.; Kim, H.T.; Lee, J.S. Associations of ABCB1, ABCC2, and ABCG2 polymorphisms with irinotecan-pharmacokinetics and clinical outcome in patients with advanced non-small cell lung cancer. Cancer 2007, 110, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-X.; Yang, Y.; Zeng, L.; Patel, H.; Bo, L.; Lin, L.; Chen, Z.-S. Establishment and characterization of an irinotecan-resistant human colon cancer cell line. Front. Oncol. 2020, 10, 3371. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef] [PubMed]
- Espelt, M.V.; Bacigalupo, M.L.; Carabias, P.; Troncoso, M.F. MicroRNAs contribute to ATP-binding cassette transporter-and autophagy-mediated chemoresistance in hepatocellular carcinoma. World J. Hepatol. 2019, 11, 344. [Google Scholar] [CrossRef]
- Hussain, S.A.; Marouf, B.H. Silibinin improves the cytotoxicity of methotrexate in chemo resistant human rhabdomyosarcoma cell lines. Saudi. Med. J. 2013, 34, 1145–1150. [Google Scholar]
- Pandey, A.; Mai, J.; Tripathi, S.C.; Hanash, S.M.; Shen, H.; Mitra, S. Inhibition of PLK1 Abrogates Side Population and Increases Radiation-Induced DNA Damage in Human Glioblastoma. Cancer Res. 2018, 78 (Suppl. S13), 4811. [Google Scholar]
- Wu, Z.; Wei, N. Knockdown of PLK1 inhibits invasion and promotes apoptosis in glioma cells through regulating autophagy. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2723–2733. [Google Scholar]
- Abbou, S.; Lanvers-Kaminsky, C.; Daudigeos-Dubus, E.; Le Dret, L.; Laplace-Builhe, C.; Molenaar, J.; Vassal, G.; Geoerger, B. Polo-like kinase inhibitor volasertib exhibits antitumor activity and synergy with vincristine in pediatric malignancies. Anticancer Res. 2016, 36, 599–609. [Google Scholar]
- Hugle, M.; Belz, K.; Fulda, S. Identification of synthetic lethality of PLK1 inhibition and microtubule-destabilizing drugs. Cell Death Differ. 2015, 22, 1946–1956. [Google Scholar] [CrossRef]
- Stehle, A.; Hugle, M.; Fulda, S. Eribulin synergizes with Polo-like kinase 1 inhibitors to induce apoptosis in rhabdomyosarcoma. Cancer Lett. 2015, 365, 37–46. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Ma, W.; Gething, M.J.; Sambrook, J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell 1993, 74, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Shamu, C.E.; Walter, P. Oligomerization and phosphorylation of the Ire1p kinase during intracellular signaling from the endoplasmic reticulum to the nucleus. EMBO J. 1996, 15, 3028–3039. [Google Scholar] [CrossRef]
- Sidrauski, C.; Walter, P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell 1997, 90, 1031–1039. [Google Scholar] [CrossRef]
- Liu, C.Y.; Schroder, M.; Kaufman, R.J. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J. Biol. Chem. 2000, 275, 24881–24885. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Sharma, P.; Alizadeh, J.; Juarez, M.; Samali, A.; Halayko, A.J.; Kenyon, N.J.; Ghavami, S.; Zeki, A.A. Autophagy, apoptosis, the unfolded protein response, and lung function in idiopathic pulmonary fibrosis. Cells 2021, 10, 1642. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Lu, Y.; Liang, F.X.; Wang, X. A synthetic biology approach identifies the mammalian UPR RNA ligase RtcB. Mol. Cell 2014, 55, 758–770. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Hollien, J.; Weissman, J.S. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science 2006, 313, 104–107. [Google Scholar] [CrossRef]
- Hollien, J.; Lin, J.H.; Li, H.; Stevens, N.; Walter, P.; Weissman, J.S. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009, 186, 323–3231. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef] [PubMed]
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef]
- Iurlaro, R.; Munoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef]
- McCarthy, N.; Dolgikh, N.; Logue, S.; Patterson, J.B.; Zeng, Q.; Gorman, A.M.; Samali, A.; Fulda, S. The IRE1 and PERK arms of the unfolded protein response promote survival of rhabdomyosarcoma cells. Cancer Lett. 2020, 490, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Aghaei, M.; Nasimian, A.; Rahmati, M.; Kawalec, P.; Machaj, F.; Rosik, J.; Bhushan, B.; Bathaie, S.Z.; Azarpira, N.; Los, M.J.; et al. The Role of BiP and the IRE1alpha-XBP1 Axis in Rhabdomyosarcoma Pathology. Cancers 2021, 13, 4927. [Google Scholar] [CrossRef] [PubMed]
- Sabnis, A.J.; Guerriero, C.J.; Olivas, V.; Sayana, A.; Shue, J.; Flanagan, J.; Asthana, S.; Paton, A.W.; Paton, J.C.; Gestwicki, J.E.; et al. Combined chemical-genetic approach identifies cytosolic HSP70 dependence in rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2016, 113, 9015–9020. [Google Scholar] [CrossRef] [PubMed]
- Sannino, S.; Guerriero, C.J.; Sabnis, A.J.; Stolz, D.B.; Wallace, C.T.; Wipf, P.; Watkins, S.C.; Bivona, T.G.; Brodsky, J.L. Compensatory increases of select proteostasis networks after Hsp70 inhibition in cancer cells. J. Cell. Sci. 2018, 131, 217760. [Google Scholar] [CrossRef]
- Zhao, N.; Cao, J.; Xu, L.; Tang, Q.; Dobrolecki, L.E.; Lv, X.; Talukdar, M.; Lu, Y.; Wang, X.; Hu, D.Z.; et al. Pharmacological targeting of MYC-regulated IRE1/XBP1 pathway suppresses MYC-driven breast cancer. J. Clin. Investig. 2018, 128, 1283–1299. [Google Scholar] [CrossRef] [PubMed]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnow, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1alpha-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019, 10, 323. [Google Scholar] [CrossRef]
- Mixon, B.A.; Eckrich, M.J.; Lowas, S.; Engel, M.E. Vincristine, irinotecan, and temozolomide for treatment of relapsed alveolar rhabdomyosarcoma. J. Pediatr. Hematol. Oncol. 2013, 35, e163–e166. [Google Scholar] [CrossRef]
- Manzella, G.; Schreck, L.D.; Breunis, W.B.; Molenaar, J.; Merks, H.; Barr, F.G.; Sun, W.; Rommele, M.; Zhang, L.; Tchinda, J.; et al. Phenotypic profiling with a living biobank of primary rhabdomyosarcoma unravels disease heterogeneity and AKT sensitivity. Nat. Commun. 2020, 11, 4629. [Google Scholar] [CrossRef]
- Gengenbacher, N.; Singhal, M.; Augustin, H.G. Preclinical mouse solid tumour models: Status quo, challenges and perspectives. Nat. Rev. Cancer 2017, 17, 751–765. [Google Scholar] [CrossRef]
- Kendsersky, N.M.; Lindsay, J.; Kolb, E.A.; Smith, M.A.; Teicher, B.A.; Erickson, S.W.; Earley, E.J.; Mosse, Y.P.; Martinez, D.; Pogoriler, J.; et al. The B7-H3-targeting antibody-drug conjugate m276-SL-PBD is potently effective against pediatric cancer preclinical solid tumor models. Clin. Cancer Res. 2021, 27, 2938–2946. [Google Scholar] [CrossRef]
- Risbridger, G.P.; Lawrence, M.G.; Taylor, R.A. PDX: Moving beyond drug screening to versatile models for research discovery. J. Endocr. Soc. 2020, 4, bvaa132. [Google Scholar] [CrossRef]
- Imle, R.; Kommoss, F.K.F.; Banito, A. Preclinical in vivo modeling of pediatric sarcoma-promises and limitations. J. Clin. Med. 2021, 10, 1578. [Google Scholar] [CrossRef]
- Grobner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef]
- Bhimani, J.; Ball, K.; Stebbing, J. Patient-derived xenograft models-the future of personalised cancer treatment. Br. J. Cancer 2020, 122, 601–602. [Google Scholar] [CrossRef]
- Pham, T.Q.; Robinson, K.; Xu, L.; Pavlova, M.N.; Skapek, S.X.; Chen, E.Y. HDAC6 promotes growth, migration/invasion, and self-renewal of rhabdomyosarcoma. Oncogene 2021, 40, 578–591. [Google Scholar] [CrossRef]
- Ignatius, M.S.; Hayes, M.N.; Moore, F.E.; Tang, Q.; Garcia, S.P.; Blackburn, P.R.; Baxi, K.; Wang, L.; Jin, A.; Ramakrishnan, A.; et al. tp53 deficiency causes a wide tumor spectrum and increases embryonal rhabdomyosarcoma metastasis in zebrafish. Elife 2018, 7, e37202. [Google Scholar] [CrossRef]
- Hayes, M.N.; McCarthy, K.; Jin, A.; Oliveira, M.L.; Iyer, S.; Garcia, S.P.; Sindiri, S.; Gryder, B.; Motala, Z.; Nielsen, G.P.; et al. Vangl2/RhoA signaling pathway regulates stem cell self-renewal programs and growth in rhabdomyosarcoma. Cell Stem Cell 2018, 22, 414–427.e6. [Google Scholar] [CrossRef]
- Ignatius, M.S.; Hayes, M.N.; Lobbardi, R.; Chen, E.Y.; McCarthy, K.M.; Sreenivas, P.; Motala, Z.; Durbin, A.D.; Molodtsov, A.; Reeder, S.; et al. The NOTCH1/SNAIL1/MEF2C pathway regulates growth and self-renewal in embryonal rhabdomyosarcoma. Cell Rep. 2017, 19, 2304–2318. [Google Scholar] [CrossRef]
- Tenente, I.M.; Hayes, M.N.; Ignatius, M.S.; McCarthy, K.; Yohe, M.; Sindiri, S.; Gryder, B.; Oliveira, M.L.; Ramakrishnan, A.; Tang, Q.; et al. Myogenic regulatory transcription factors regulate growth in rhabdomyosarcoma. Elife 2017, 6, e19214. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; DeRan, M.T.; Ignatius, M.S.; Grandinetti, K.B.; Clagg, R.; McCarthy, K.M.; Lobbardi, R.M.; Brockmann, J.; Keller, C.; Wu, X.; et al. Glycogen synthase kinase 3 inhibitors induce the canonical WNT/beta-catenin pathway to suppress growth and self-renewal in embryonal rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, 5349–5354. [Google Scholar] [CrossRef] [PubMed]
- Le, X.; Pugach, E.K.; Hettmer, S.; Storer, N.Y.; Liu, J.; Wills, A.A.; DiBiase, A.; Chen, E.Y.; Ignatius, M.S.; Poss, K.D.; et al. A novel chemical screening strategy in zebrafish identifies common pathways in embryogenesis and rhabdomyosarcoma development. Development 2013, 140, 2354–2364. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.E.; Cantor, E.L.; Ehlen, M.S.; Banerjee, A.; Malempati, S.; Stenzel, P.; Woltjer, R.L.; Gandour-Edwards, R.; Goodwin, N.C.; Yang, Y.; et al. A Patient-derived xenograft model of parameningeal embryonal rhabdomyosarcoma for preclinical studies. Sarcoma 2015, 2015, 826124. [Google Scholar] [CrossRef] [PubMed]
- Stewart, E.; Federico, S.; Karlstrom, A.; Shelat, A.; Sablauer, A.; Pappo, A.; Dyer, M.A. The childhood solid tumor network: A new resource for the developmental biology and oncology research communities. Dev. Biol. 2016, 411, 287–293. [Google Scholar] [CrossRef]
- Comiskey, D.F., Jr.; Jacob, A.G.; Sanford, B.L.; Montes, M.; Goodwin, A.K.; Steiner, H.; Matsa, E.; Tapia-Santos, A.S.; Bebee, T.W.; Grieves, J.; et al. A novel mouse model of rhabdomyosarcoma underscores the dichotomy of MDM2-ALT1 function in vivo. Oncogene 2018, 37, 95–106. [Google Scholar] [CrossRef]
- Grunewald, T.G.; Alonso, M.; Avnet, S.; Banito, A.; Burdach, S.; Cidre-Aranaz, F.; Di Pompo, G.; Distel, M.; Dorado-Garcia, H.; Garcia-Castro, J.; et al. Sarcoma treatment in the era of molecular medicine. EMBO Mol. Med. 2020, 12, e11131. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Roveri, M.; Pfohl, A.; Jaaks, P.; Alijaj, N.; Leroux, J.C.; Luciani, P.; Bernasconi, M. Prolonged circulation and increased tumor accumulation of liposomal vincristine in a mouse model of rhabdomyosarcoma. Nanomedicine 2017, 12, 1135–1151. [Google Scholar] [CrossRef]
- Kemp, C.J. Animal models of chemical carcinogenesis: Driving breakthroughs in cancer research for 100 years. Cold Spring Harb. Protoc. 2015, 2015, 865–874. [Google Scholar] [CrossRef]
- Li, J.J.; Kovach, A.R.; DeMonia, M.; Slemmons, K.K.; Oristian, K.M.; Chen, C.; Linardic, C.M. Expression of oncogenic HRAS in human Rh28 and RMS-YM rhabdomyosarcoma cells leads to oncogene-induced senescence. Sci. Rep. 2021, 11, 16505. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Kawagoe, R.; Sasai, K.; Li, Y.; Russell, H.R.; Curran, T.; McKinnon, P.J. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 2007, 26, 6442–6447. [Google Scholar] [CrossRef]
- Gutierrez, W.R.; Scherer, A.; McGivney, G.R.; Brockman, Q.R.; Knepper-Adrian, V.; Laverty, E.A.; Roughton, G.A.; Dodd, R.D. Divergent immune landscapes of primary and syngeneic Kras-driven mouse tumor models. Sci. Rep. 2021, 11, 1098. [Google Scholar] [CrossRef] [PubMed]
- Zanola, A.; Rossi, S.; Faggi, F.; Monti, E.; Fanzani, A. Rhabdomyosarcomas: An overview on the experimental animal models. J. Cell. Mol. Med. 2012, 16, 1377–1391. [Google Scholar] [CrossRef]
- Rokita, J.L.; Rathi, K.S.; Cardenas, M.F.; Upton, K.A.; Jayaseelan, J.; Cross, K.L.; Pfeil, J.; Egolf, L.E.; Way, G.P.; Farrel, A.; et al. Genomic profiling of childhood tumor patient-derived xenograft models to enable rational clinical trial design. Cell Rep. 2019, 29, 1675–1689.e9. [Google Scholar] [CrossRef]
- Lu, W.; Chao, T.; Ruiqi, C.; Juan, S.; Zhihong, L. Patient-derived xenograft models in musculoskeletal malignancies. J. Transl. Med. 2018, 16, 107. [Google Scholar] [CrossRef]
- Chen, E.Y.; Langenau, D.M. Zebrafish models of rhabdomyosarcoma. Methods Cell Biol. 2011, 105, 383–402. [Google Scholar] [CrossRef]
- Casey, M.J.; Stewart, R.A. Pediatric cancer models in zebrafish. Trends Cancer 2020, 6, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, F.; Fahs, A.; Ghayad, S.E.; Saab, R. Signaling pathways in Rhabdomyosarcoma invasion and metastasis. Cancer Metastasis. Rev. 2020, 39, 287–301. [Google Scholar] [CrossRef]
- Zibat, A.; Missiaglia, E.; Rosenberger, A.; Pritchard-Jones, K.; Shipley, J.; Hahn, H.; Fulda, S. Activation of the hedgehog pathway confers a poor prognosis in embryonal and fusion gene-negative alveolar rhabdomyosarcoma. Oncogene 2010, 29, 6323–6330. [Google Scholar] [CrossRef]
- Ali, S.; Champagne, D.L.; Spaink, H.P.; Richardson, M.K. Zebrafish embryos and larvae: A new generation of disease models and drug screens. Birth Defects Res. C Embryo Today 2011, 93, 115–133. [Google Scholar] [CrossRef]
- Etchin, J.; Kanki, J.; Look, A.T. Zebrafish as a model for the study of human cancer. In The Zebrafish: Disease Models and Chemical Screens; Detrich, H.W., Weterfield, M., Zon, L.I.B., Eds.; Academic Press: Cambridge, MA, USA, 2011; Volume 105, pp. 309–337. [Google Scholar]
- Yan, C.; Brunson, D.C.; Tang, Q.; Do, D.; Iftimia, N.A.; Moore, J.C.; Hayes, M.N.; Welker, A.M.; Garcia, E.G.; Dubash, T.D.; et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell 2019, 177, 1903–1914.e14. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, S.; Ratnakumar, K.; Langdon, E.; Kansler, E.; Kim, I.; Campbell, N.R.; Perry, E.; McMahon, A.; Kaufman, C.; van Rooijen, E.; et al. A quantitative system for studying metastasis using transparent zebrafish. Cancer Res. 2015, 75, 4272–4282. [Google Scholar] [CrossRef] [PubMed]
- Scientists see human cancer in zebrafish. Cancer Discov. 2019, 9, 819–820. [CrossRef]
- Ignatius, M.S.; Chen, E.; Elpek, N.M.; Fuller, A.Z.; Tenente, I.M.; Clagg, R.; Liu, S.; Blackburn, J.S.; Linardic, C.M.; Rosenberg, A.E.; et al. In vivo imaging of tumor-propagating cells, regional tumor heterogeneity, and dynamic cell movements in embryonal rhabdomyosarcoma. Cancer Cell 2012, 21, 680–693. [Google Scholar] [CrossRef] [PubMed]
- Kendall, G.C.; Watson, S.; Xu, L.; LaVigne, C.A.; Murchison, W.; Rakheja, D.; Skapek, S.X.; Tirode, F.; Delattre, O.; Amatruda, J.F. PAX3-FOXO1 transgenic zebrafish models identify HES3 as a mediator of rhabdomyosarcoma tumorigenesis. Elife 2018, 7, e33800. [Google Scholar] [CrossRef]
- Le, X.; Langenau, D.M.; Keefe, M.D.; Kutok, J.L.; Neuberg, D.S.; Zon, L.I. Heat shock-inducible Cre/Lox approaches to induce diverse types of tumors and hyperplasia in transgenic zebrafish. Proc. Natl. Acad. Sci. USA 2007, 104, 9410–9415. [Google Scholar] [CrossRef]
- Tang, Q.; Moore, J.C.; Ignatius, M.S.; Tenente, I.M.; Hayes, M.N.; Garcia, E.G.; Torres Yordan, N.; Bourque, C.; He, S.; Blackburn, J.S.; et al. Imaging tumour cell heterogeneity following cell transplantation into optically clear immune-deficient zebrafish. Nat. Commun. 2016, 7, 10358. [Google Scholar] [CrossRef]
- Hayes, M.N.; Langenau, D.M. Discovering novel oncogenic pathways and new therapies using zebrafish models of sarcoma. In Zebrafish; Detrich, H.W., Westerfield, M., Zon, L., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 138, pp. 525–561. [Google Scholar]
- Storer, N.Y.; White, R.M.; Uong, A.; Price, E.; Nielsen, G.P.; Langenau, D.M.; Zon, L.I. Zebrafish rhabdomyosarcoma reflects the developmental stage of oncogene expression during myogenesis. Development 2013, 140, 3040–3050. [Google Scholar] [CrossRef]
- Tenente, I.M.; Tang, Q.; Moore, J.C.; Langenau, D.M. Normal and malignant muscle cell transplantation into immune compromised adult zebrafish. J. Vis. Exp. 2014, 94, e52597. [Google Scholar] [CrossRef]
- Rai, R.; Raval, R.; Khandeparker, R.V.; Chidrawar, S.K.; Khan, A.A.; Ganpat, M.S. Tissue engineering: Step ahead in maxillofacial reconstruction. J. Int. Oral Health JIOH 2015, 7, 138–142. [Google Scholar]
- Kang, H.-W.; Lee, S.J.; Ko, I.K.; Kengla, C.; Yoo, J.J.; Atala, A. A 3D bioprinting system to produce human-scale tissue constructs with structural integrity. Nat. Biotechnol. 2016, 34, 312–319. [Google Scholar] [CrossRef]
- Ma, X.; Qu, X.; Zhu, W.; Li, Y.-S.; Yuan, S.; Zhang, H.; Liu, J.; Wang, P.; Lai, C.S.E.; Zanella, F. Deterministically patterned biomimetic human iPSC-derived hepatic model via rapid 3D bioprinting. Proc. Natl. Acad. Sci. USA 2016, 113, 2206–2211. [Google Scholar] [CrossRef]
- Zhu, W.; Qu, X.; Zhu, J.; Ma, X.; Patel, S.; Liu, J.; Wang, P.; Lai, C.S.E.; Gou, M.; Xu, Y. Direct 3D bioprinting of prevascularized tissue constructs with complex microarchitecture. Biomater 2017, 124, 106–115. [Google Scholar] [CrossRef]
- Jammalamadaka, U.; Tappa, K. Recent advances in biomaterials for 3D printing and tissue engineering. J. Funct. Biomater. 2018, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Ma, X.; Zhu, W.; Wang, P.; Miller, K.L.; Stupin, J.; Koroleva-Maharajh, A.; Hairabedian, A.; Chen, S. Scanningless and continuous 3D bioprinting of human tissues with decellularized extracellular matrix. J. Biomater. 2019, 194, 1–13. [Google Scholar] [CrossRef]
- Wang, P.; Berry, D.; Moran, A.; He, F.; Tam, T.; Chen, L.; Chen, S. Controlled growth factor release in 3D-Printed hydrogels. Adv. Healthc. Mater. 2020, 9, e1900977. [Google Scholar] [CrossRef]
- Sinha, S.K. Additive manufacturing (AM) of medical devices and scaffolds for tissue engineering based on 3D and 4D printing. In 3D and 4D Printing of Polymer Nanocomposite Materials; Elsevier: Amsterdam, The Netherlands, 2020; pp. 119–160. [Google Scholar] [CrossRef]
- Chen, A.; Su, J.; Li, Y.; Zhang, H.; Shi, Y.; Yan, C.; Lu, J. 3D/4D printed bio-piezoelectric smart scaffolds for next-generation bone tissue engineering. Int. J. Extrem. Manuf. 2023, 5, 032007. [Google Scholar] [CrossRef]
- Mohapatra, S.; Kar, R.K.; Biswal, P.K.; Bindhani, S. Approaches of 3D printing in current drug delivery. Sens. Int. 2022, 3, 100146. [Google Scholar] [CrossRef]
- Pavan Kalyan, B.G.; Kumar, L. 3D Printing: Applications in Tissue Engineering, Medical Devices, and Drug Delivery. AAPS PharmSciTech 2022, 23, 92. [Google Scholar] [CrossRef]
- Ramezani, M.; Mohd Ripin, Z. 4D Printing in Biomedical Engineering: Advancements, Challenges, and Future Directions. J. Funct. Biomater. 2023, 14, 347. [Google Scholar] [CrossRef]
- Kim, J.H.; Seol, Y.-J.; Ko, I.K.; Kang, H.-W.; Lee, Y.K.; Yoo, J.J.; Atala, A.; Lee, S.J. 3D bioprinted human skeletal muscle constructs for muscle function restoration. Sci. Rep. 2018, 8, 12307. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-J.; Jun, Y.-J.; Kim, D.Y.; Yi, H.-G.; Chae, S.-H.; Kang, J.; Lee, J.; Gao, G.; Kong, J.-S.; Jang, J. A 3D cell printed muscle construct with tissue-derived bioink for the treatment of volumetric muscle loss. Biomater 2019, 206, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Mozetic, P.; Giannitelli, S.M.; Gori, M.; Trombetta, M.; Rainer, A. Engineering muscle cell alignment through 3D bioprinting. J. Biom. Mater. Res. A 2017, 105, 2582–2588. [Google Scholar] [CrossRef] [PubMed]
- Mendonça, G.; Edwards, S.P.; Mayers, C.A.; Meneghetti, P.C.; Liu, F. Digital immediate complete denture for a patient with rhabdomyosarcoma: A clinical report. J. Prosthodont. 2021, 30, 196–201. [Google Scholar] [CrossRef]
- O’Sullivan, A.; Duffy, E.; O’Sullivan, K.; Cronin, U.; Lyons, E.; O’Sullivan, L.; Twomey, F. Bespoke 3D printed eye cover for teen with rhabdomyosarcoma. BMJ Support Palliat. Care 2021, 2021, 002900. [Google Scholar] [CrossRef]
- Tejo-Otero, A.; Fenollosa-Artés, F.; Uceda, R.; Castellví-Fernández, A.; Lustig-Gainza, P.; Valls-Esteve, A.; Ayats-Soler, M.; Munuera, J.; Buj-Corral, I.; Krauel, L. 3D printed prototype of a complex neuroblastoma for preoperative surgical planning. Ann. 3D Print. Med. 2021, 2, 100014. [Google Scholar] [CrossRef]
- Tejo-Otero, A.; Lustig-Gainza, P.; Fenollosa-Artés, F.; Valls, A.; Krauel, L.; Buj-Corral, I. 3D printed soft surgical planning prototype for a biliary tract rhabdomyosarcoma. J. Mech. Behav. Biomed. Mater. 2020, 109, 103844. [Google Scholar] [CrossRef]
- Shin, Y.C.; Kim, C.; Song, S.-J.; Jun, S.; Kim, C.-S.; Hong, S.W.; Hyon, S.-H.; Han, D.-W.; Oh, J.-W. Ternary aligned nanofibers of RGD peptide-displaying M13 bacteriophage/PLGA/graphene oxide for facilitated myogenesis. Nanotheranostics 2018, 2, 144. [Google Scholar] [CrossRef]
- Zhang, Z.; Klausen, L.H.; Chen, M.; Dong, M. Electroactive scaffolds for neurogenesis and myogenesis: Graphene-based nanomaterials. Small 2018, 14, 1801983. [Google Scholar] [CrossRef]
- Kim, W.; Kim, G. A functional bioink and its application in myoblast alignment and differentiation. J. Chem. Eng. 2019, 366, 150–162. [Google Scholar] [CrossRef]
- Kim, Y.; Pagan-Diaz, G.; Gapinske, L.; Kim, Y.; Suh, J.; Solomon, E.; Harris, J.F.; Nam, S.; Bashir, R. Integration of graphene electrodes with 3D skeletal muscle tissue models. Adv. Healthc. Mater. 2020, 9, 1901137. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.S.; Kang, J.I.; Le Thi, P.; Park, K.M.; Hong, S.W.; Choi, Y.S.; Han, D.-W.; Park, K.D. Three-dimensional printable gelatin hydrogels incorporating Graphene Oxide to enable spontaneous myogenic differentiation. ACS Macro Lett. 2021, 10, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Bilge, S.; Ergene, E.; Talak, E.; Gokyer, S.; Donar, Y.O.; Sınağ, A.; Huri, P.Y. Recycled algae-based carbon materials as electroconductive 3D printed skeletal muscle tissue engineering scaffolds. J. Mater. Sci. Mater. Med. 2021, 32, 73. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.S.; Mostafavi, A.; Quint, J.P.; Panayi, A.C.; Baldino, K.; Williams, T.J.; Daubendiek, J.G.; Hugo Sánchez, V.; Bonick, Z.; Trujillo-Miranda, M.; et al. In Situ Printing of Adhesive Hydrogel Scaffolds for the Treatment of Skeletal Muscle Injuries. ACS Appl. Bio. Mater. 2020, 3, 1568–1579. [Google Scholar] [CrossRef]
- Lee, V.K.; Lanzi, A.M.; Ngo, H.; Yoo, S.-S.; Vincent, P.A.; Dai, G. Generation of multi-scale vascular network system within 3D hydrogel using 3D bio-printing technology. Cell Mol. Bioeng. 2014, 7, 460–472. [Google Scholar] [CrossRef]
- Hakimi, N.; Cheng, R.; Leng, L.; Sotoudehfar, M.; Ba, P.Q.; Bakhtyar, N.; Amini-Nik, S.; Jeschke, M.G.; Günther, A. Handheld skin printer: In situ formation of planar biomaterials and tissues. Lab. Chip. 2018, 18, 1440–1451. [Google Scholar] [CrossRef]
- D O’Connell, C.; Di Bella, C.; Thompson, F.; Augustine, C.; Beirne, S.; Cornock, R.; Richards, C.J.; Chung, J.; Gambhir, S.; Yue, Z. Development of the Biopen: A handheld device for surgical printing of adipose stem cells at a chondral wound site. Biofabrication 2016, 8, 015019. [Google Scholar] [CrossRef] [PubMed]
- Quint, J.P.; Mostafavi, A.; Endo, Y.; Panayi, A.; Russell, C.S.; Nourmahnad, A.; Wiseman, C.; Abbasi, L.; Samandari, M.; Sheikhi, A. In vivo printing of Nanoenabled scaffolds for the treatment of skeletal muscle injuries. Adv. Healthc. Mater. 2021, 10, 2002152. [Google Scholar] [CrossRef]
- Chae, M.P.; Rozen, W.M.; McMenamin, P.G.; Findlay, M.W.; Spychal, R.T.; Hunter-Smith, D.J. Emerging applications of bedside 3D printing in plastic surgery. Front. Surg. 2015, 2, 25. [Google Scholar] [CrossRef]
- Krauel, L.; Fenollosa, F.; Riaza, L.; Pérez, M.; Tarrado, X.; Morales, A.; Gomà, J.; Mora, J. Use of 3D prototypes for complex surgical oncologic cases. World J. Surg. 2016, 40, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Rebong, R.E.; Stewart, K.T.; Utreja, A.; Ghoneima, A.A. Accuracy of three-dimensional dental resin models created by fused deposition modeling, stereolithography, and Polyjet prototype technologies: A comparative study. Angle Orthod. 2018, 88, 363–369. [Google Scholar] [CrossRef]
- Adams, F.; Qiu, T.; Mark, A.; Fritz, B.; Kramer, L.; Schlager, D.; Wetterauer, U.; Miernik, A.; Fischer, P. Soft 3D-printed phantom of the human kidney with collecting system. Ann. Biomed. Eng. 2017, 45, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Dini, D.; y Baena, F.R.; Forte, A.E. Composite hydrogel: A high fidelity soft tissue mimic for surgery. Mater. Des. 2018, 160, 886–894. [Google Scholar] [CrossRef]
- Tejo-Otero, A.; Buj-Corral, I.; Fenollosa-Artés, F. 3D printing in medicine for preoperative surgical planning: A review. Ann. Biomed. Eng. 2020, 48, 536–555. [Google Scholar] [CrossRef]
- van de Belt, T.H.; Nijmeijer, H.; Grim, D.; Engelen, L.J.; Vreeken, R.; van Gelder, M.M.; Ter Laan, M. Patient-specific actual-size three-dimensional printed models for patient education in glioma treatment: First experiences. World Neurosurg 2018, 117, e99–e105. [Google Scholar] [CrossRef]
- Biglino, G.; Koniordou, D.; Gasparini, M.; Capelli, C.; Leaver, L.-K.; Khambadkone, S.; Schievano, S.; Taylor, A.M.; Wray, J. Piloting the use of patient-specific cardiac models as a novel tool to facilitate communication during cinical consultations. Pediatr. Cardiol. 2017, 38, 813–818. [Google Scholar] [CrossRef]
- Bernhard, J.-C.; Isotani, S.; Matsugasumi, T.; Duddalwar, V.; Hung, A.J.; Suer, E.; Baco, E.; Satkunasivam, R.; Djaladat, H.; Metcalfe, C. Personalized 3D printed model of kidney and tumor anatomy: A useful tool for patient education. World J. Urol. 2016, 34, 337–345. [Google Scholar] [CrossRef]
- Agarwal, T.; Hann, S.Y.; Chiesa, I.; Cui, H.; Celikkin, N.; Micalizzi, S.; Barbetta, A.; Costantini, M.; Esworthy, T.; Zhang, L.G.; et al. 4D printing in biomedical applications: Emerging trends and technologies. J. Mater. Chem. B 2021, 9, 7608–7632. [Google Scholar] [CrossRef] [PubMed]
- Saska, S.; Pilatti, L.; Blay, A.; Shibli, J.A. Bioresorbable polymers: Advanced materials and 4D printing for tissue engineering. Polymers 2021, 13, 563. [Google Scholar] [CrossRef] [PubMed]
- Wan, Z.; Zhang, P.; Liu, Y.; Lv, L.; Zhou, Y. Four-dimensional bioprinting: Current developments and applications in bone tissue engineering. Acta Biomater. 2020, 101, 26–42. [Google Scholar] [CrossRef]
- Apsite, I.; Constante, G.; Dulle, M.; Vogt, L.; Caspari, A.; Boccaccini, A.R.; Synytska, A.; Salehi, S.; Ionov, L. 4D Biofabrication of fibrous artificial nerve graft for neuron regeneration. Biofabrication 2020, 12, 035027. [Google Scholar] [CrossRef]
- Aigner, T.; Scheibel, T. Self-rolling refillable tubular enzyme containers made of recombinant spider silk and chitosan. ACS Appl. Bio. Mater. Interfaces 2019, 11, 15290–15297. [Google Scholar] [CrossRef] [PubMed]
- Kirillova, A.; Maxson, R.; Stoychev, G.; Gomillion, C.T.; Ionov, L. 4D biofabrication using shape-morphing hydrogels. Adv. Mater. 2017, 29, 1703443. [Google Scholar] [CrossRef]
- Zhang, L.; Xiang, Y.; Zhang, H.; Cheng, L.; Mao, X.; An, N.; Zhang, L.; Zhou, J.; Deng, L.; Zhang, Y. A Biomimetic 3D-Self-Forming Approach for Microvascular Scaffolds. Adv. Sci. 2020, 7, 1903553. [Google Scholar] [CrossRef] [PubMed]
- Stroganov, V. 4D Biofabrication Using Self-Folding Polymers. Doctoral Dissertation, University of Bayreuth, Bayreuth, Germany, 2018. [Google Scholar]
- Hendrikson, W.J.; Rouwkema, J.; Clementi, F.; Van Blitterswijk, C.A.; Farè, S.; Moroni, L. Towards 4D printed scaffolds for tissue engineering: Exploiting 3D shape memory polymers to deliver time-controlled stimulus on cultured cells. Biofabrication 2017, 9, 031001. [Google Scholar] [CrossRef]
- Miao, S.; Nowicki, M.; Cui, H.; Lee, S.-J.; Zhou, X.; Mills, D.K.; Zhang, L.G. 4D anisotropic skeletal muscle tissue constructs fabricated by staircase effect strategy. Biofabrication 2019, 11, 035030. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-Y.; Xie, H.; Xu, Z.-y.; Li, L.; Jiang, M.-N.; Tang, L.; Yang, K.-K.; Wang, Y.-Z. 4D printing of shape memory aliphatic copolyester via UV-assisted FDM strategy for medical protective devices. J. Chem. Eng. 2020, 396, 125242. [Google Scholar] [CrossRef]
- Wan, X.; Wei, H.; Zhang, F.; Liu, Y.; Leng, J. 3D printing of shape memory poly(d,l-lactide-co-trimethylene carbonate) by direct ink writing for shape-changing structures. J. Appl. Polym. Sci. 2019, 136, 48177. [Google Scholar] [CrossRef]
- Podstawczyk, D.; Nizioł, M.; Szymczyk-Ziółkowska, P.; Fiedot-Toboła, M. Development of thermoinks for 4D direct printing of temperature-induced self-rolling hydrogel actuators. Adv. Funct. Mater. 2021, 31, 2009664. [Google Scholar] [CrossRef]
- Nishiguchi, A.; Zhang, H.; Schweizerhof, S.; Schulte, M.F.; Mourran, A.; Möller, M. 4D Printing of a Light-Driven Soft Actuator with Programmed Printing Density. ACS. Appl. Bio. Mater. Interfaces 2020, 12, 12176–12185. [Google Scholar] [CrossRef]
- Constante, G.; Apsite, I.; Alkhamis, H.; Dulle, M.; Schwarzer, M.; Caspari, A.; Synytska, A.; Salehi, S.; Ionov, L. 4D Biofabrication Using a Combination of 3D Printing and Melt-Electrowriting of Shape-Morphing Polymers. ACS Appl. Bio. Mater. Interfaces 2021, 13, 12767–12776. [Google Scholar] [CrossRef] [PubMed]
- Miao, S.; Castro, N.; Nowicki, M.; Xia, L.; Cui, H.; Zhou, X.; Zhu, W.; Lee, S.-j.; Sarkar, K.; Vozzi, G.; et al. 4D printing of polymeric materials for tissue and organ regeneration. Mater. Today 2017, 20, 577–591. [Google Scholar] [CrossRef]
- Li, M.; Nagamori, E.; Kino-oka, M. Disruption of myoblast alignment by highly motile rhabdomyosarcoma cell in tissue structure. J. Biosci. Bioeng. 2017, 123, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Stefanek, E.; Samiei, E.; Kavoosi, M.; Esmaeillou, M.; Roustai Geraylow, K.; Emami, A.; Ashrafizadeh, M.; Perrin, D.; Gordon, J.W.; Akbari, M.; et al. A bioengineering method for modeling alveolar rhabdomyosarcoma and assessing chemotherapy responses. MethodsX 2021, 8, 101473. [Google Scholar] [CrossRef]
- Roll, W.; Dirksen, U.; Weckesser, M. Update PET in der pädiatrischen onkologie. Der. Nuklearmediziner. 2018, 41, 211–221. [Google Scholar] [CrossRef][Green Version]
- Van Winkle, P.; Angiolillo, A.; Krailo, M.; Cheung, Y.K.; Anderson, B.; Davenport, V.; Reaman, G.; Cairo, M.S. Ifosfamide, carboplatin, and etoposide (ICE) reinduction chemotherapy in a large cohort of children and adolescents with recurrent/refractory sarcoma: The Children’s Cancer Group (CCG) experience. Pediatr. Blood Cancer 2005, 44, 338–347. [Google Scholar] [CrossRef]
- Breitfeld, P.P.; Lyden, E.; Raney, R.B.; Teot, L.A.; Wharam, M.; Lobe, T.; Crist, W.M.; Maurer, H.M.; Donaldson, S.S.; Ruymann, F.B. Ifosfamide and etoposide are superior to vincristine and melphalan for pediatric metastatic rhabdomyosarcoma when administered with irradiation and combination chemotherapy: A report from the Intergroup Rhabdomyosarcoma Study Group. J. Pediatr. Hematol. Oncol. 2001, 23, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Defachelles, A.-S.; Bogart, E.; Casanova, M.; Merks, J.H.; Bisogno, G.; Calareso, G.; Gallego Melcon, S.; Gatz, S.A.; Le Deley, M.-C.; McHugh, K. Randomized phase II trial of vincristine-irinotecan with or without temozolomide, in children and adults with relapsed or refractory rhabdomyosarcoma: A European Paediatric Soft tissue Sarcoma Study Group and Innovative Therapies for Children With Cancer trial. J. Clin. Oncol. 2021, 39, 2979–2990. [Google Scholar] [PubMed]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef]
- Sasaki, K.; Tsuno, N.H.; Sunami, E.; Tsurita, G.; Kawai, K.; Okaji, Y.; Nishikawa, T.; Shuno, Y.; Hongo, K.; Hiyoshi, M. Chloroquine potentiates the anti-cancer effect of 5-fluorouracil on colon cancer cells. BMC Cancer 2010, 10, 370. [Google Scholar] [CrossRef]
- Yang, H.-Z.; Ma, Y.; Zhou, Y.; Xu, L.-M.; Chen, X.-J.; Ding, W.-B.; Zou, H.-B. Autophagy contributes to the enrichment and survival of colorectal cancer stem cells under oxaliplatin treatment. Cancer Lett. 2015, 361, 128–136. [Google Scholar] [CrossRef]
- Han, W.; Sun, J.; Feng, L.; Wang, K.; Li, D.; Pan, Q.; Chen, Y.; Jin, W.; Wang, X.; Pan, H. Autophagy inhibition enhances daunorubicin-induced apoptosis in K562 cells. PLoS ONE 2011, 6, e28491. [Google Scholar] [CrossRef]
- Peng, Q.; Qin, J.; Zhang, Y.; Cheng, X.; Wang, X.; Lu, W.; Xie, X.; Zhang, S. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J. Exp. Clin. Cancer Res. 2017, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.-C.; Wang, H.-C.; Hou, Y.-C.; Tung, H.-L.; Chiu, T.-J.; Shan, Y.-S. Blockade of autophagy reduces pancreatic cancer stem cell activity and potentiates the tumoricidal effect of gemcitabine. Mol. Cancer 2015, 14, 179. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, D.; Bläuer, M.; Hirota, M.; Ikonen, N.H.; Sand, J.; Laukkarinen, J. Autophagy is needed for the growth of pancreatic adenocarcinoma and has a cytoprotective effect against anticancer drugs. Eur. J. Cancer 2014, 50, 1382–1390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Si, S.; Schoen, S.; Chen, J.; Jin, X.-B.; Wu, G. Suppression of autophagy enhances preferential toxicity of paclitaxel to folliculin-deficient renal cancer cells. J. Exp. Clin. Cancer Res. 2013, 32, 99. [Google Scholar] [CrossRef] [PubMed]
- Quan, Y.; Lei, H.; Wahafu, W.; Liu, Y.; Ping, H.; Zhang, X. Inhibition of autophagy enhances the anticancer effect of enzalutamide on bladder cancer. Biomed. Pharmacother. 2019, 120, 109490. [Google Scholar] [CrossRef]
- Wang, F.; Tang, J.; Li, P.; Si, S.; Yu, H.; Yang, X.; Tao, J.; Lv, Q.; Gu, M.; Yang, H. Chloroquine enhances the radiosensitivity of bladder cancer cells by inhibiting autophagy and activating apoptosis. Cell. Physiol. Biochem. 2018, 45, 54–66. [Google Scholar] [CrossRef]
- Domagala, A.; Stachura, J.; Gabrysiak, M.; Muchowicz, A.; Zagozdzon, R.; Golab, J.; Firczuk, M. Inhibition of autophagy sensitizes cancer cells to Photofrin-based photodynamic therapy. BMC. Cancer 2018, 18, 210. [Google Scholar] [CrossRef]
- Hao, C.; Liu, G.; Tian, G. Autophagy inhibition of cancer stem cells promotes the efficacy of cisplatin against non-small cell lung carcinoma. Ther. Adv. Respir. Dis. 2019, 13, 1753466619866097. [Google Scholar] [CrossRef]
- Liu, F.; Liu, D.; Yang, Y.; Zhao, S. Effect of autophagy inhibition on chemotherapy-induced apoptosis in A549 lung cancer cells. Oncol. Lett. 2013, 5, 1261–1265. [Google Scholar] [CrossRef]
- Ren, J.-H.; He, W.-S.; Nong, L.; Zhu, Q.-Y.; Hu, K.; Zhang, R.-G.; Huang, L.-L.; Zhu, F.; Wu, G. Acquired cisplatin resistance in human lung adenocarcinoma cells is associated with enhanced autophagy. Cancer Biother. Radiopharm. 2010, 25, 75–80. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, S.; Tombolan, L.; Saggioro, M.; Frasson, C.; Rampazzo, E.; Pellegrini, S.; Favaretto, F.; Biz, C.; Ruggieri, P.; Gamba, P.; et al. Rhabdomyosarcoma cells produce their own extracellular matrix with minimal involvement of cancer-associated fibroblasts: A preliminary study. Front. Oncol. 2020, 10, 600980. [Google Scholar] [CrossRef]
- Liu, P.; Fan, J.; Wang, Z.; Zai, W.; Song, P.; Li, Y.; Ju, D. The role of autophagy in the cytotoxicity induced by trastuzumab emtansine (T-DM1) in HER2-positive breast cancer cells. AMB Express 2020, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- McCreery, K.P.; Xu, X.; Scott, A.K.; Fajrial, A.K.; Calve, S.; Ding, X.; Neu, C.P. Nuclear stiffness decreases with disruption of the extracellular matrix in living tissues. Small 2021, 17, e2006699. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
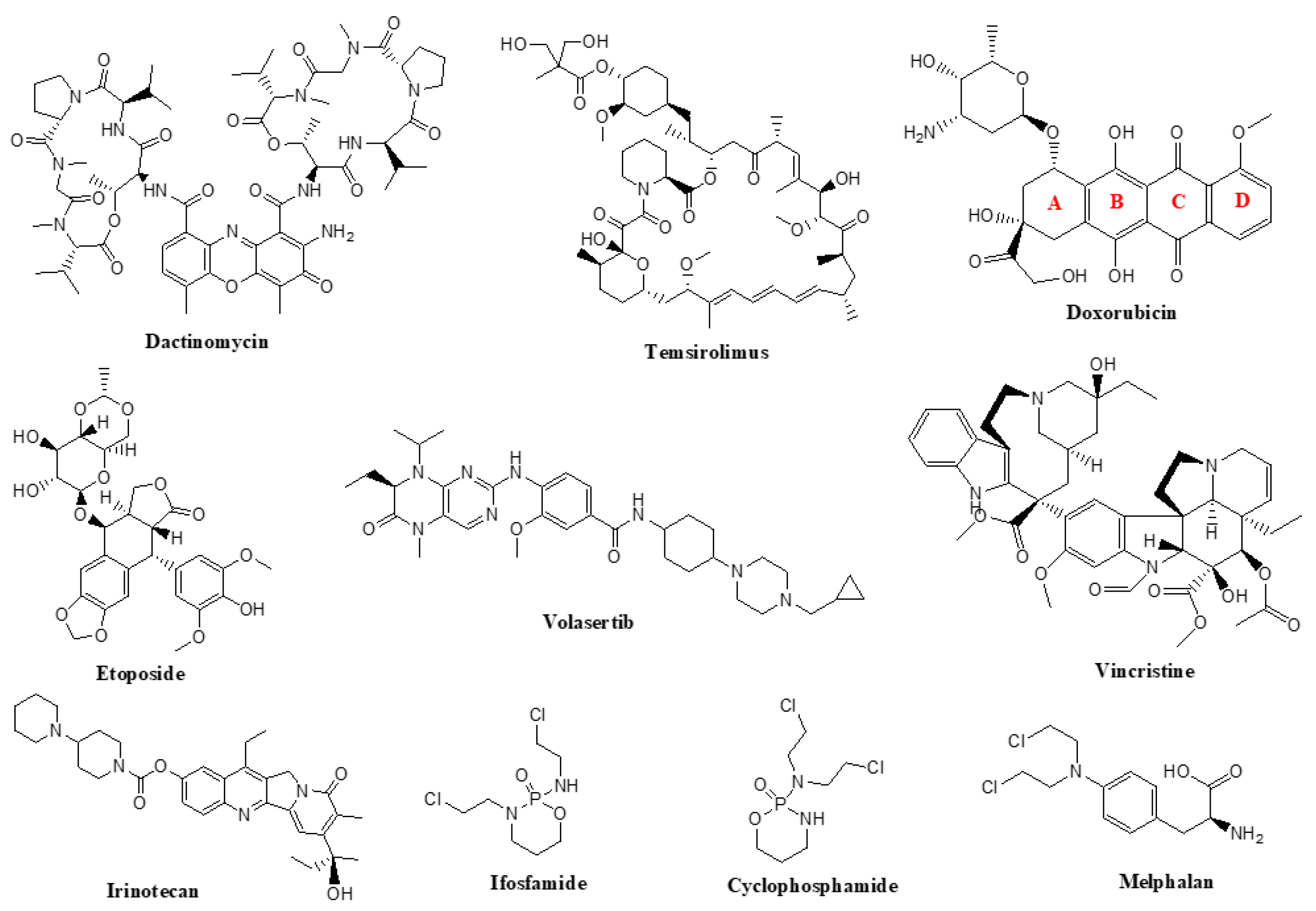{kind=link}
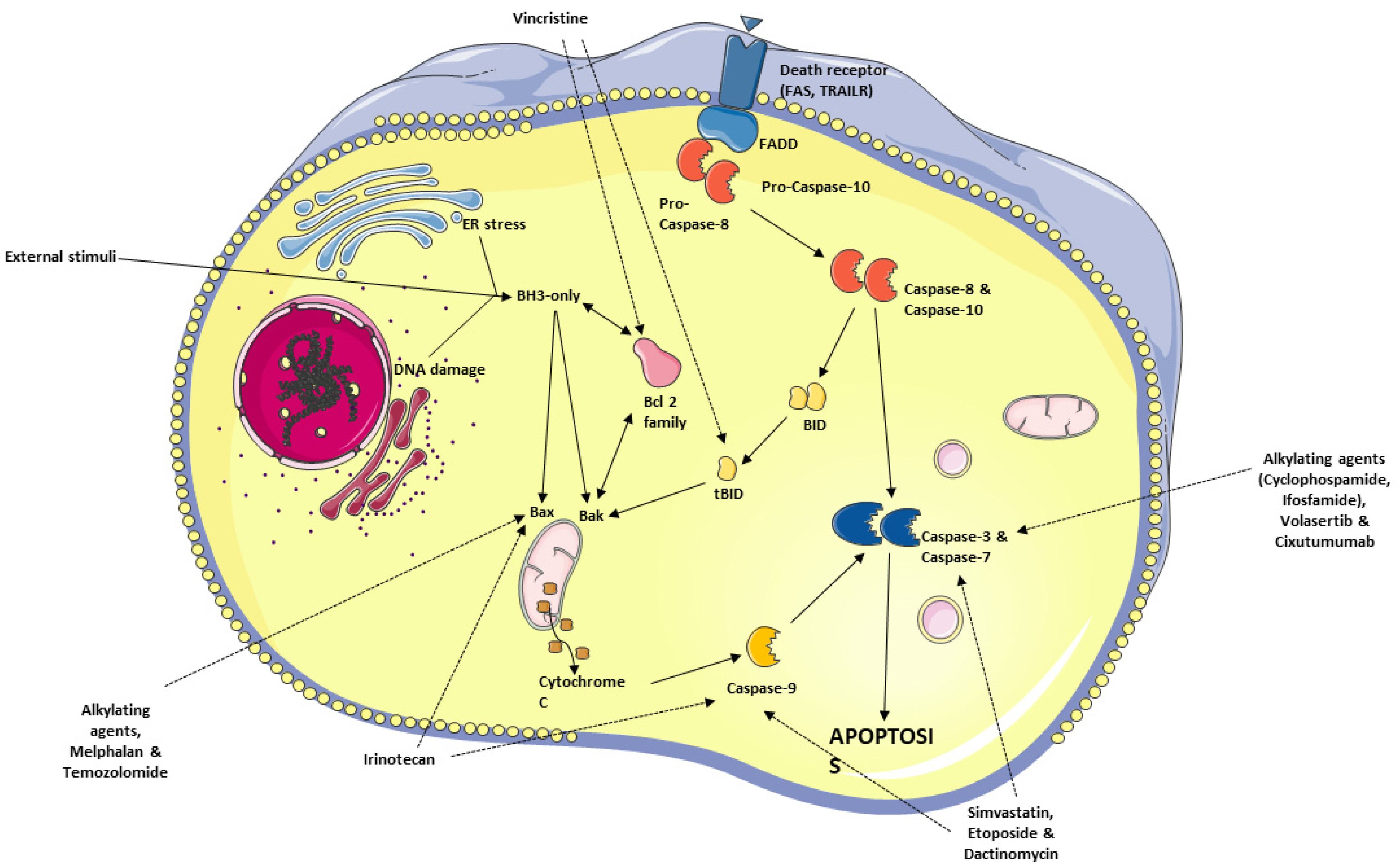{kind=link}
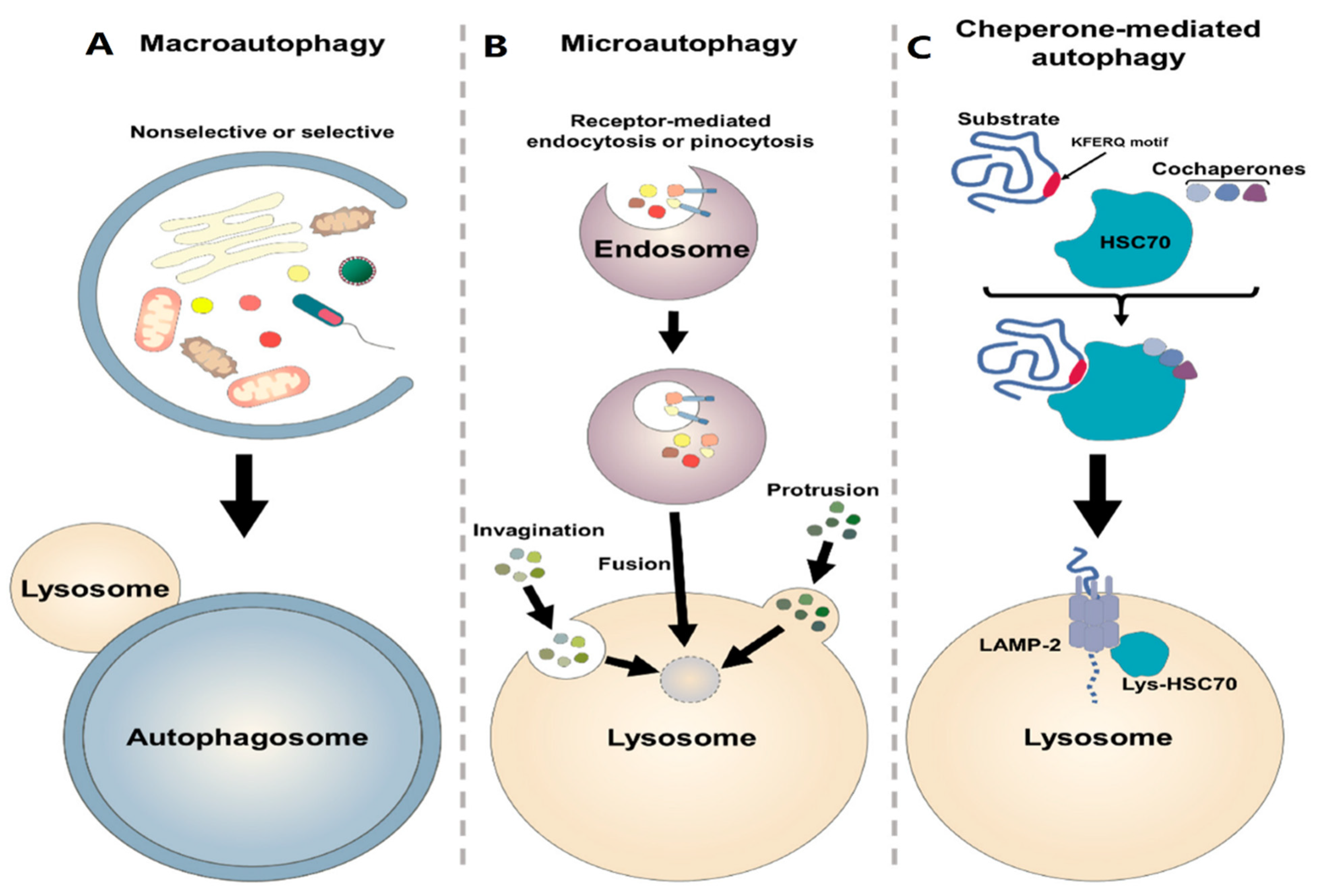{kind=link}
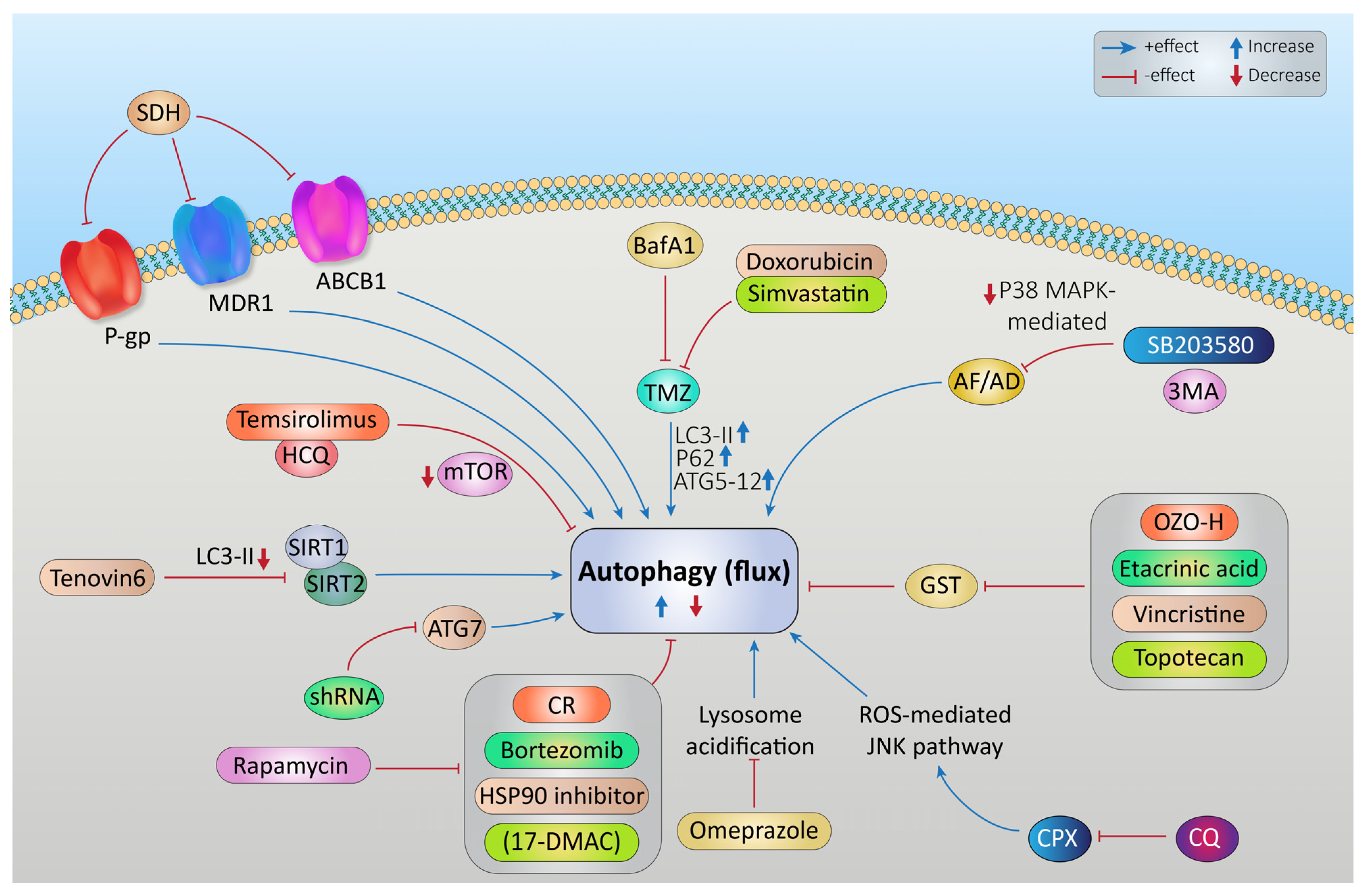{kind=link}
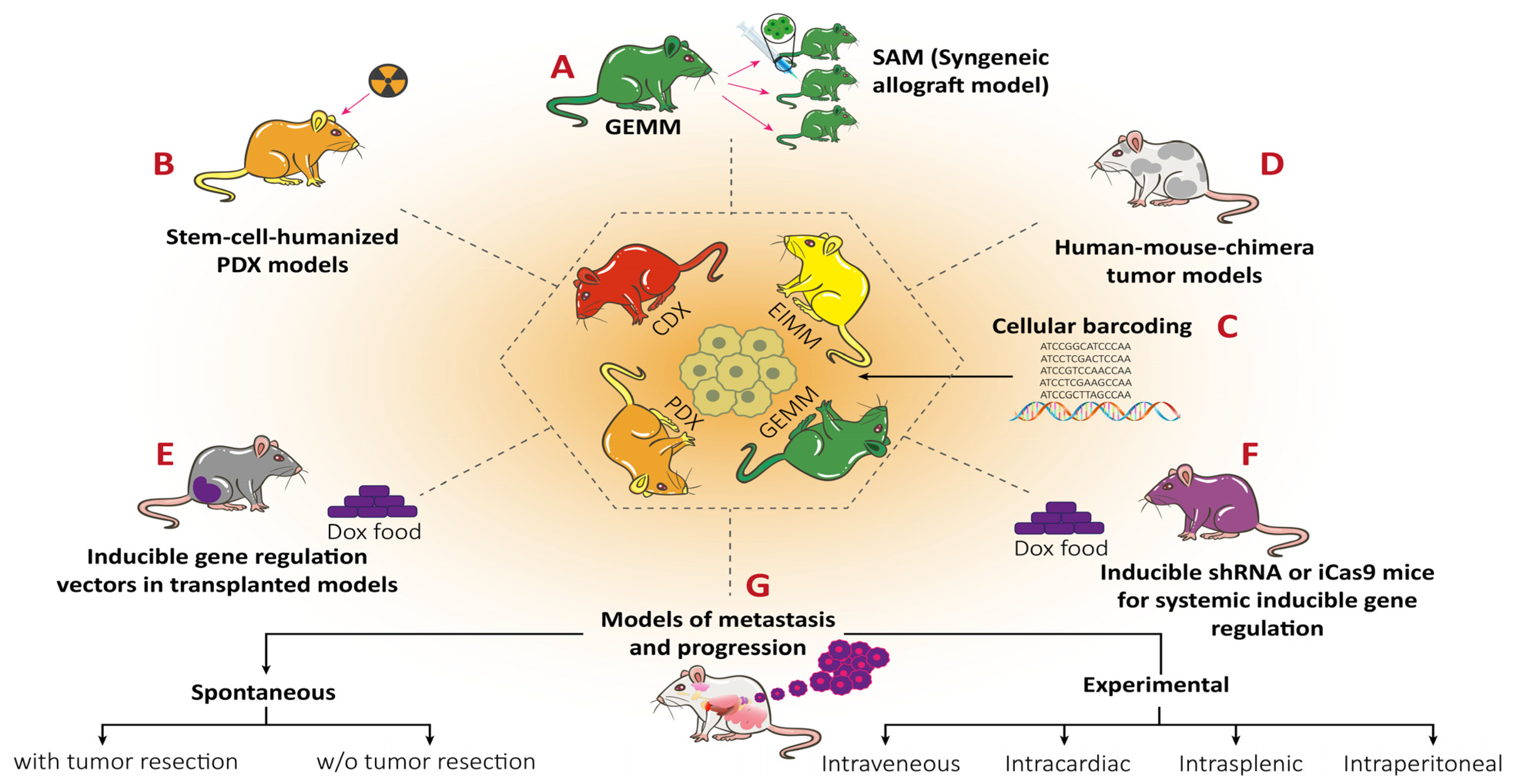{kind=link}
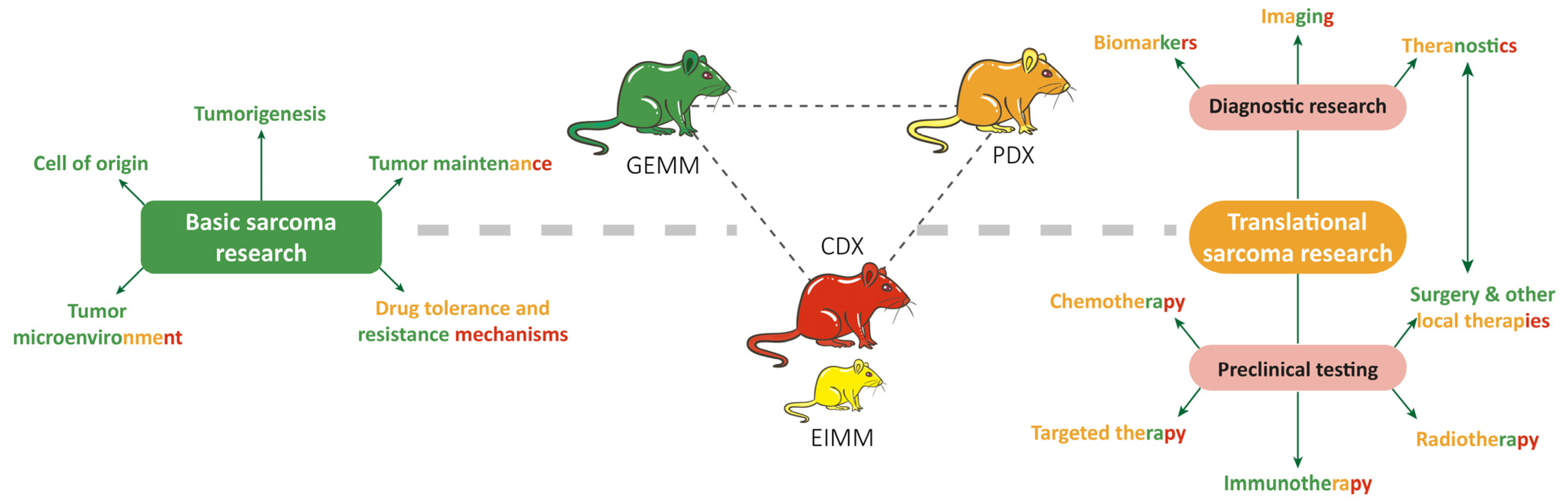{kind=link}
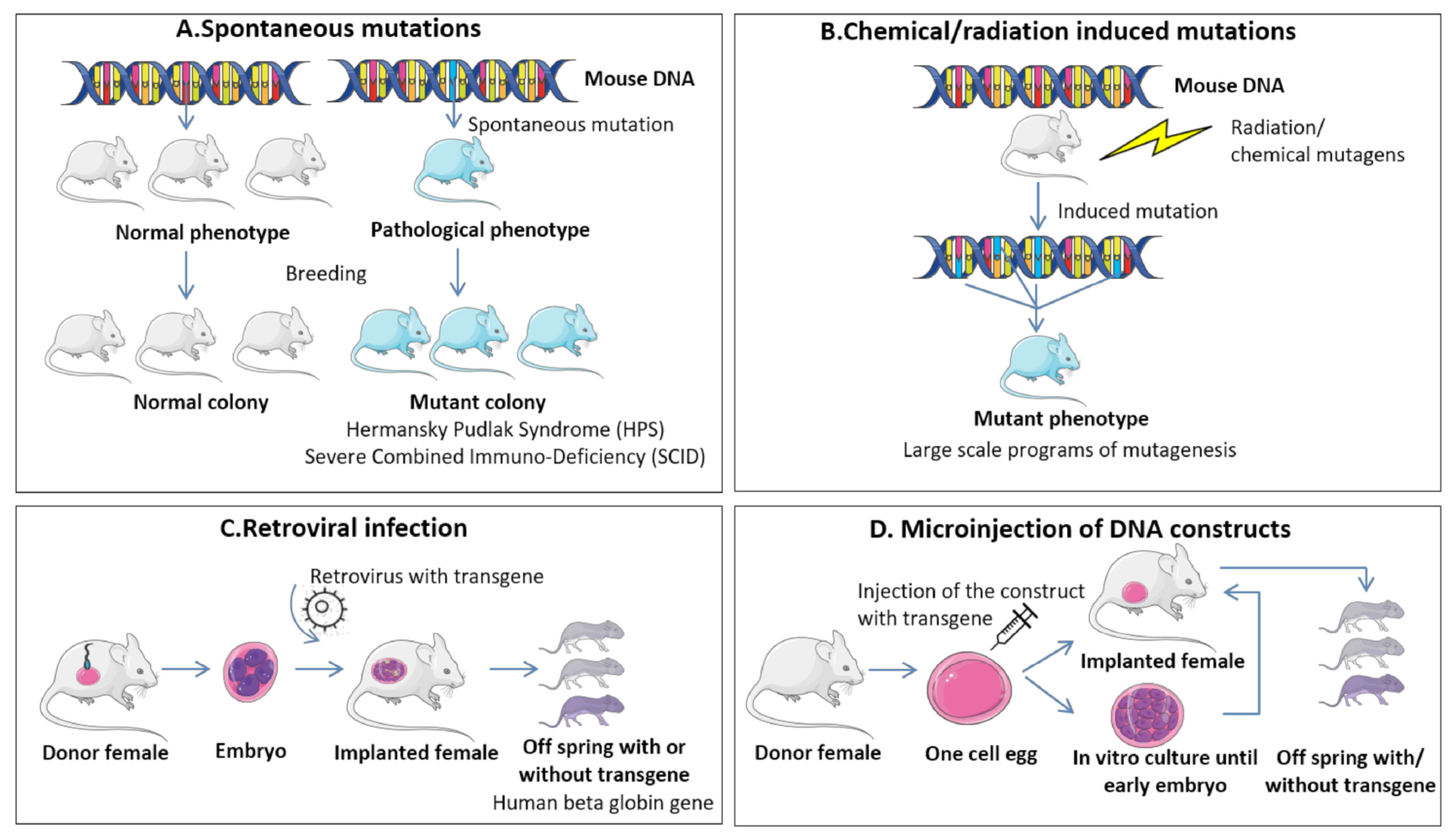{kind=link}
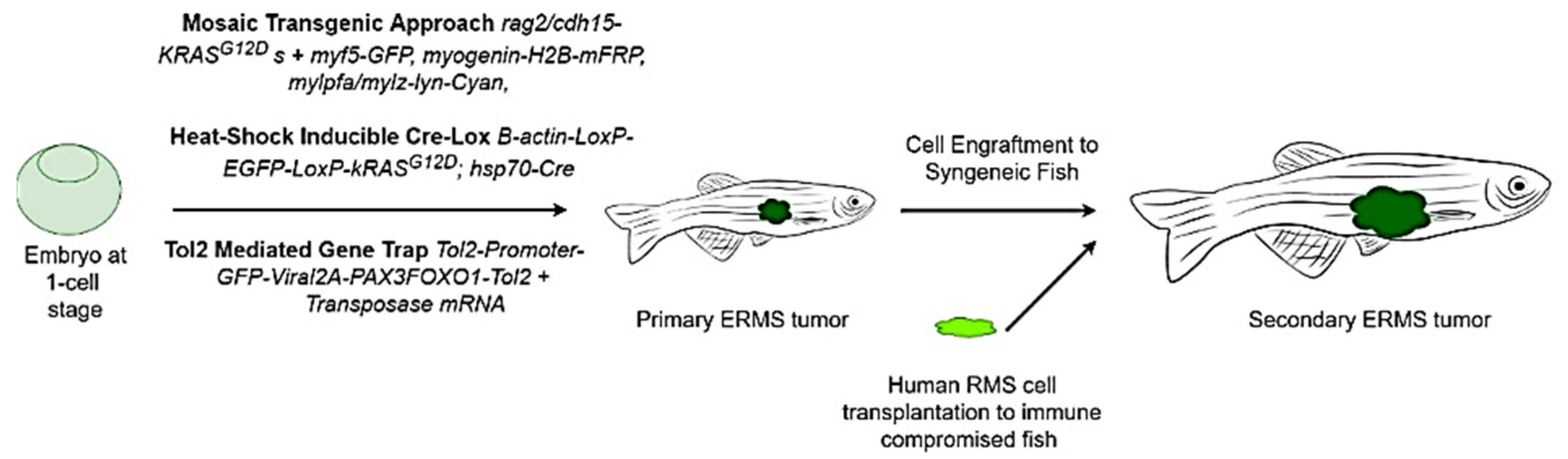{kind=link}
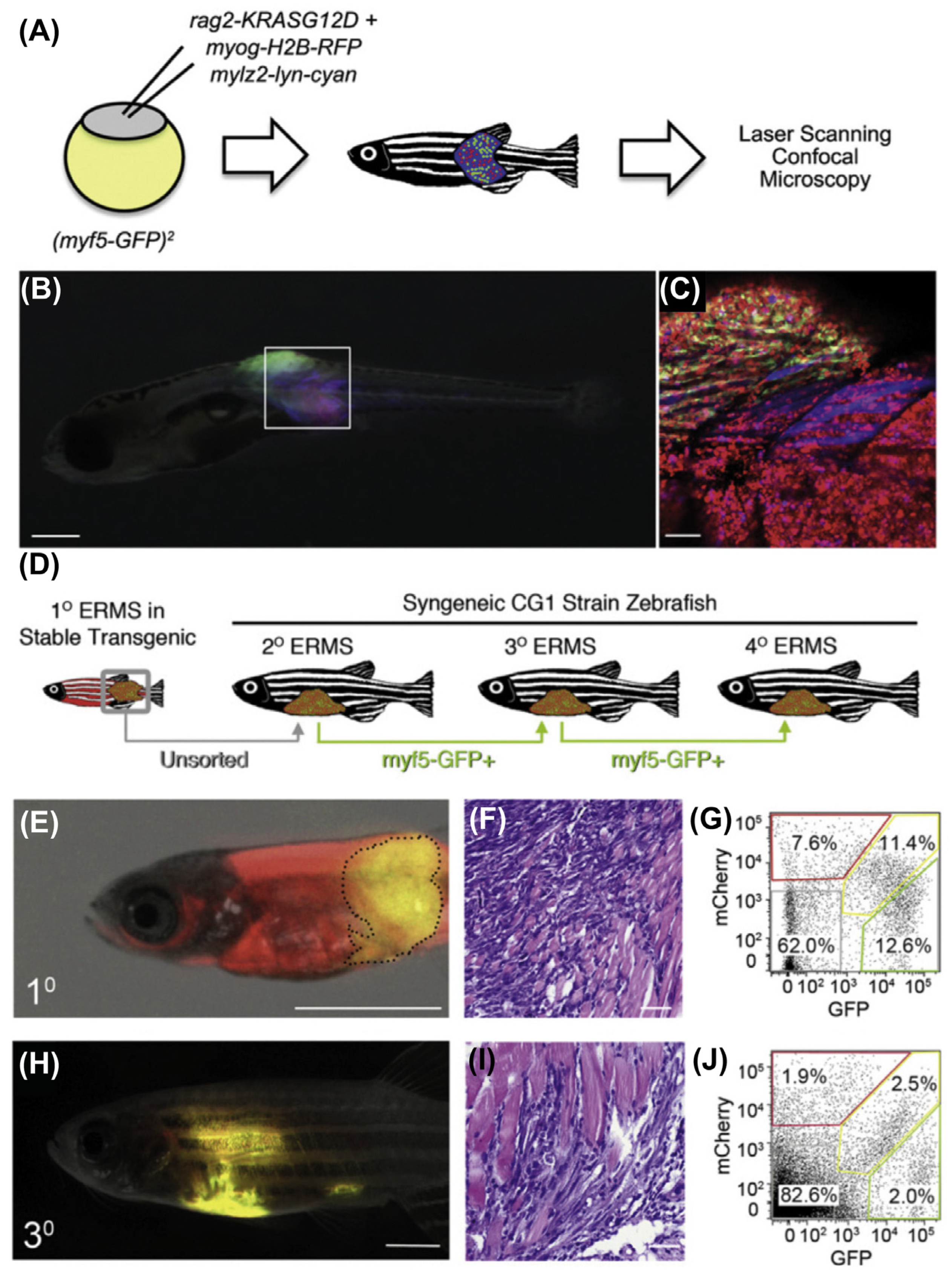{kind=link}
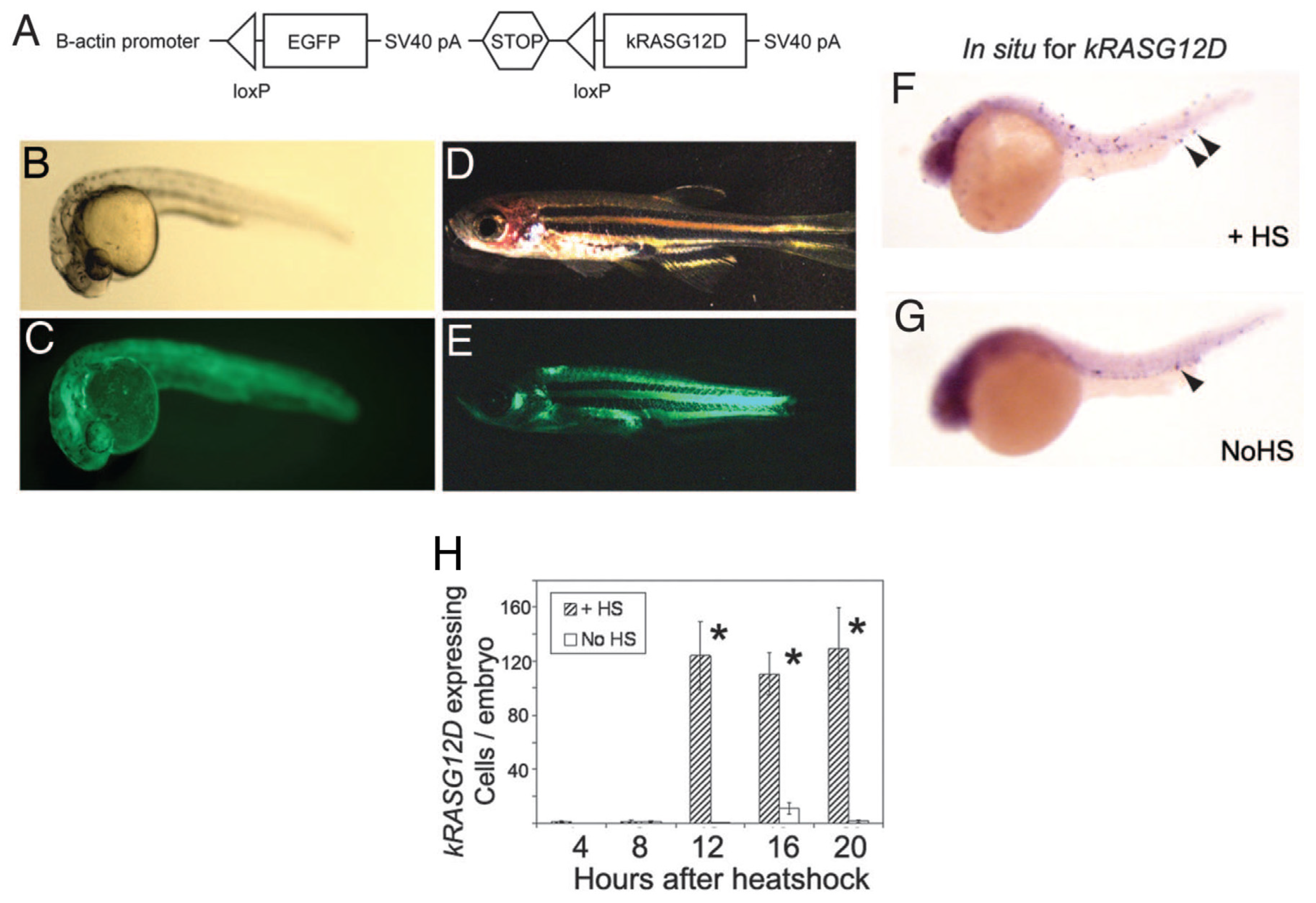{kind=link}
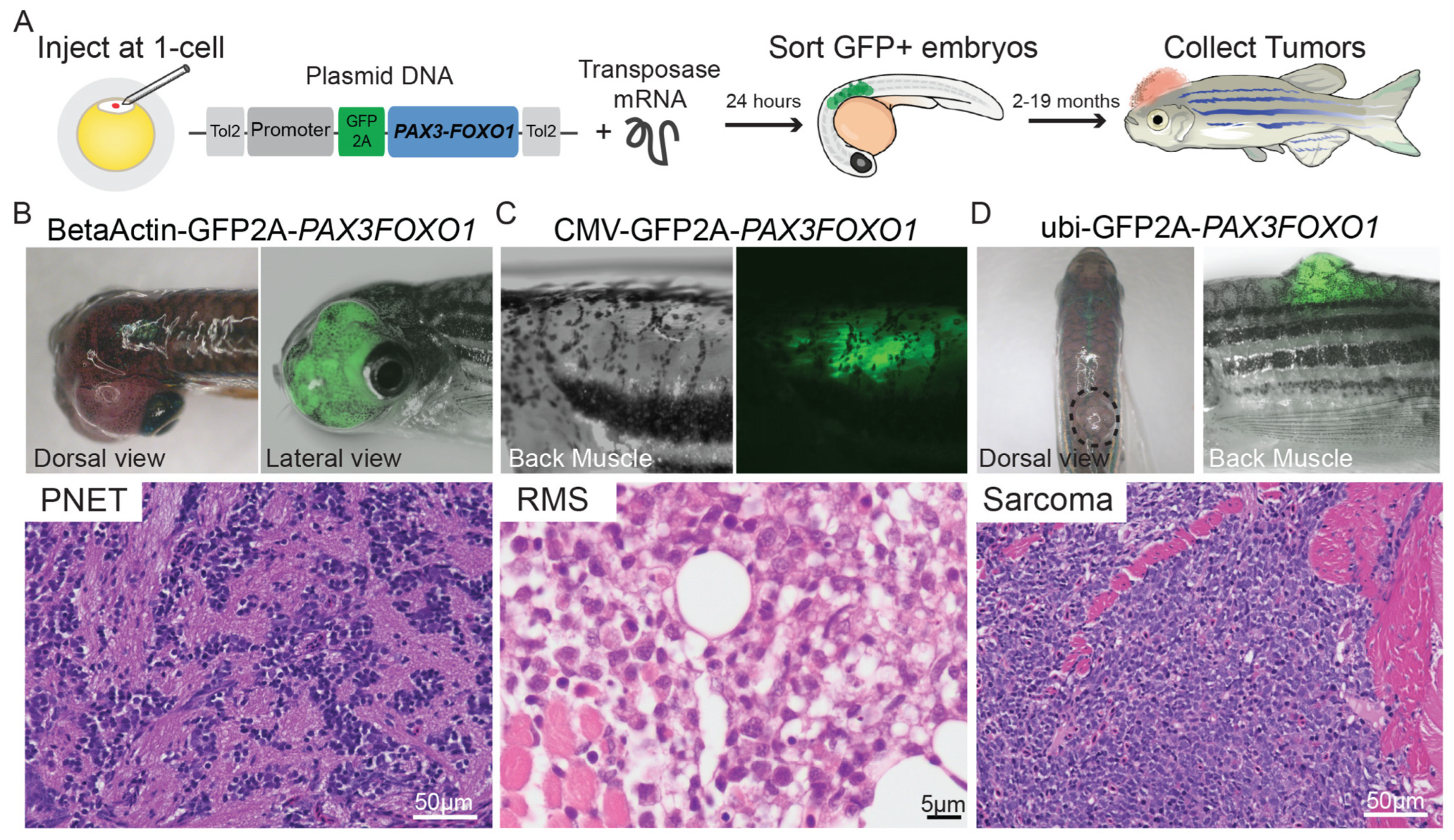{kind=link}
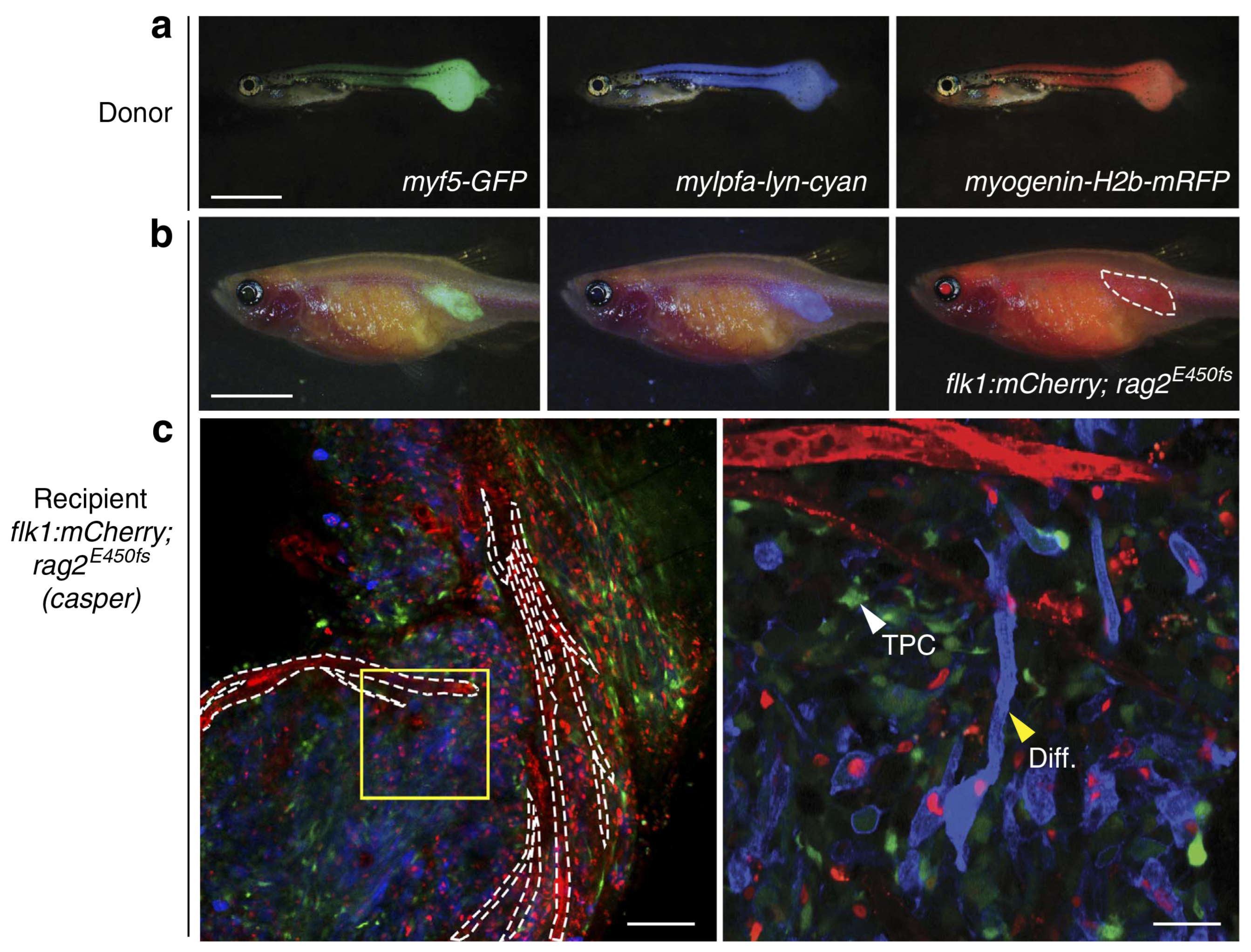{kind=link}
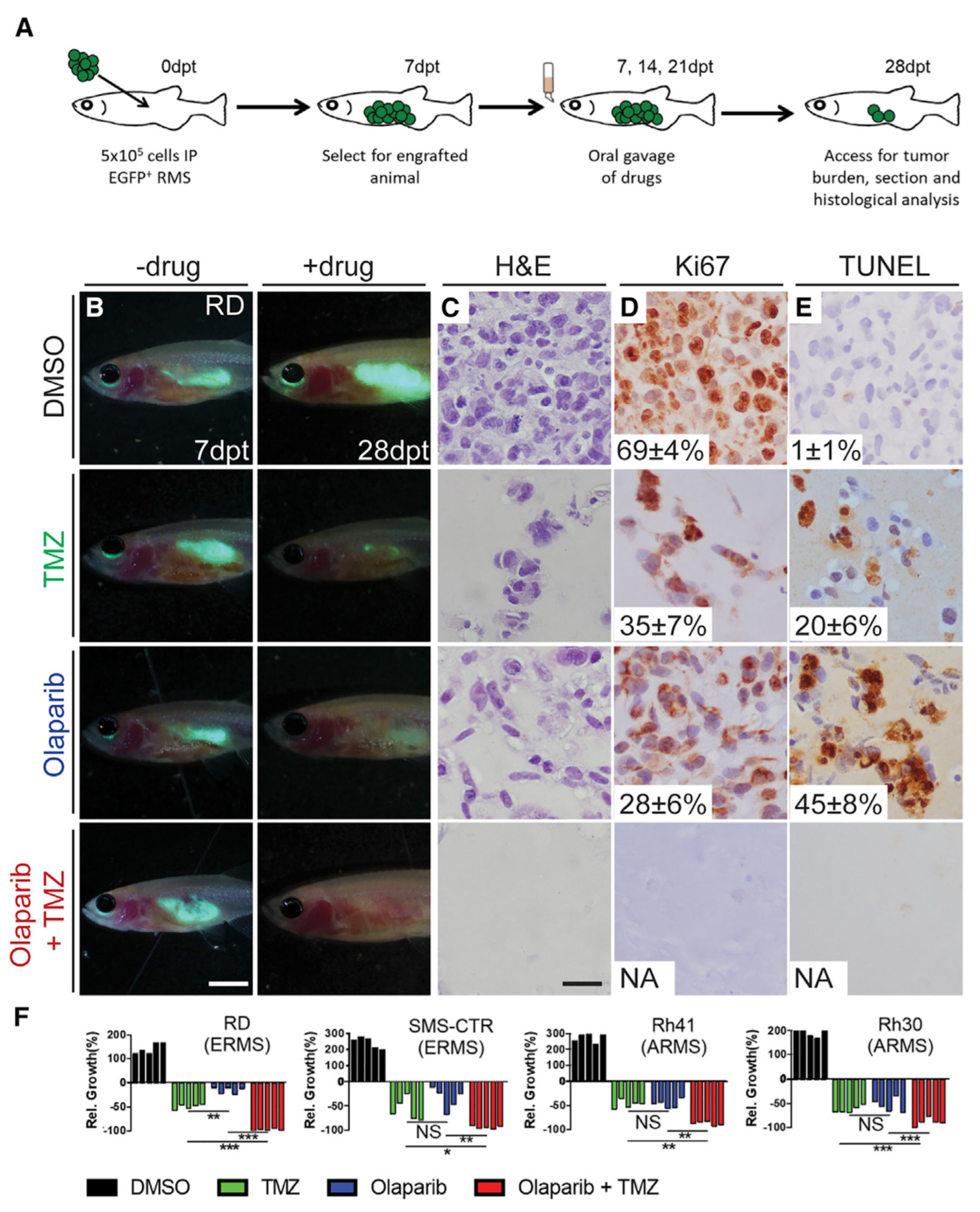{kind=link}
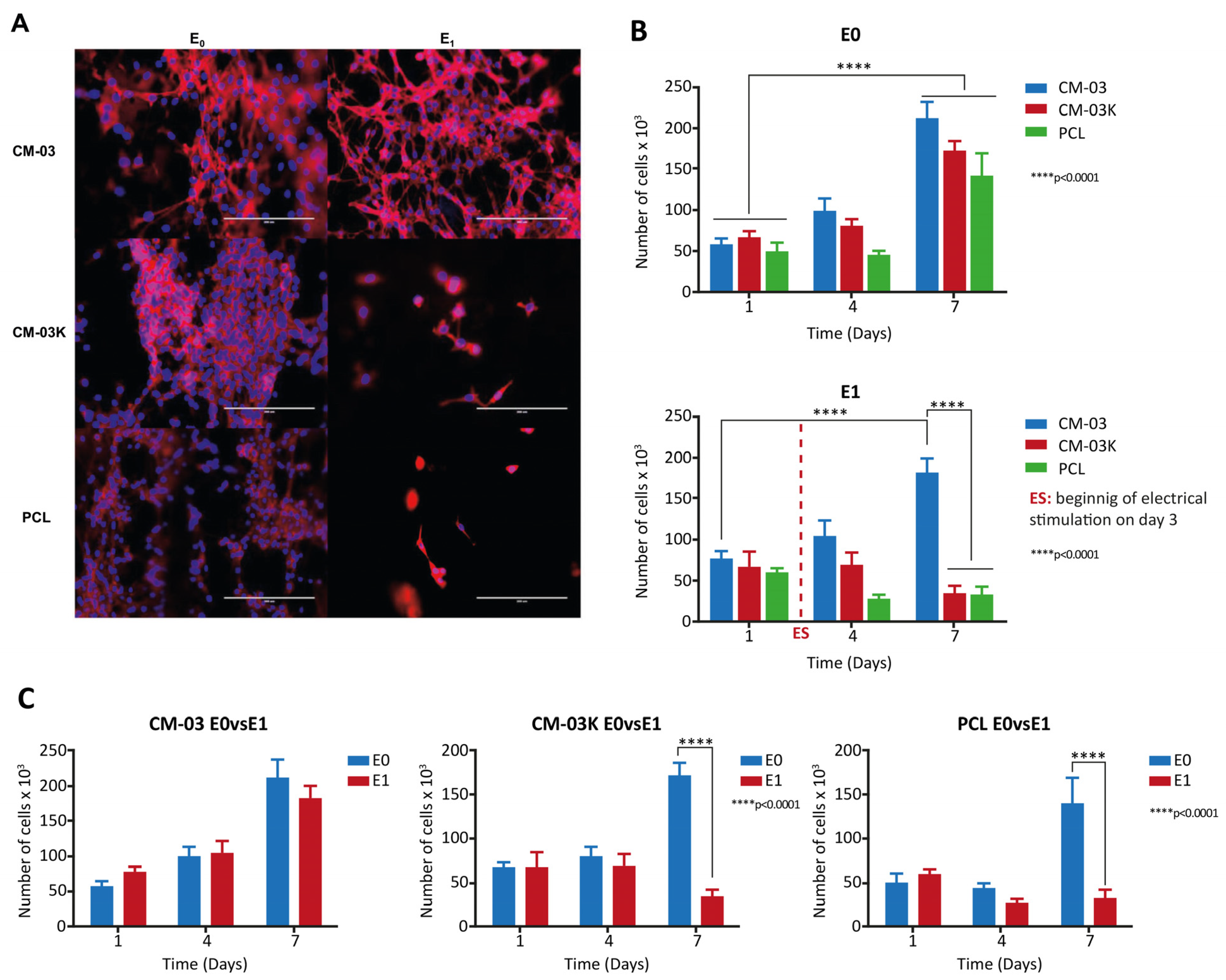{kind=link}
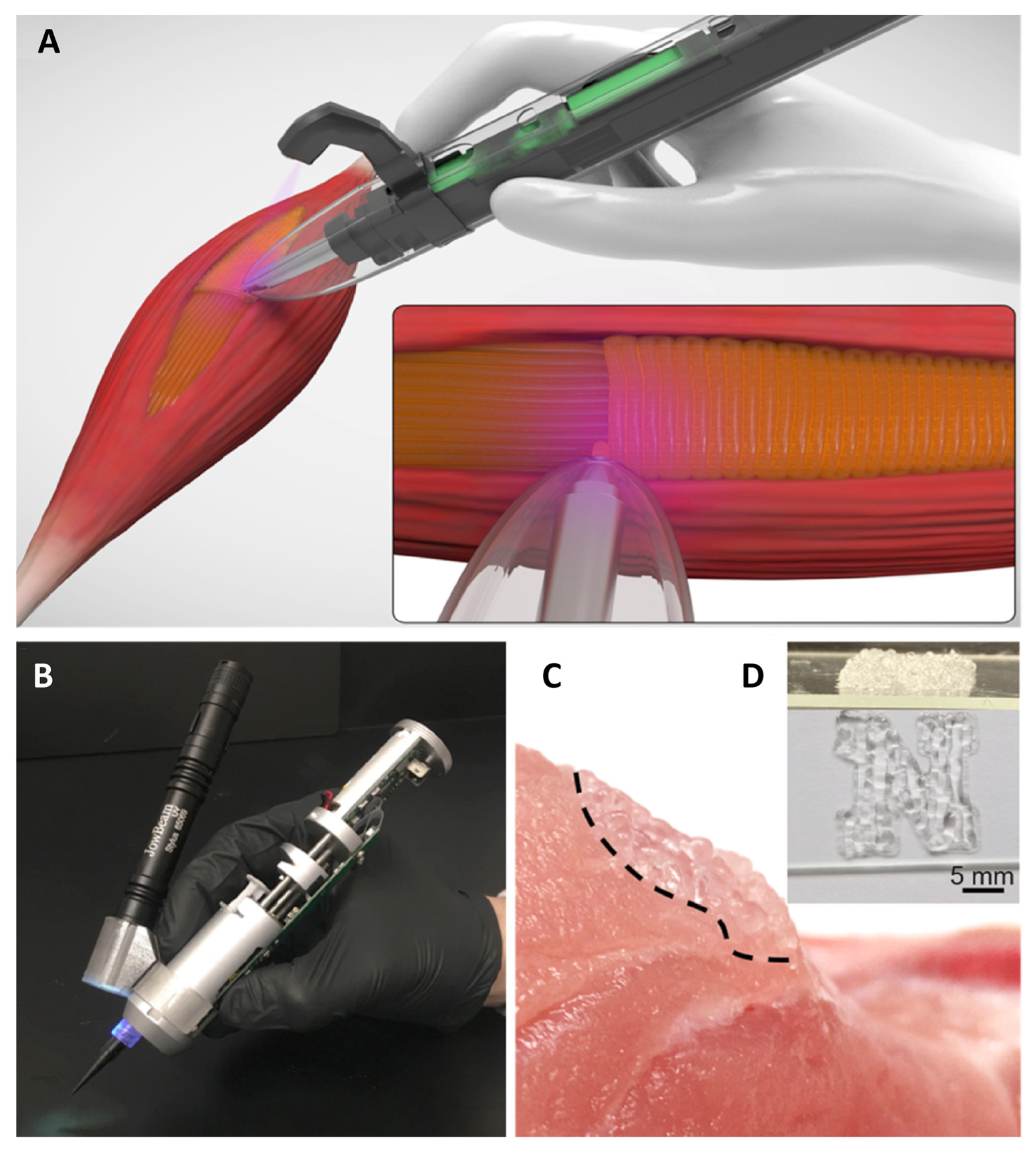{kind=link}
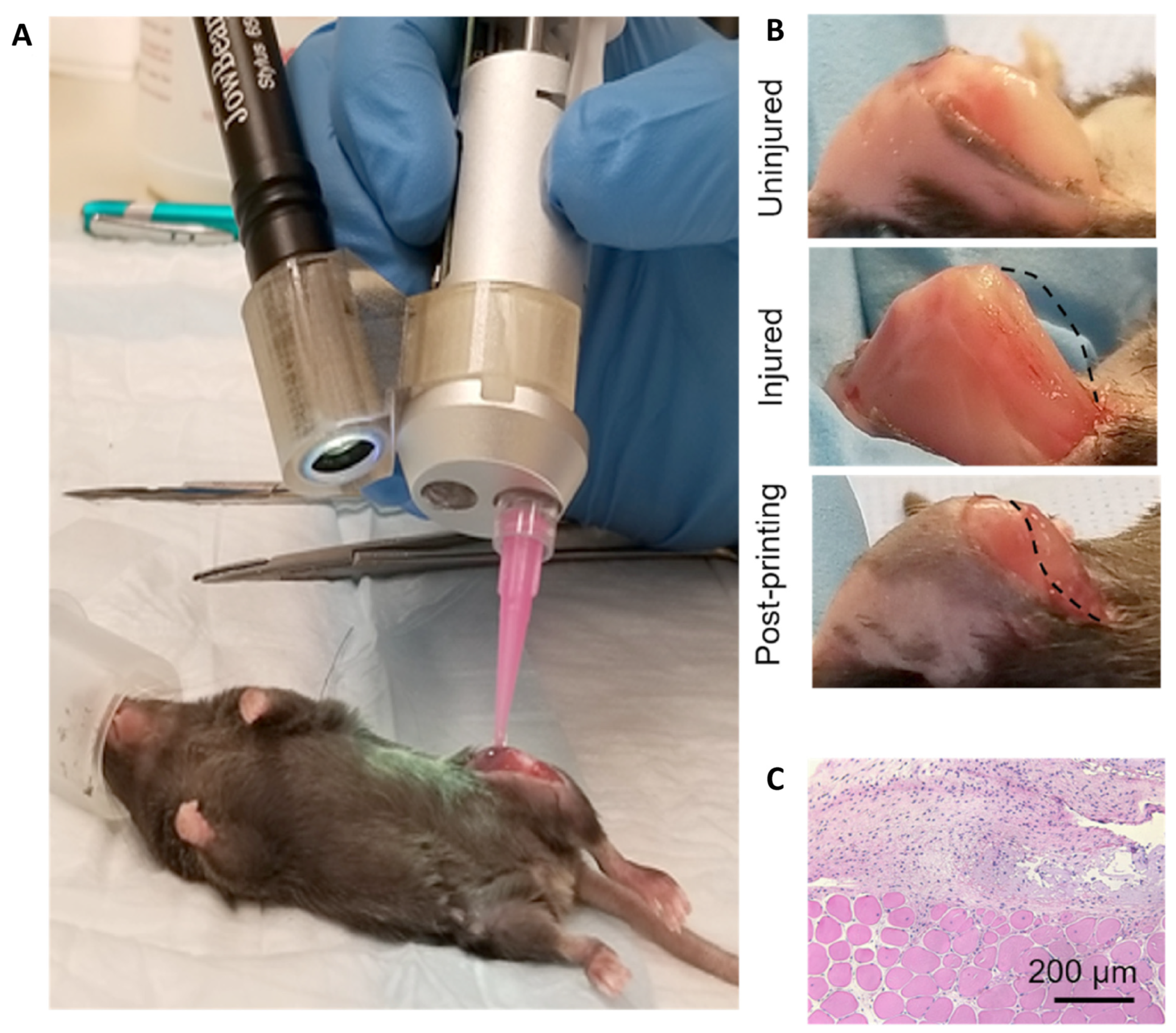{kind=link}
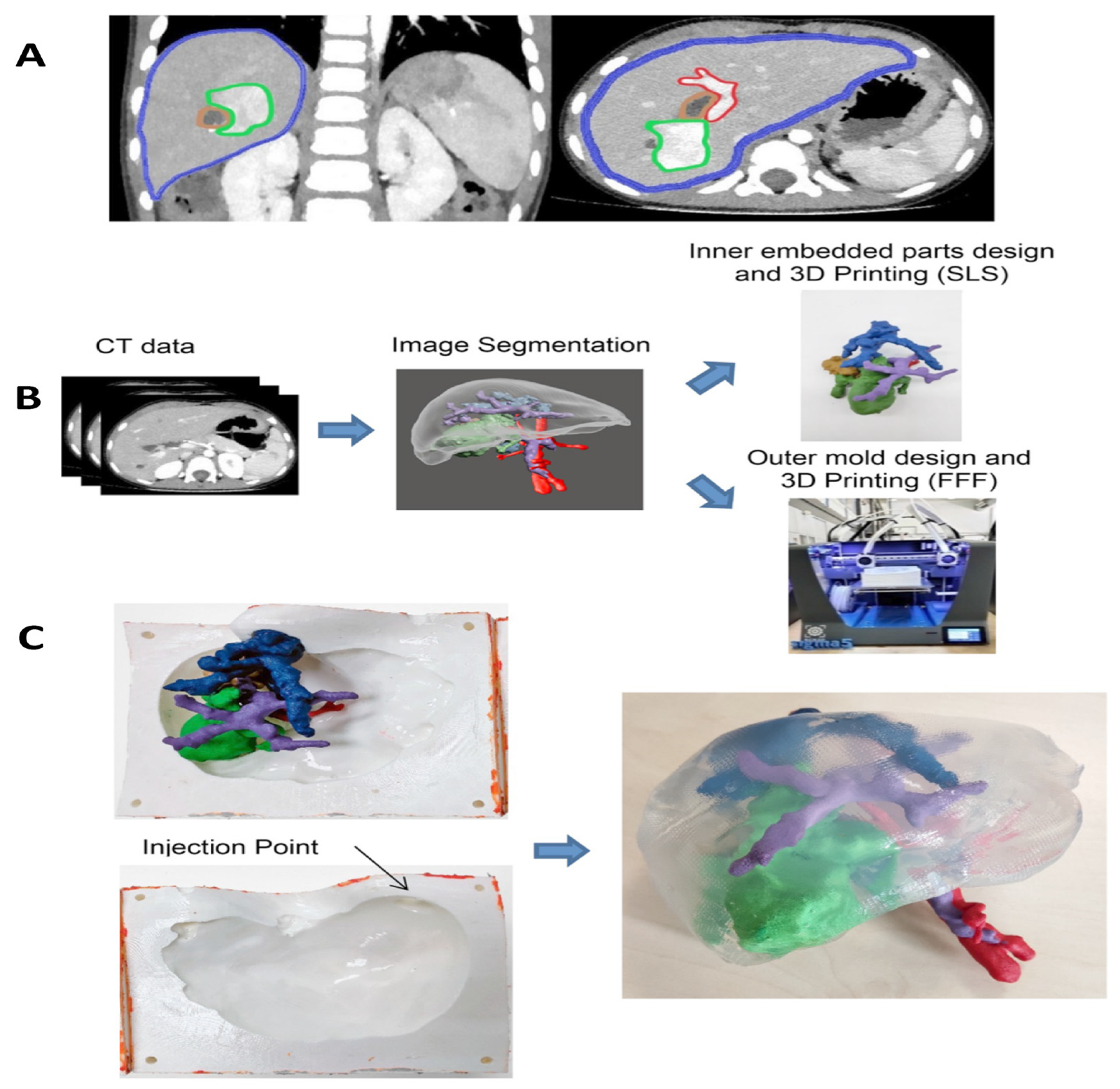{kind=link}
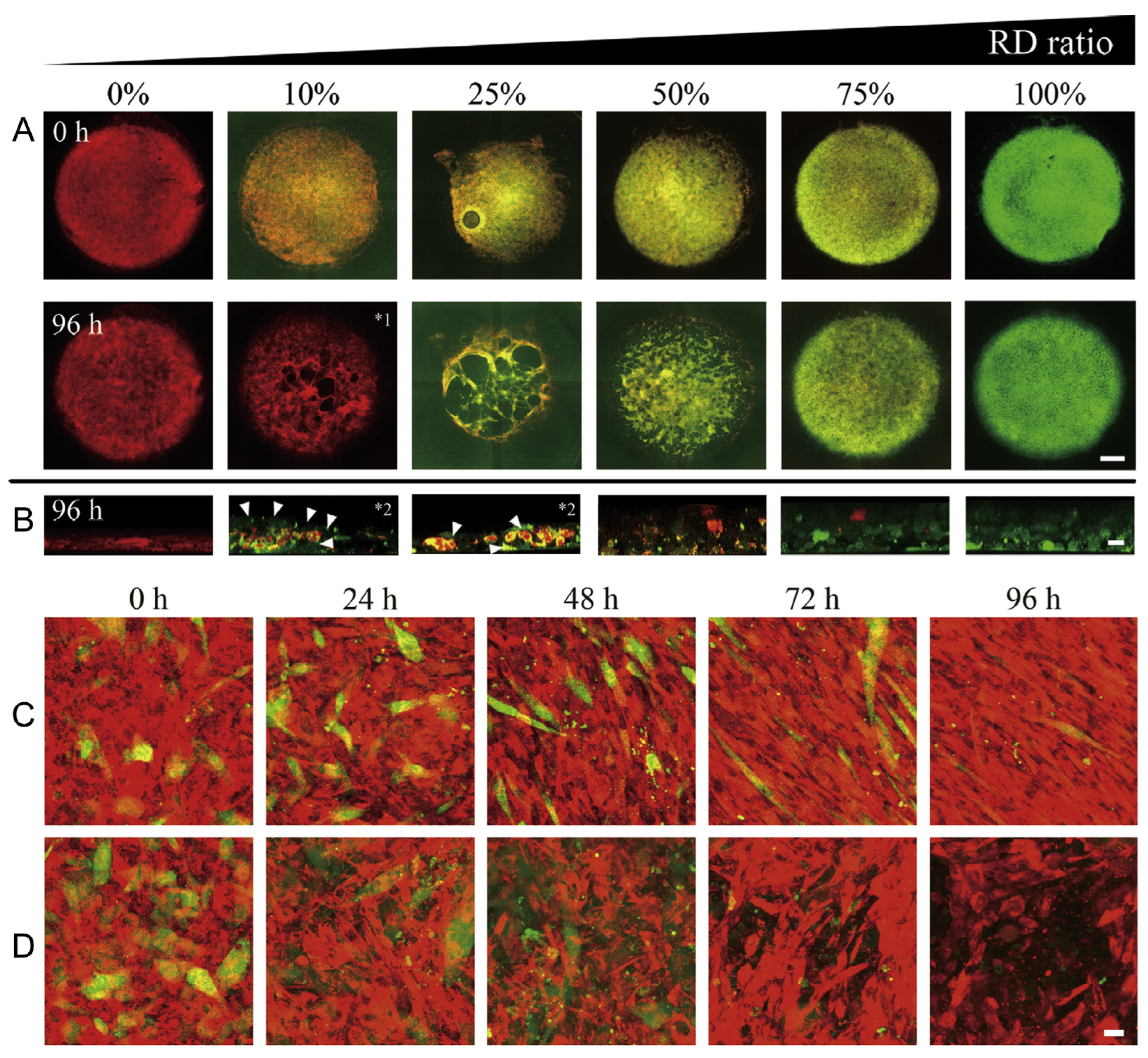{kind=link}
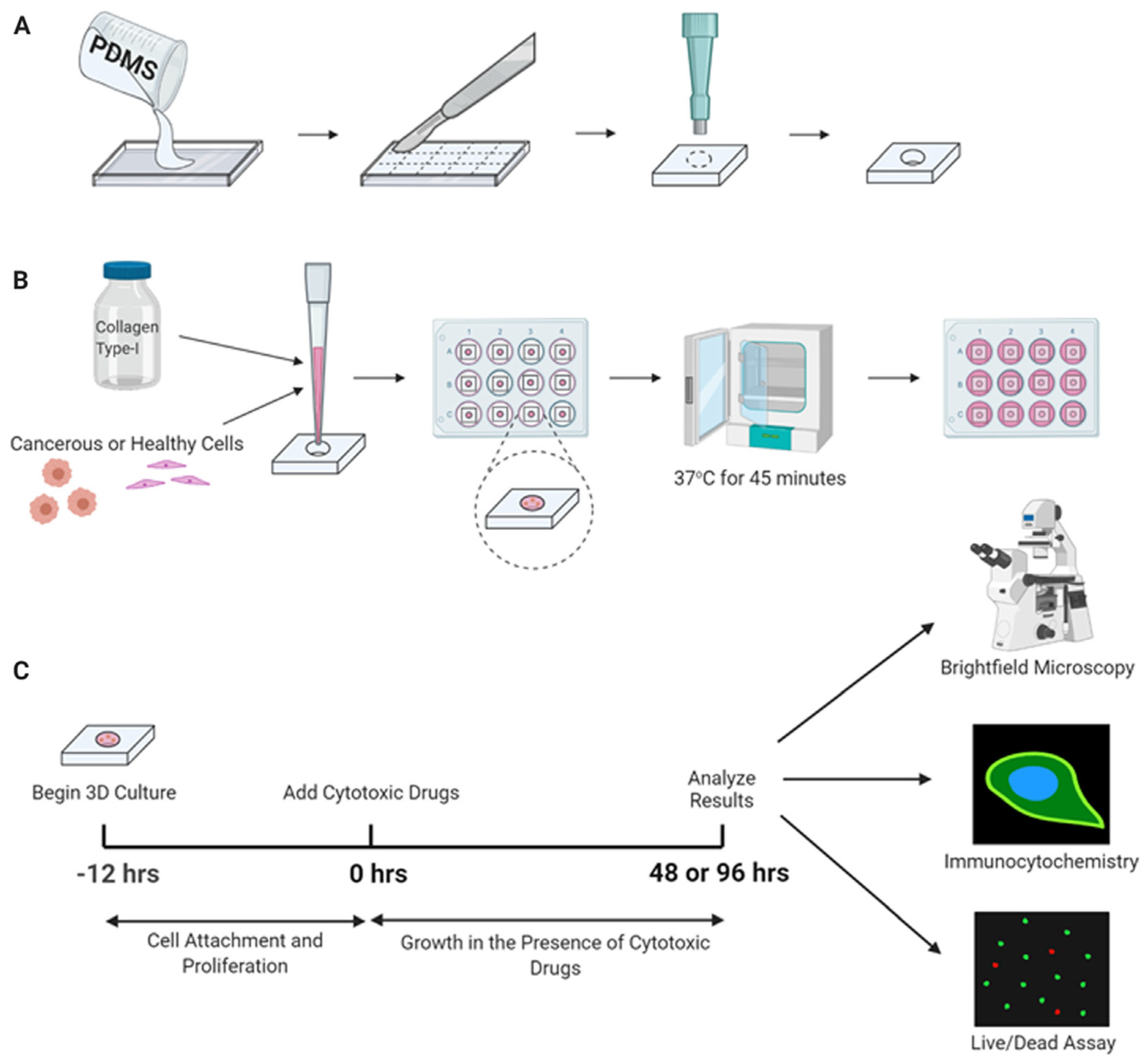{kind=link}











| Treatment | Clinical Trial Phase | Reference |
|---|---|---|
| Pazopanib | II | [64] |
| Pazopanib or placebo | III | [65] |
| Sorafenib | II | [66] |
| Sorafenib | II | [67] |
| Crizotinib | II | [68] |
| Temsirolimus | II | [69] |
| Cixutumumab | II | [70] |
| Cixutumumab | II | [71] |
| Therapeutic Agents | Clinical Trial ID | Number of Participants | Study Phase | Comments |
|---|---|---|---|---|
| Abemaciclib | NCT04238819 | 60 | I | Study recruiting |
| Temsirolimus or bevacizumab | NCT01222715 | 87 | II | Study completed, has results |
| Cixutumumab | NCT00668148 | 113 | II | Study completed, has results |
| Cixutumumab | NCT00831844 | 116 | II | Study completed, has results |
| Cixutumumab or temozolomide | NCT01055314 | 175 | II | Study completed, has results |
| Cixutumumab and Ttemsirolimus | NCT01614795 | 46 | II | Study completed, has results |
| Crizotinib | NCT01524926 | 582 | II | Study active, not recruiting |
| Onivyde and talazoparib or temozolomide | NCT04901702 | 160 | I/II | Study not yet recruiting |
| Palbociclib | NCT03709680 | 133 | I | Study recruiting |
| Pazopanib | NCT01532687 | 54 | II | Study completed, has results |
| Regorafenib | NCT02048371 | 150 | II | Study recruiting |
| Regorafenib | NCT02085148 | 62 | I | Study completed, has results |
| Sorafenib | NCT01502410 | 20 | II | Study completed, has results |
| Sorafenib | NCT02050919 | 20 | II | Study completed, has results |
| Temozolomide | NCT01355445 | 120 | II | Study completed, has results |
| Temsirolimus | NCT02567435 | 397 | III | Study recruiting |
| Temsirolimus | NCT00106353 | 71 | I and II | Study completed, has results |
| Temsirolimus | NCT00949325 | 24 | I and II | Study completed, has results |
| Trabectedin | NCT00070109 | 50 | II | Study completed, has results |
| Vinorelbine | NCT04994132 | 100 | III | Study not yet recruiting |
| Vinorelbine | NCT00003234 | 50 | II | Study completed, has results |
| Vinorelbine | NCT04994132 | 100 | III | Study not yet recruiting |
| Model | Therapeutic Agent | Autophagy Inhibitor | Act | Outcomes/Effects | Ref. | |
|---|---|---|---|---|---|---|
| Pharmacologic | Genetic | |||||
| ARMS cell lines (RH30 and RH4) | Temozolomide | Bafilomycin A1 | - | Inhibition of V-ATPase/ATG7 | Promoted chemotherapy efficacy | [250] |
| Human RMS cell line (hRD) | Doxorubicin | Simvastatin | - | Activation of mitochondrial apoptotic pathway (BAX) | Improved the sensitivity of cancer cells towards Dox and improved antitumor activity | [219] |
| ERMS CSC cell lines | Doxorubicin | Omeprazole | V0c siRNA | Inhibition of V-ATPase/lysosomal pH | Enhanced cytotoxic effect of chemotherapy and reduced the invasive potential of ERMS CSCs | [290] |
| Human RMS cell lines (RH30 and hRD) | Ciclopirox Olamine | Chloroquine | - | Inhibition of lysosomal pH | Improved antitumor activity | [291] |
| Human RMS cell lines (RH30 and hRD) | Bortezomib and 17-DMAG | Chloroquine | - | Inhibition of lysosomal pH/UPS and HSR systems | Enhanced drug-induced apoptosis | [292] |
| ERMS (RD) and ARMS (RMS13) cell lines | Bortezomib | Bafilomycin A1/ST80 | BAG3 siRNA | Inhibition of V-ATPase/ATG7 | Impaired cancer cell growth and increased cell death | [293] |
| ERMS cell lines (RD, RH30 and RMS) | Tenovin-6 | - | SIRT1 and SIRT2 siRNA | Inhibition of Sirtuins | Impaired cancer cell growth and increased apoptosis | [301] |
| Human RMS cell line (hRD) | Methotrexate and SDH | - | - | Inhibition of P-gp | Enhanced methotrexate-mediated cytotoxicity | [328] |
| ARMS RH30 (FG+) | Vincristine | Etoposide | - | Inhibition of PLK1/activation of mitochondrial apoptotic pathway (BAX/BAK) | Improved antitumor activity and increased apoptosis | [332] |
| ARMS RH30 and ERMS RD, TE381.T (FG+) | Vincristine | Volasertib | - | |||
| ARMS RH30 (FG+) | Doxorubicin | Etoposide | - | |||
| ARMS RH30 (FG+) | Eribulin | Etoposide | - | [333] | ||
| ARMS RMS1 (FG+) | Etoposide | Volasertib | - | [331] | ||
| Parameter | Method | Tumor Onset | Outcomes | Ref |
|---|---|---|---|---|
| HDAC6 | CRISPR/Cas9 method for deletion of HDAC6*, constructs containing rag2-KRASG12D-U6-hdac6 guide RNA, rag2-Cas9 and myogenin-H2B-RFP injected in 1st-cell stage | 15–20 days of post-fertilization | HDAC6 was found to have significant role in ERMS tumorigenesis, promoting tumor growth, metastasis, and self-renewal. | [371] |
| RAC1 | Engraftment of KRAS-driven zebrafish ERMS tumors co-expressing GFP and mutant RAC1** (RAC1V12), dorsal subcutaneous way | Tumor harvest after 3 weeks | Zebrafish expressing RAC1V12 exhibited more aggressive tumor growth and invasiveness compared to the control group (empty vector). | [371] |
| tp53 | KRAS-induced ERMS generated in tp53del/del zebrafish | Tumors were tracked 90 days | Deletion of tp53 increased metastasis and invasion of ERMS cells, but not the total frequency of tumor cells. | [372] |
| Van Gogh-like 2 (Vangl2) | KRAS-induced ERMS is generated in fish with additional Vangl2 gene | 15 days of post-fertilization, 90 days after transplantation | Expression of Vangl2 supports TPCs and has positive effect for their self-renewal. No effect of Vangl2 was found on the size, penetrability, and latency of the ERMS tumors. | [373] |
| Intracellular NOTCH1 (ICN1) | KRAS-induced ERMS (KRASG12D and KRASG12D-ICN1) was generated in transgenic zebrafish expressing myf5-GFP and mylz2-mCherry | Tumors imaged over 100 days after transplantation to the recipient fish | ICN1 enhanced the number of tumor-propagating cells in zebrafish ERMS, by blocking the differentiation of zebrafish ERMS cells into self-renewing myf5 positive TPCs. | [374] |
| myf5 | KRAS-induced ERMS was generated in zebrafish with rag2-KRASG12D, with additional mylpfa-mCherry, myf5-GFP injection | Animals were imaged after 35 days post-fertilization | Re-expression of myf5 enhanced tumor formation and penetration, thus had a role in reprogramming of ERMS cells into TPCs. | [375] |
| GSK3 inhibitors screening | KRAS-induced ERMS in myf5-GFP and/or mylz2-mCherry transgenic fish | Tumor engraftment was monitored from 10 to 120 days after drug treatment | GSK3*** inhibitors suppressed ERMS growth, depleted TPCs, and blocked self-renewal while activating the WNT/β-catenin pathway. | [376] |
| Screening of PD98059 and TPCK drugs | rag2-KRASG12D and rag2-DsRed transgenic zebrafish | Tumors were observed after 7–10 days of post-fertilization | Tumor growth was reduced with the drug treatments, showing anticancer potential. | [377] |
| Animal Model | Injection Types | Pros | Cons |
|---|---|---|---|
| CDX | Heterotopic (subcutaneous) engraftment—Easy to apply and used to monitor tumor growth. In therapeutic applications, drug response may differ from the orthotopic engraftment. Orthotopic engraftment—The most preferable injection type for clinical applications due to high prediction value. Technically, this injection technique is challenging and difficult to monitor the tumor growth. |
|
|
| PDX |
|
| |
| EIMM |
|
| |
| GEMM |
|
|
| Phantom Production Method | Advantages | Disadvantages |
|---|---|---|
| FFF |
|
|
| SLA |
|
|
| Material jetting |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zarrabi, A.; Perrin, D.; Kavoosi, M.; Sommer, M.; Sezen, S.; Mehrbod, P.; Bhushan, B.; Machaj, F.; Rosik, J.; Kawalec, P.; et al. Rhabdomyosarcoma: Current Therapy, Challenges, and Future Approaches to Treatment Strategies. Cancers 2023, 15, 5269. https://doi.org/10.3390/cancers15215269
Zarrabi A, Perrin D, Kavoosi M, Sommer M, Sezen S, Mehrbod P, Bhushan B, Machaj F, Rosik J, Kawalec P, et al. Rhabdomyosarcoma: Current Therapy, Challenges, and Future Approaches to Treatment Strategies. Cancers. 2023; 15(21):5269. https://doi.org/10.3390/cancers15215269
Chicago/Turabian StyleZarrabi, Ali, David Perrin, Mahboubeh Kavoosi, Micah Sommer, Serap Sezen, Parvaneh Mehrbod, Bhavya Bhushan, Filip Machaj, Jakub Rosik, Philip Kawalec, and et al. 2023. "Rhabdomyosarcoma: Current Therapy, Challenges, and Future Approaches to Treatment Strategies" Cancers 15, no. 21: 5269. https://doi.org/10.3390/cancers15215269
APA StyleZarrabi, A., Perrin, D., Kavoosi, M., Sommer, M., Sezen, S., Mehrbod, P., Bhushan, B., Machaj, F., Rosik, J., Kawalec, P., Afifi, S., Bolandi, S. M., Koleini, P., Taheri, M., Madrakian, T., Łos, M. J., Lindsey, B., Cakir, N., Zarepour, A., ... Ghavami, S. (2023). Rhabdomyosarcoma: Current Therapy, Challenges, and Future Approaches to Treatment Strategies. Cancers, 15(21), 5269. https://doi.org/10.3390/cancers15215269