
Identification of a Complex Karyotype Signature with Clinical Implications in AML and MDS-EB Using Gene Expression Profiling

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
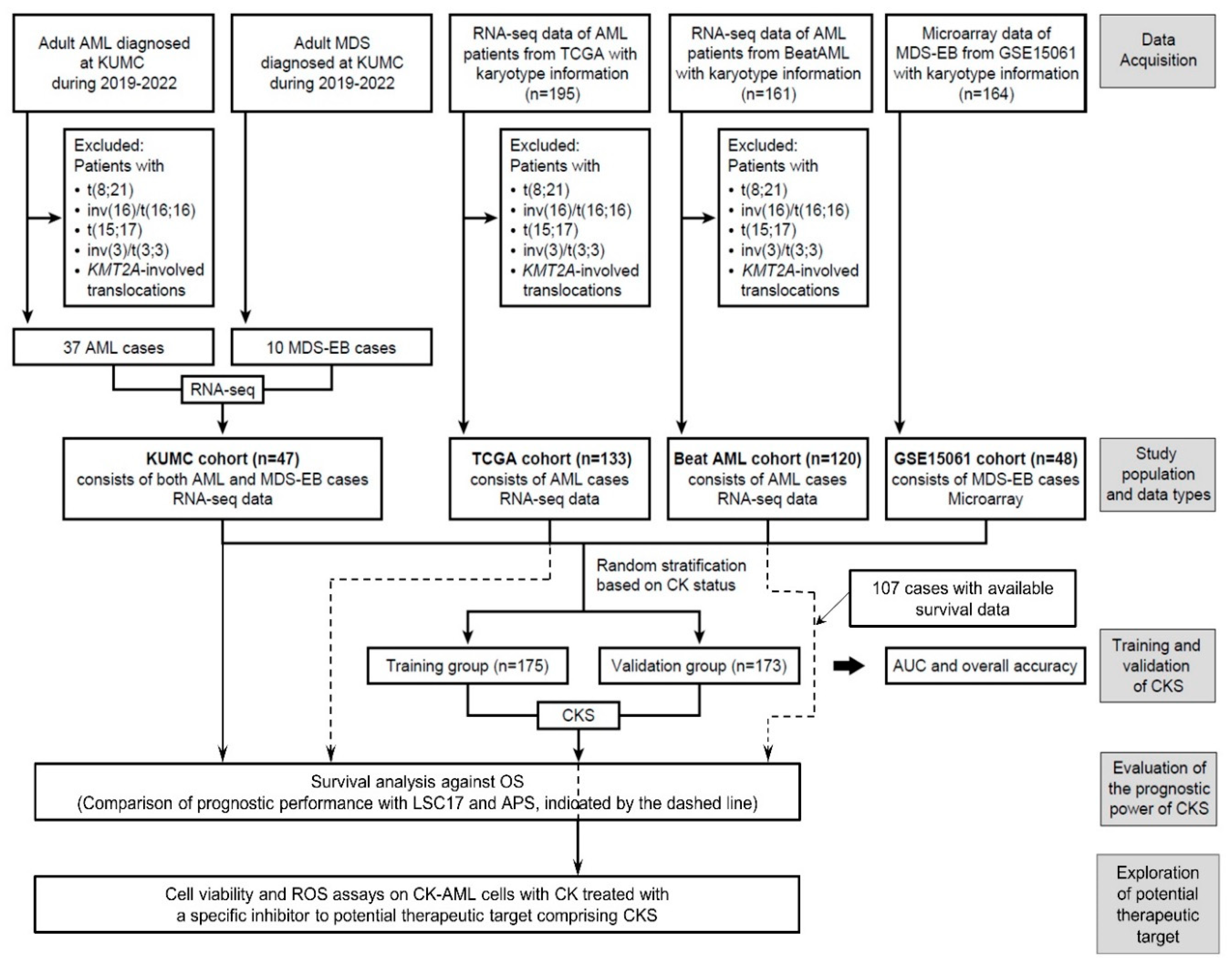
2. Materials and Methods
2.1. Patient Samples
2.2. RNA Extraction, Library Preparation, and RNA-Seq
2.3. Differential Gene Expression and Pathway Analysis
2.4. Signature Development for CK Prediction
2.5. Cell Viability and ROS Assays on AML Cells with CK Treated with LCS-1, a Specific Inhibitor of Superoxide Dismutase 1 (SOD1)
2.6. Statistical Analysis
3. Results
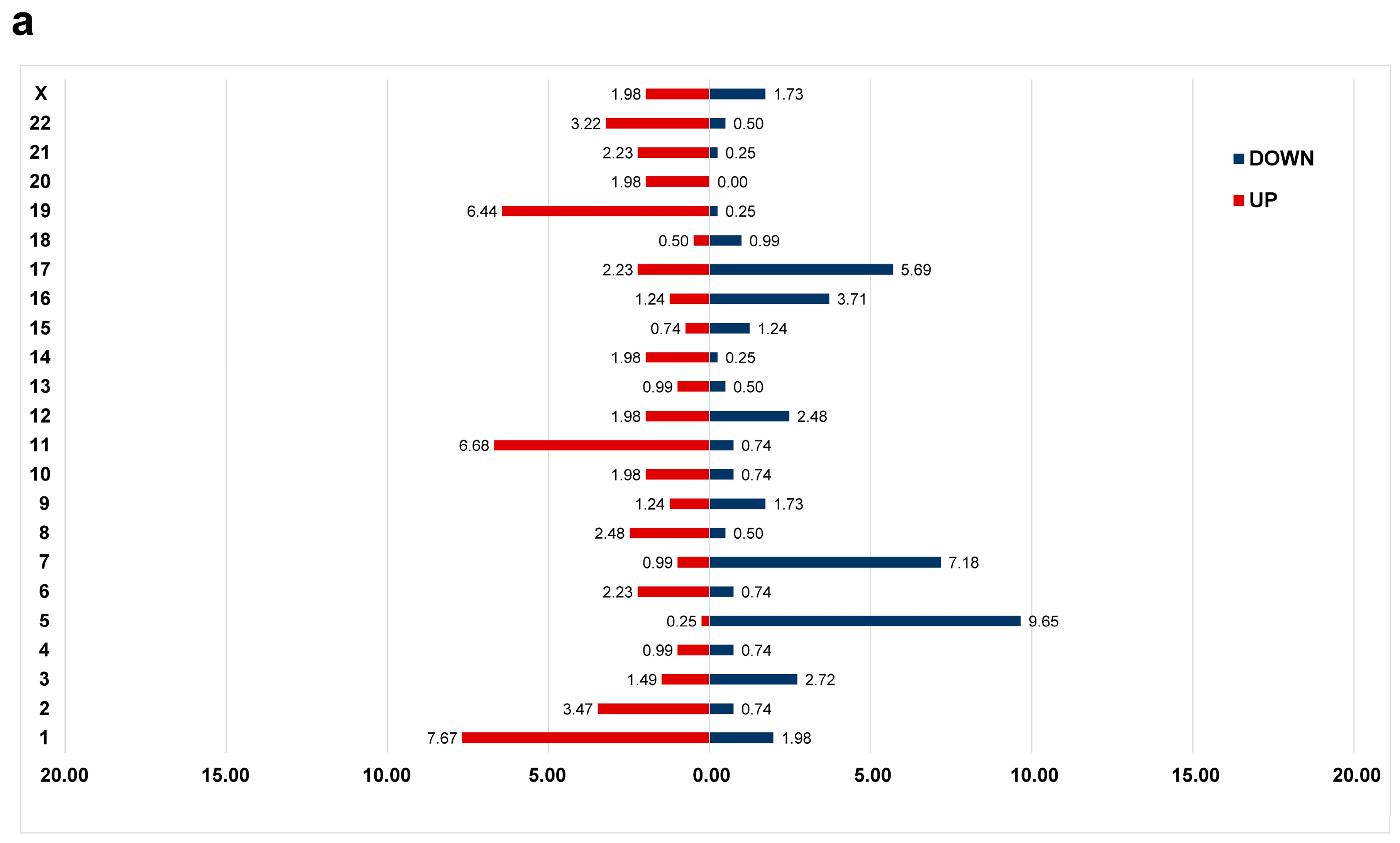
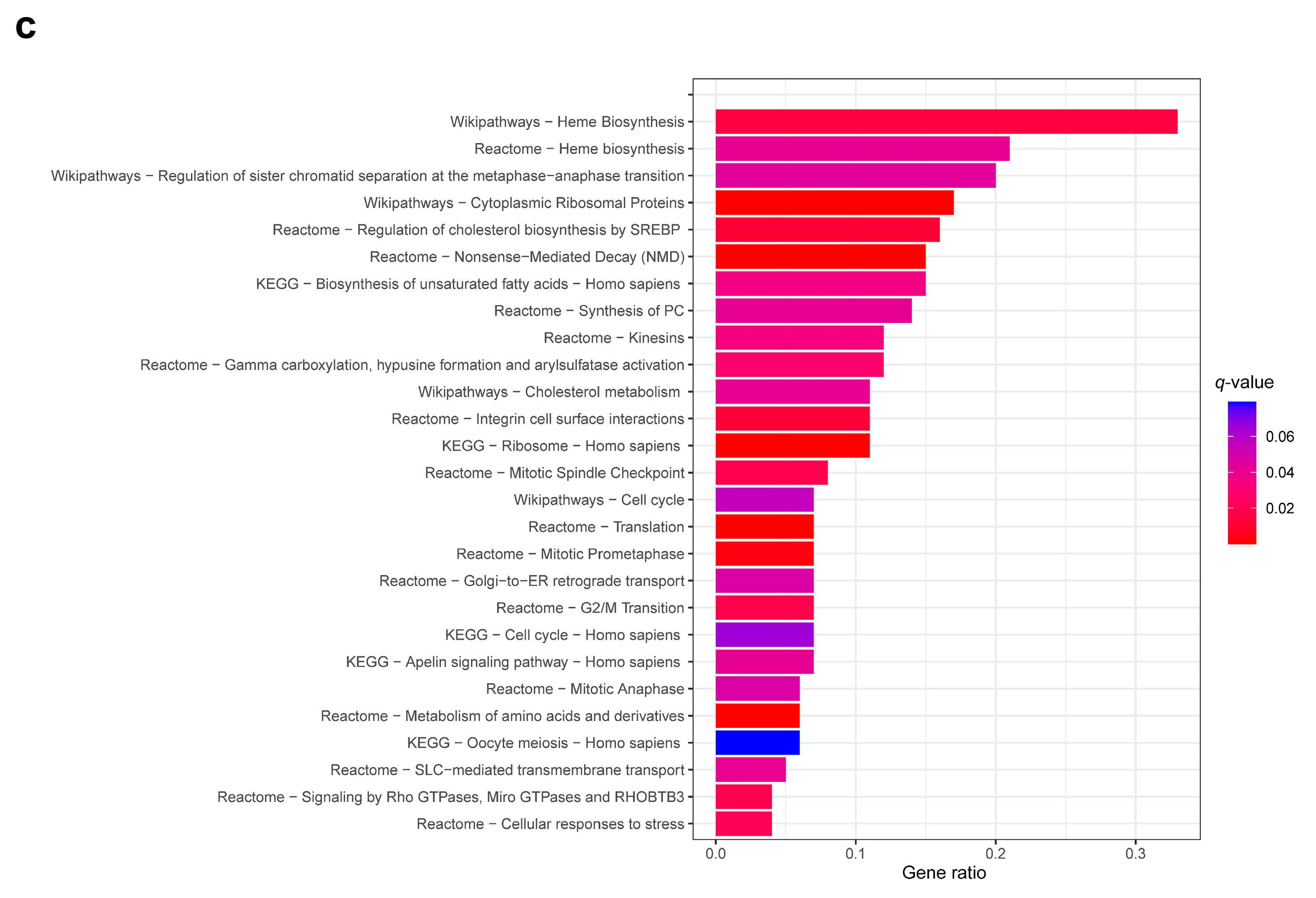
3.1. Differential Gene Expression Profile between CK and Non-CK Group
3.2. CKS Training and Validation
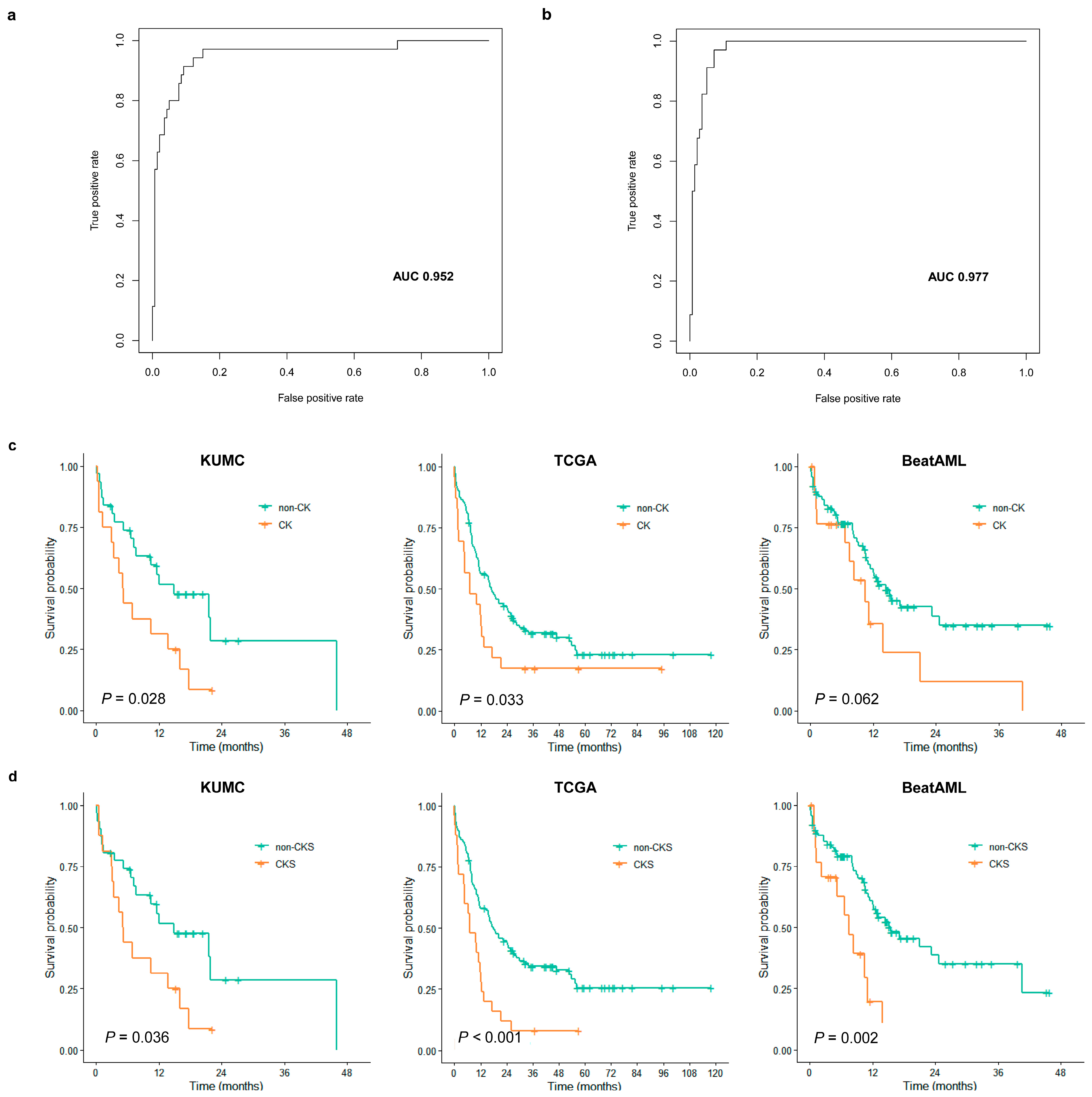
3.3. Assessment of CKS Prediction for CK
3.4. Evaluation of CKS as a Prognostic Score
3.5. Comparison of Prognostic Performance with Other Models
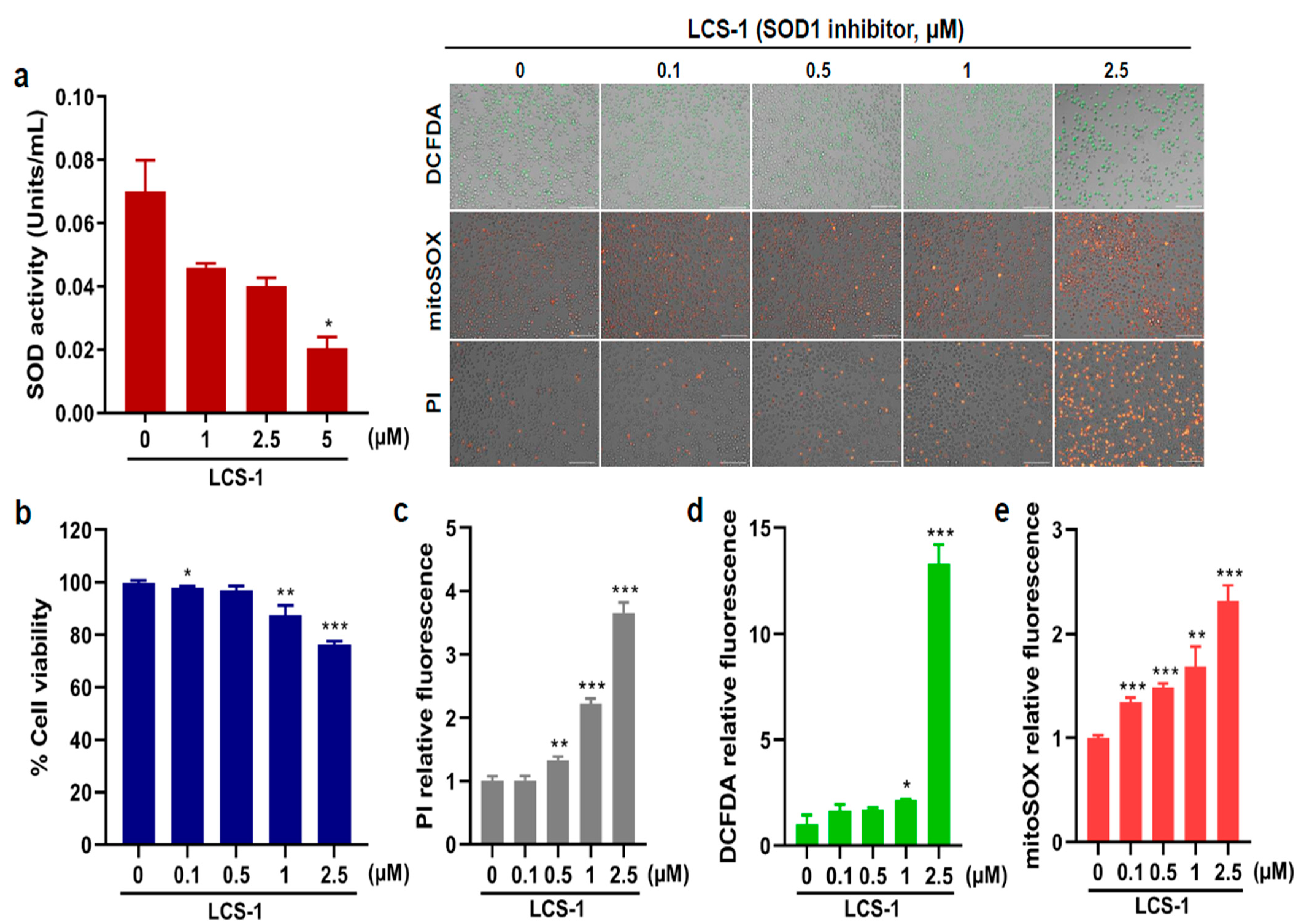
3.6. Effects of SOD1 Inhibition on AML Cell Proliferation and ROS Production
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.R.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.; Hasserjian, R.P.; Döhner, H. Distinguishing AML from MDS: A fixed blast percentage may no longer be optimal. Blood 2022, 139, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Menssen, A.J.; Walter, M.J. Genetics of progression from MDS to secondary leukemia. Blood 2020, 136, 50–60. [Google Scholar] [CrossRef]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017, 129, 424–447. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Schoch, C.; Haferlach, T.; Haase, D.; Fonatsch, C.; Löffler, H.; Schlegelberger, B.; Staib, P.; Sauerland, M.C.; Heinecke, A.; Büchner, T.; et al. Patients with de novo acute myeloid leukaemia and complex karyotype aberrations show a poor prognosis despite intensive treatment: A study of 90 patients. Br. J. Haematol. 2001, 112, 118–126. [Google Scholar] [CrossRef]
- Byrd, J.C.; Mrózek, K.; Dodge, R.K.; Carroll, A.J.; Edwards, C.G.; Arthur, D.C.; Pettenati, M.J.; Patil, S.R.; Rao, K.W.; Watson, M.S.; et al. Pretreatment cytogenetic abnormalities are predictive of induction success, cumulative incidence of relapse, and overall survival in adult patients with de novo acute myeloid leukemia: Results from Cancer and Leukemia Group B (CALGB 8461). Blood 2002, 100, 4325–4336. [Google Scholar] [CrossRef] [PubMed]
- Stölzel, F.; Mohr, B.; Kramer, M.; Oelschlägel, U.; Bochtler, T.; Berdel, W.E.; Kaufmann, M.; Baldus, C.D.; Schäfer-Eckart, K.; Stulmann, R.; et al. Karyotype complexity and prognosis in acute myeloid leukemia. Blood Cancer J. 2016, 6, e386. [Google Scholar] [CrossRef]
- Mrózek, K.; Eisfeld, A.-K.; Kohlschmidt, J.; Carroll, A.J.; Walker, C.J.; Nicolet, D.; Blachly, J.S.; Bill, M.; Papaioannou, D.; Wang, E.S.; et al. Complex karyotype in de novo acute myeloid leukemia: Typical and atypical subtypes differ molecularly and clinically. Leukemia 2019, 33, 1620–1634. [Google Scholar] [CrossRef] [PubMed]
- Haase, D.; Stevenson, K.E.; Neuberg, D.; Maciejewski, J.P.; Nazha, A.; Sekeres, M.A.; Ebert, B.L.; Garcia-Manero, G.; Haferlach, C.; Haferlach, T.; et al. TP53 mutation status divides myelodysplastic syndromes with complex karyotypes into distinct prognostic subgroups. Leukemia 2019, 33, 1747–1758. [Google Scholar] [CrossRef] [PubMed]
- Gerstung, M.; Pellagatti, A.; Malcovati, L.; Giagounidis, A.; Della Porta, M.G.; Jädersten, M.; Dolatshad, H.; Verma, A.; Cross, N.C.P.; Vyas, P.; et al. Combining gene mutation with gene expression data improves outcome prediction in myelodysplastic syndromes. Nat. Commun. 2015, 6, 5901. [Google Scholar] [CrossRef]
- Griffith, M.; Griffith, O.L.; Krysiak, K.; Skidmore, Z.L.; Christopher, M.J.; Klco, J.M.; Ramu, A.; Lamprecht, T.L.; Wagner, A.H.; Campbell, K.M.; et al. Comprehensive genomic analysis reveals FLT3 activation and a therapeutic strategy for a patient with relapsed adult B-lymphoblastic leukemia. Exp. Hematol. 2016, 44, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Docking, T.R.; Parker, J.D.K.; Jädersten, M.; Duns, G.; Chang, L.; Jiang, J.; Pilsworth, J.A.; Swanson, L.A.; Chan, S.K.; Chiu, R.; et al. A clinical transcriptome approach to patient stratification and therapy selection in acute myeloid leukemia. Nat. Commun. 2021, 12, 2474. [Google Scholar] [CrossRef]
- Ng, S.W.; Mitchell, A.; Kennedy, J.A.; Chen, W.C.; McLeod, J.; Ibrahimova, N.; Arruda, A.; Popescu, A.; Gupta, V.; Schimmer, A.D.; et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature 2016, 540, 433–437. [Google Scholar] [CrossRef]
- Kim, D.D.H.; Basso, I.N.; Kim, T.S.; Yi, S.Y.; Kim, K.H.; Murphy, T.; Chan, S.; Minden, M.; Pasic, I.; Lam, W.; et al. The 17-gene stemness score associates with relapse risk and long-term outcomes following allogeneic haematopoietic cell transplantation in acute myeloid leukaemia. EJHaem 2022, 3, 873–884. [Google Scholar] [CrossRef]
- Elsayed, A.H.; Rafiee, R.; Cao, X.; Raimondi, S.; Downing, J.R.; Ribeiro, R.; Fan, Y.; Guber, T.A.; Baker, S.; Klco, J.; et al. A six-gene leukemic stem cell score identifies high risk pediatric acute myeloid leukemia. Leukemia 2020, 34, 735–745. [Google Scholar] [CrossRef]
- Huang, B.J.; Smith, J.L.; Farrar, J.E.; Wang, Y.-C.; Umeda, M.; Ries, R.E.; Leonti, A.R.; Crowgey, E.; Furlan, S.N.; Tarlock, K.; et al. Integrated stem cell signature and cytomolecular risk determination in pediatric acute myeloid leukemia. Nat. Commun. 2022, 13, 5487. [Google Scholar] [CrossRef] [PubMed]
- Duployez, N.; Marceau-Renaut, A.; Villenet, C.; Petit, A.; Rousseau, A.; Ng, S.W.K.; Paquet, A.; Gonzales, F.; Barthélémy, A.; Leprêtre, F.; et al. The stem cell-associated gene expression signature allows risk stratification in pediatric acute myeloid leukemia. Leukemia 2019, 33, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.I.; Kohlmann, A.; Williams, P.M.; Wieczorek, L.; Liu, W.-M.; Li, R.; Wei, W.; Bowen, D.T.; Loeffler, H.; Hernandez, J.M.; et al. Microarray-based classifiers and prognosis models identify subgroups with distinct clinical outcomes and high risk of AML transformation of myelodysplastic syndrome. Blood 2009, 114, 1063–1072. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Herwig, R.; Hardt, C.; Lienhard, M.; Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 2016, 11, 1889–1907. [Google Scholar] [CrossRef]
- Liu, W.-M.; Li, R.; Sun, J.Z.; Wang, J.; Tsai, J.; Wen, W.; Kohlmann, A.; Williams, P.M. PQN and DQN: Algorithms for expression microarrays. J. Theor. Biol. 2006, 243, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Foltz, S.M.; Greene, C.S.; Taroni, J.N. Cross-platform normalization enables machine learning model training on microarray and RNA-seq data simultaneously. Commun. Biol. 2023, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.H.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Fujibayashi, Y.; Isa, R.; Nishiyama, D.; Sakamoto-Inada, N.; Kawasumi, N.; Yamaguchi, J.; Kuwahara-Ota, S.; Matsumura-Kimoto, Y.; Tsukamoto, T.; Chinen, Y.; et al. Aberrant BUB1 Overexpression Promotes Mitotic Segregation Errors and Chromosomal Instability in Multiple Myeloma. Cancers 2020, 12, 2206. [Google Scholar] [CrossRef] [PubMed]
- Bruno, S.; di Rorà, A.G.L.; Napolitano, R.; Soverini, S.; Martinelli, G.; Simonetti, G. CDC20 in and out of mitosis: A prognostic factor and therapeutic target in hematological malignancies. J. Exp. Clin. Cancer Res. 2022, 41, 159. [Google Scholar] [CrossRef]
- Bill, M.; Nicolet, D.; Kohlschmidt, J.; Walker, C.J.; Mrózek, K.; Eisfeld, A.-K.; Papaioannou, D.; Rong-Mullins, X.; Brannan, Z.; Kolitz, J.E.; et al. Mutations associated with a 17-gene leukemia stem cell score and the score’s prognostic relevance in the context of the European LeukemiaNet classification of acute myeloid leukemia. Haematologica 2020, 105, 721–729. [Google Scholar] [CrossRef]
- Ng, S.W.K.; Murphy, T.; King, I.; Zhang, T.; Mah, M.; Lu, Z.; Stickle, N.; Ibrahimova, N.; Arruda, A.; Mitchell, A.; et al. A clinical laboratory–developed LSC17 stemness score assay for rapid risk assessment of patients with acute myeloid leukemia. Blood Adv. 2022, 6, 1064–1073. [Google Scholar] [CrossRef]
- Mrózek, K. Cytogenetic, Molecular Genetic, and Clinical Characteristics of Acute Myeloid Leukemia with a Complex Karyotype. Semin. Oncol. 2008, 35, 365–377. [Google Scholar] [CrossRef]
- Giagounidis, A.A.; Germing, U.; Aul, C. Biological and Prognostic Significance of Chromosome 5q Deletions in Myeloid Malignancies. Clin. Cancer Res. 2006, 12, 5–10. [Google Scholar] [CrossRef]
- Mori, M.; Adema, V.; Gurnari, C.; Pagliuca, S.; Terkawi, L.; Bahaj, W.; Kewan, T.; Barnard, J.; Visconte, V.; Haferlach, T.; et al. Novel synthetic lethal targets for myeloid neoplasms with loss of chromosome 7. Blood 2021, 138 (Suppl. 1), 3346. [Google Scholar] [CrossRef]
- Itzhar, N.; Dessen, P.; Toujani, S.; Auger, N.; Preudhomme, C.; Richon, C.; Lazar, V.; Saada, V.; Bennaceur, A.; Bourhis, J.H.; et al. Chromosomal Minimal Critical Regions in Therapy-Related Leukemia Appear Different from Those of De Novo Leukemia by High-Resolution aCGH. PLoS ONE 2011, 6, e16623. [Google Scholar] [CrossRef] [PubMed]
- Jerez, A.; Sugimoto, Y.; Makishima, H.; Verma, A.; Jankowska, A.M.; Przychodzen, B.; Visconte, V.; Tiu, R.V.; O’Keefe, C.L.; Mohamedali, A.M.; et al. Loss of heterozygosity in 7q myeloid disorders: Clinical associations and genomic pathogenesis. Blood 2012, 119, 6109–6117. [Google Scholar] [CrossRef] [PubMed]
- Adema, V.; Palomo, L.; Walter, W.; Mallo, M.; Hutter, S.; La Framboise, T.; Arenillas, L.; Meggendorfer, M.; Radivoyevitch, T.; Xicoy, B.; et al. Pathophysiologic and clinical implications of molecular profiles resultant from deletion 5q. EBioMedicine 2022, 80, 104059. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Kim, Y.M.; Wang, X.; Li, Y.; Lu, X.; Sternenberger, A.R.; Li, S.; Lee, J.-Y. Genomic Copy Number Variations in the Myelodysplastic Syndrome and Acute Myeloid Leukemia Patients with del(5q) and/or -7/del(7q). Int. J. Med. Sci. 2015, 12, 719–726. [Google Scholar] [CrossRef]
- Pourrajab, F.; Zare-Khormizi, M.R.; Hashemi, A.S.; Hekmatimoghaddam, S. Genetic Characterization and Risk Stratification of Acute Myeloid Leukemia. Cancer Manag. Res. 2020, 12, 2231–2253. [Google Scholar] [CrossRef]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.-M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef]
- Schoch, C.; Kern, W.; Kohlmann, A.; Hiddemann, W.; Schnittger, S.; Haferlach, T. Acute myeloid leukemia with a complex aberrant karyotype is a distinct biological entity characterized by genomic imbalances and a specific gene expression profile. Genes, Chromosom. Cancer 2005, 43, 227–238. [Google Scholar] [CrossRef]
- Daneshbod, Y.; Kohan, L.; Taghadosi, V.; Weinberg, O.K.; Arber, D.A. Prognostic Significance of Complex Karyotypes in Acute Myeloid Leukemia. Curr. Treat. Options Oncol. 2019, 20, 15. [Google Scholar] [CrossRef]
- Moison, C.; Lavallée, V.P.; Thiollier, C.; Lehnertz, B.; Boivin, I.; Mayotte, N.; Gareau, Y.; Fréchette, M.; Blouin-Chagnon, V.; Corneau, S.; et al. Complex karyotype AML displays G2/M signature and hypersensitivity to PLK1 inhibition. Blood Adv. 2019, 3, 552–563. [Google Scholar] [CrossRef]
- Nguyen, L.X.T.; Troadec, E.; Kalvala, A.; Kumar, B.; Hoang, D.H.; Viola, D.; Zhang, B.; Nguyen, D.Q.; Aldoss, I.; Ghoda, L.; et al. The Bcl-2 inhibitor venetoclax inhibits Nrf2 antioxidant pathway activation induced by hypomethylating agents in AML. J. Cell. Physiol. 2019, 234, 14040–14049. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Wang, J.; Pan, C.; Zheng, L.; Shang, Q. Nrf2 Overexpression Increases Risk of Venetoclax Resistance in Acute Myeloid Leukemia by Promoting Glycolysis. Blood 2022, 140 (Suppl. 1), 8340. [Google Scholar] [CrossRef]
- Kontro, M.; Kumar, A.; Majumder, M.M.; Eldfors, S.; Parsons, A.; Pemovska, T.; Saarela, J.; Yadav, B.; Malani, D.; Fløisand, Y.; et al. HOX gene expression predicts response to BCL-2 inhibition in acute myeloid leukemia. Leukemia 2017, 31, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Wang, R.; Li, X.; Wang, H.-Y.; Zheng, X.S. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov. Today 2016, 21, 143–149. [Google Scholar] [CrossRef]
- Eleutherio, E.C.A.; Magalhães, R.S.S.; de Araujo Brasil, A.; Neto, J.R.M.; de Holanda Paranhos, L. SOD1, more than just an antioxidant. Arch. Biochem. Biophys. 2021, 697, 108701. [Google Scholar] [CrossRef]
- Somwar, R.; Erdjument-Bromage, H.; Larsson, E.; Shum, D.; Lockwood, W.W.; Yang, G.; Sander, C.; Ouerfelli, O.; Tempst, P.J.; Djaballah, H.; et al. Superoxide dismutase 1 (SOD1) is a target for a small molecule identified in a screen for inhibitors of the growth of lung adenocarcinoma cell lines. Proc. Natl. Acad. Sci. USA 2011, 108, 16375–16380. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, A.; Sena, L.A.; Diebold, L.P.; Mazar, A.P.; Chandel, N.S. Targeting SOD1 reduces experimental non–small-cell lung cancer. J. Clin. Investig. 2014, 124, 117–128. [Google Scholar] [CrossRef]
- Lin, J.; Beer, T.M.; Ryan, C.J.; Mathew, P.; Wilding, G.; Morris, M.; Callahan, J.A.; Gordon, G.; Reich, S.; Carducci, M.A. A randomized, phase II study of ATN-224 in patients with biochemically relapsed, hormone-naive prostate cancer: A DOD/PCF Prostate Cancer Clinical Trials Consortium trial. J. Clin. Oncol. 2009, 27, 5135. [Google Scholar] [CrossRef]
- Yu, X.; Chen, C.; Hu, Y.; Li, K.; Zhang, Y.; Chen, Z.; Nie, D.; Gao, R.; Huang, Y.; Zhong, M.; et al. High expression of LOC541471, GDAP1, SOD1, and STK25 is associated with poor overall survival of patients with acute myeloid leukemia. Cancer Med. 2023, 12, 9055–9067. [Google Scholar] [CrossRef]
- Huang, P.; Feng, L.; Oldham, E.A.; Keating, M.J.; Plunkett, W. Superoxide dismutase as a target for the selective killing of cancer cells. Nature 2000, 407, 390–395. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KUMC (n = 47) | |
|---|---|
| Sex (%) | |
| Male | 29 (61.7) |
| Female | 18 (38.3) |
| Age (years) | |
| Median (range) | 67.0 (31.0–86.0) |
| Hb (g/dL) | |
| Median (range) | 7.9 (5.9–12.1) |
| WBC count (×109/L) | |
| Median (range) | 3.8 (0.6–247.2) |
| Platelet count (×109/L) | |
| Median (range) | 51.0 (3.0–240.0) |
| BM blasts (%) | |
| Median (range) | 40.0 (0.8–97.4) |
| Diagnosis | |
| AML with recurrent genetic abnormalities | 17 (36.2) |
| AML with mutated NPM1 | 8 (17.0) |
| AML with biallelic mutation of CEBPA | 3 (6.4) |
| AML with mutated RUNX1 | 6 (12.8) |
| AML with MRC | 11 (23.4) |
| AML, NOS | 9 (19.1) |
| MDS-EB | 10 (21.3) |
| Risk stratification by 2022 ELN | |
| Favorable | 7 (18.9) |
| Intermediate | 9 (24.3) |
| Adverse | 21 (56.8) |
| Risk stratificaton by IPSS-M | 8 (17.0) |
| High | 2 (20.0) |
| Very high | 80 (80.0) |
| Number of chromosome abnormalities | |
| Mean ± SD | 5.2 ± 8.2 |
| Total (n = 47) | CK (n = 16) | Non-CK (n = 31) | p | |
|---|---|---|---|---|
| Sex (%) | 0.691 | |||
| Male | 29 (61.7) | 11 (68.8) | 18 (58.1) | |
| Female | 18 (38.3) | 5 (31.2) | 13 (41.9) | |
| Age (years) | 0.661 | |||
| Median (range) | 67.0 (31.0–86.0) | 66.0 (31.0–84.0) | 67.0 (46.0–86.0) | |
| Hb (g/dL) | 0.875 | |||
| Median (range) | 7.9 (5.9–12.1) | 8.1 (6.5–9.7) | 7.8 (5.9–12.1) | |
| WBC count (× 109/L) | 0.021 | |||
| Median (range) | 3.8 (0.6–247.2) | 2.6 (1.4–30.4) | 5.4 (0.6–247.2) | |
| Platelet count (× 109/L) | 0.148 | |||
| Median (range) | 51.0 (3.0–240.0) | 34.0 (9.0–240.0) | 56.0 (3.0–177.0) | |
| BM blasts (%) | 0.116 | |||
| Median (range) | 40.0 (0.8–97.4) | 26.5 (0.8–96) | 48.1 (11.0–97.4) | |
| Risk stratification by genetics | ||||
| FLT3-ITD | 0.104 | |||
| without | 40 (85.1) | 16 (100.0) | 24 (77.4) | |
| with | 7 (14.9) | 0 (0.0) | 7 (22.6) | |
| RUNX1 | 0.155 | |||
| without | 39 (83.0) | 16 (100.0) | 23 (74.2) | |
| with | 8 (17.0) | 0 (0.0) | 8 (25.8) | |
| Overall survival (months) | ||||
| Median (range) | 10.3 (0.1–46.1) | 5.1 (0.2–22.3) | 11.6 (0.1–46.1) | |
| AML Total (n = 37) | CK (n = 27) | Non-CK (n = 10) | ||
| Risk stratification by 2022 ELN | 0.005 | |||
| Favorable | 7 (18.9) | 0 (0.0) | 7 (25.9) | |
| Intermediate | 9 (24.3) | 0 (0.0) | 9 (33.3) | |
| Adverse | 21 (56.8) | 10 (100.0) | 11 (40.7) |
| Cohort | KUMC | TCGA | BeatAML | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Univariate Analysis | |||||||||
| HR (95% CI) | p | HR (95% CI) | p | HR (95% CI) | p | ||||
| Age (>65 years) | 1.1584 (0.5548–2.4185) | 0.6969 | 2.8149 (1.8637–4.2516) | <0.0001 | 2.7475 (1.5945–4.7343) | 0.0003 | |||
| PB WBC count | 1.0000 (1.0000–1.0000) | 0.9775 | 1.0026 (0.9984–1.0068) | 0.2347 | 1.0002 (0.9960–1.0045) | 0.9114 | |||
| BM blast count | 0.9970 (0.9849–1.0093) | 0.6364 | 0.9950 (0.9849–1.0053) | 0.3428 | 0.9948 (0.9851–1.0046) | 0.2978 | |||
| FLT3-ITD mutation | 0.9978 (0.3839–2.5937) | 0.9965 | 1.0012 (0.5922- 1.6927) | 0.9963 | 1.8201 (0.9591–3.4542) | 0.0683 | |||
| RUNX1 mutation | 1.6715 (0.5755–4.8550) | 0.3475 | 1.4898 (0.8118–2.7339) | 0.2004 | 1.2844 (0.6556–2.5161) | 0.4680 | |||
| CK | 2.1812 (1.0741–4.4293) | 0.0318 | 1.7175 (1.0407–2.8343) | 0.0353 | 1.8371 (0.9644–3.4994) | 0.0657 | |||
| CKS | 2.1098 (1.0376–4.2902) | 0.0402 | 2.2926 (1.4286–3.6793) | 0.0006 | 2.8349 (1.4498–5.5433) | 0.0024 | |||
| LSC17 | - | - | 1.7483 (1.1692–2.6144) | 0.0068 | 2.1387 (1.2059–3.7931) | 0.0097 | |||
| APS | - | - | 1.4957 (1.0045–2.2271) | 0.0486 | 2.2187 (1.2529–3.9290) | 0.0065 | |||
| Multivariate analysis | |||||||||
| HR (95% CI) | p | Overall p | HR (95% CI) | p | Overall p | HR (95% CI) | p | Overall p | |
| CK + Age | - | - | - | 1.5819 (0.9562–2.6170) | 0.0756 | <0.0001 | 2.0341 (1.0605–3.9017) | 0.0335 | 0.0003 |
| CKS + Age | - | - | - | 1.8671 (1.1495–3.0326) | 0.0121 | <0.0001 | 2.6512 (1.3458–5.2227) | 0.0050 | <0.0001 |
| LSC17 + Age | - | - | - | 1.5503 (1.0332–2.3262) | 0.0351 | <0.0001 | 2.0246 (1.1371–3.6047) | 0.0171 | 0.0001 |
| APS + Age | - | - | - | 1.5428 (1.0360–2.2975) | 0.0337 | <0.0001 | 1.8607 (1.0336–3.3496) | 0.0394 | 0.0003 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, C.; Kim, H.N.; Kwon, J.A.; Hwang, J.; Park, J.-Y.; Shin, O.S.; Yoon, S.-Y.; Yoon, J. Identification of a Complex Karyotype Signature with Clinical Implications in AML and MDS-EB Using Gene Expression Profiling. Cancers 2023, 15, 5289. https://doi.org/10.3390/cancers15215289
Lee C, Kim HN, Kwon JA, Hwang J, Park J-Y, Shin OS, Yoon S-Y, Yoon J. Identification of a Complex Karyotype Signature with Clinical Implications in AML and MDS-EB Using Gene Expression Profiling. Cancers. 2023; 15(21):5289. https://doi.org/10.3390/cancers15215289
Chicago/Turabian StyleLee, Cheonghwa, Ha Nui Kim, Jung Ah Kwon, Jinha Hwang, Ji-Ye Park, Ok Sarah Shin, Soo-Young Yoon, and Jung Yoon. 2023. "Identification of a Complex Karyotype Signature with Clinical Implications in AML and MDS-EB Using Gene Expression Profiling" Cancers 15, no. 21: 5289. https://doi.org/10.3390/cancers15215289
APA StyleLee, C., Kim, H. N., Kwon, J. A., Hwang, J., Park, J. -Y., Shin, O. S., Yoon, S. -Y., & Yoon, J. (2023). Identification of a Complex Karyotype Signature with Clinical Implications in AML and MDS-EB Using Gene Expression Profiling. Cancers, 15(21), 5289. https://doi.org/10.3390/cancers15215289