

The Role of Macrophages in Sarcoma Tumor Microenvironment and Treatment


Abstract
Simple Summary
Abstract
1. Introduction
2. M1 and M2 Macrophages
2.1. M1 and M2 Characteristics

{kind=link}
{kind=link}
| M1 | M2a | M2b | M2c | M2d | Reference | |
|---|---|---|---|---|---|---|
| Stimulation/ activation | IFN-γ LPS GM-CSF | IL-4 IL-13 Fungal and Helminth infection M-CSF | Immune complexes IL-1R | IL-10 TGF-β GCs | IL-6 Adenosine | [3,8,11,12,13,17] |
| Marker expression | CD11c CD1a CD16 CD32 CD40 CD54 CD68 CD86 CD80 CD197 MHC-II IL-1R IL2RA TLR2 TLR4 iNOS SOCS3 | Arg1 CD36 CD163 * CD200R CD204 CD209 (DC-SIGN) CSF1R MHC-II SR-A MMR (CD206) MGL1/2 TGM2 DcR3 IL-1R II | Arg1 CD80 CD86 MHC-II | Arg1 CCR2 CD36 CD163 TLR1 TLR8 SLAM | VEGF | [11,12,13,15,17,18,19,20,21,22] |
| Cytokine secretion | TNF-α IL-1 β IL-6 IL-12 IL-23 | IL-10 TGF-β IL-1ra | IL-1 IL-6 IL-10 TNF-α | IL-10 TGF-β | IL-10 IL-12 TNF-α TGF-β | [8,13,16,17] |
| Chemokine secretion | CCL2 CCL3 CCL4 CCL5 CCL8 CCL9 CXCL8 CXCL9 CXCL10 CXCL11 CXCL16 CCR7 | CCL2 CCL17 CCL18 CCL22 CCL24 | CCL1 | CCL13 CCL16 CCL18 | CCL5 CXCL10 CXCL16 | [12,14,17,18] |
2.2. Macrophage Polarization
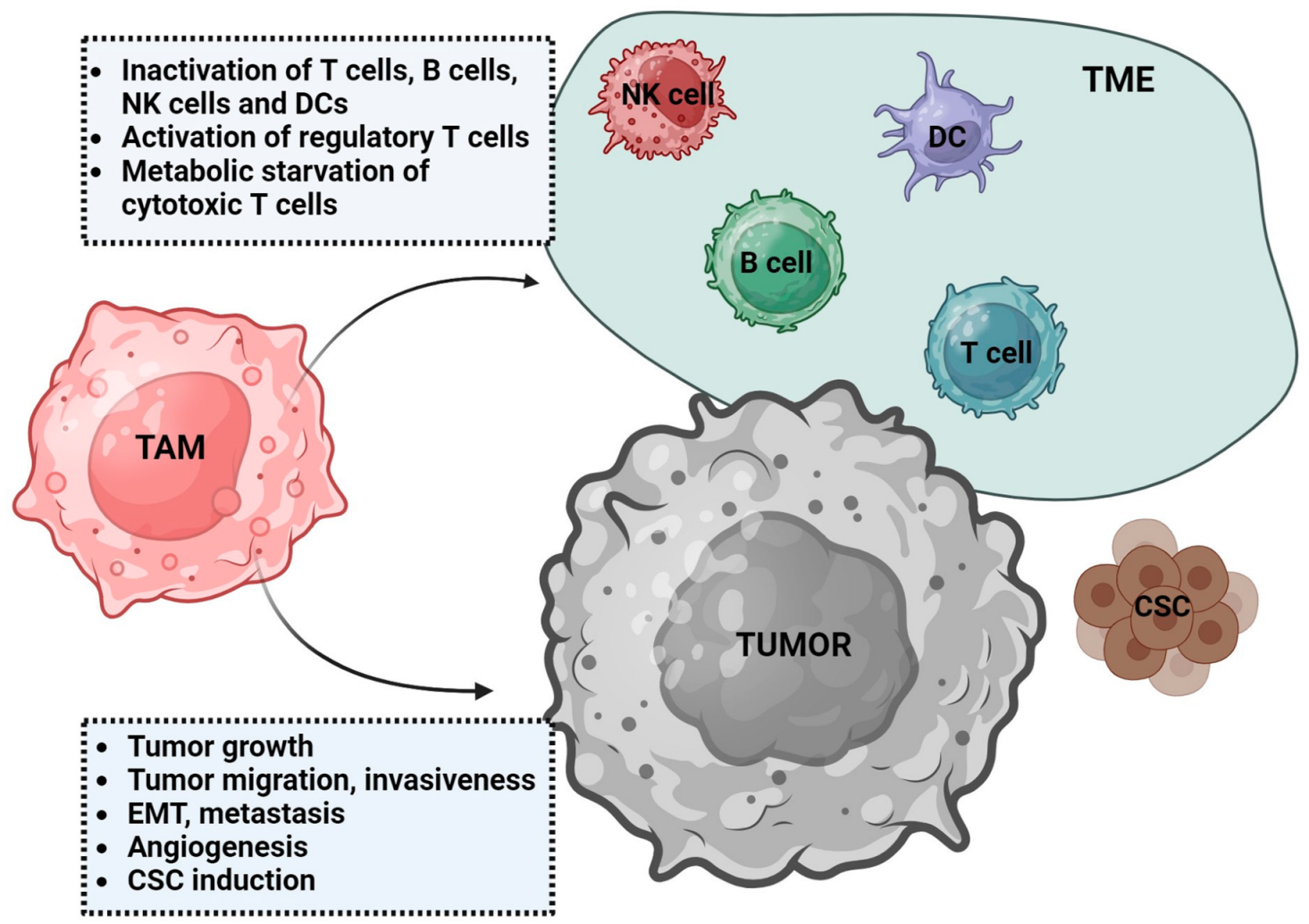
3. Tumor-Associated Macrophages (TAMs)
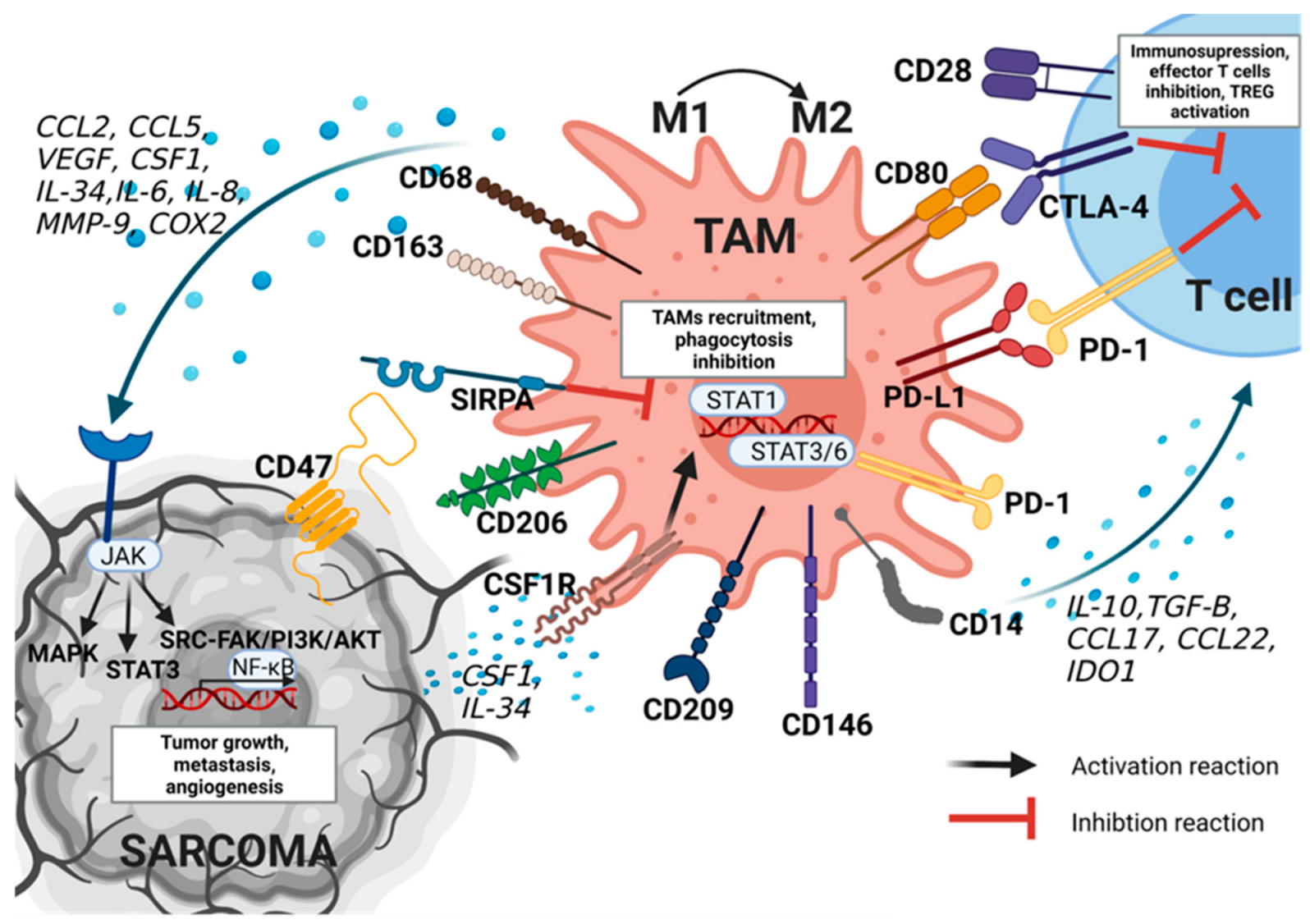
3.1. Characteristics of TAMs
3.2. TAMs Function in Tumor Development
4. TAMs in Sarcomas
4.1. TAMs Phenotype and Role in Sarcoma
4.2. The Role of TAMs as a Predictive and Prognostic Factors in Sarcoma
| Tams’ Marker | Sarcoma Subtype | Overall Survival Rate | Progression-Free Survival | Metastatic Potential | Reference |
|---|---|---|---|---|---|
| CD68 | dedifferentiated chondrosarcoma | decreased | - | increased | [35] |
| myxoid liposarcoma | decreased | - | - | [65] | |
| dedifferentiated liposarcoma | decreased | - | - | [69] | |
| Ewing sarcoma | decreased | - | - | [50] | |
| leiomyosarcoma | - | decreased | - | [33] | |
| CD163 | synovial sarcoma | decreased | decreased | - | [71] |
| undifferentiated pleomorphic sarcoma | decreased | - | - | [70] | |
| solitary fibrous tumors | - | decreased | - | [33] | |
| myxoid liposarcoma | decreased | - | - | [68] | |
| dedifferentiated liposarcoma | decreased | increased | - | [33,69] | |
| osteosarcoma | increased | - | increased | [50,75] | |
| Ewing sarcoma | increased | - | - | [51] | |
| leiomyosarcoma | - | increased | - | [33] | |
| SIRPA | dedifferentiated chondrosarcoma | decreased | - | increased | [35] |
| synovial sarcoma | decreased | - | - | [33] | |
| myxofibrosarcoma | decreased | - | - | [33] | |
| CSF1R | dedifferentiated chondrosarcoma | decreased | - | increased | [35] |
| CD14 | osteosarcoma | increased | - | - | [58] |
| myxoid liposarcoma | decreased | - | - | [62] | |
| dedifferentiated liposarcoma | decreased | - | - | [66] |
5. Macrophages as Treatment Targets in Sarcoma
6. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Fujiwara, T.; Healey, J.; Ogura, K.; Yoshida, A.; Kondo, H.; Hata, T.; Kure, M.; Tazawa, H.; Nakata, E.; Kunisada, T.; et al. Role of Tumor-Associated Macrophages in Sarcomas. Cancers 2021, 13, 1086. [Google Scholar] [CrossRef]
- Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol. Rev. 2008, 222, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef]
- Mantovani, A.; Schioppa, T.; Porta, C.; Allavena, P.; Sica, A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006, 25, 315–322. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Skubitz, K.M.; D’Adamo, D.R. Sarcoma. Mayo Clin. Proc. 2007, 82, 1409–1432. [Google Scholar] [CrossRef]
- Nacev, B.A.; Sanchez-Vega, F.; Smith, S.A.; Antonescu, C.R.; Rosenbaum, E.; Shi, H.; Tang, C.; Socci, N.D.; Rana, S.; Gularte-Mérida, R.; et al. Clinical sequencing of soft tissue and bone sarcomas delineates diverse genomic landscapes and potential therapeutic targets. Nat. Commun. 2022, 13, 1107. [Google Scholar] [CrossRef]
- Zhang, Q.; Liu, L.; Gong, C.; Shi, H.; Zeng, Y.; Wang, X.; Zhao, Y.; Wei, Y. Prognostic Significance of Tumor-Associated Macrophages in Solid Tumor: A Meta-Analysis of the Literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef]
- Li, M.; He, L.; Zhu, J.; Zhang, P.; Liang, S. Targeting tumor-associated macrophages for cancer treatment. Cell Biosci. 2022, 12, 85. [Google Scholar] [CrossRef]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2020, 9, 1512. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Geng, X.; Hou, J.; Wu, G. New insights into M1/M2 macrophages: Key modulators in cancer progression. Cancer Cell Int. 2021, 21, 389. [Google Scholar] [CrossRef] [PubMed]
- Van Dalen, F.; van Stevendaal, M.; Fennemann, F.; Verdoes, M.; Ilina, O. Molecular Repolarisation of Tumour-Associated Macrophages. Molecules 2018, 24, 9. [Google Scholar] [CrossRef]
- Chávez-Galán, L.; Olleros, M.L.; Vesin, D.; Garcia, I. Much More than M1 and M2 Macrophages, There are also CD169(+) and TCR(+) Macrophages. Front. Immunol. 2015, 6, 263. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Kumar, B.; Datta, J.; Teknos, T.N.; Kumar, P. IL-6 Promotes Head and Neck Tumor Metastasis by Inducing Epithelial–Mesenchymal Transition via the JAK-STAT3-SNAIL Signaling Pathway. Mol. Cancer Res. 2011, 9, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Spiller, K.L.; Anfang, R.R.; Spiller, K.J.; Ng, J.; Nakazawa, K.R.; Daulton, J.W.; Vunjak-Novakovic, G. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials 2014, 35, 4477–4488. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef]
- Raes, G.; De Baetselier, P.; Noël, W.; Beschin, A.; Brombacher, F.; Hassanzadeh, G.G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J. Leukoc. Biol. 2002, 71, 597–602. [Google Scholar] [CrossRef]
- Vanmeerbeek, I.; Govaerts, J.; Laureano, R.S.; Sprooten, J.; Naulaerts, S.; Borras, D.M.; Laoui, D.; Mazzone, M.; Van Ginderachter, J.A.; Garg, A.D. The Interface of Tumour-Associated Macrophages with Dying Cancer Cells in Immuno-Oncology. Cells 2022, 11, 3890. [Google Scholar] [CrossRef]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 promotes inflammatory macrophage polarization and T H1-TH17 responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef]
- Gleissner, C.A.; Erbel, C. CXCL4-Induced Macrophages: A Novel Therapeutic Target in Human Atherosclerosis? In Atherogenesis; IntechOpen: London, UK, 2012. [Google Scholar] [CrossRef][Green Version]
- Barros, M.H.M.; Hauck, F.; Dreyer, J.H.; Kempkes, B.; Niedobitek, G. Macrophage Polarisation: An Immunohistochemical Approach for Identifying M1 and M2 Macrophages. PLoS ONE 2013, 8, e80908. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.E.; Whyte, C.S.; Gordon, P.; Barker, R.N.; Rees, A.J.; Wilson, H.M. A critical role for suppressor of cytokine signalling 3 in promoting M1 macrophage activation and function in vitro and in vivo. Immunology 2014, 141, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, Y.; Hamilton, T.A. IL-4-induced STAT6 suppresses IFN-gamma-stimulated STAT1-dependent transcription in mouse macrophages. J. Immunol. 1997, 159, 5474–5482. [Google Scholar] [CrossRef]
- Sawa-Wejksza, K.; Kandefer-Szerszeń, M. Tumor-Associated Macrophages as Target for Antitumor Therapy. Arch. Immunol. Ther. Exp. 2018, 66, 97–111. [Google Scholar] [CrossRef]
- Feng, Y.; Ye, Z.; Song, F.; He, Y.; Liu, J. The Role of TAMs in Tumor Microenvironment and New Research Progress. Stem Cells Int. 2022, 2022, 5775696. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Katsuya, Y.; Horinouchi, H.; Asao, T.; Kitahara, S.; Goto, Y.; Kanda, S.; Fujiwara, Y.; Nokihara, H.; Yamamoto, N.; Watanabe, S.; et al. Expression of programmed death 1 (PD-1) and its ligand (PD-L1) in thymic epithelial tumors: Impact on treatment efficacy and alteration in expression after chemotherapy. Lung Cancer 2016, 99, 4–10. [Google Scholar] [CrossRef]
- Aramini, B.; Masciale, V.; Grisendi, G.; Banchelli, F.; D’Amico, R.; Maiorana, A.; Morandi, U.; Dominici, M.; Haider, K.H. Cancer stem cells and macrophages: Molecular connections and future perspectives against cancer. Oncotarget 2021, 12, 230–250. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Shoushtari, A.N.; Agaram, N.P.; Kuk, D.; Qin, L.-X.; Carvajal, R.D.; Dickson, M.A.; Gounder, M.; Keohan, M.L.; Schwartz, G.K.; et al. Prevalence of tumor-infiltrating lymphocytes and PD-L1 expression in the soft tissue sarcoma microenvironment. Hum. Pathol. 2015, 46, 357–365. [Google Scholar] [CrossRef]
- Dancsok, A.R.; Gao, D.; Lee, A.F.; Steigen, S.E.; Blay, J.-Y.; Thomas, D.M.; Maki, R.G.; Nielsen, T.O.; Demicco, E.G. Tumor-associated macrophages and macrophage-related immune checkpoint expression in sarcomas. Oncoimmunology 2020, 9, 1747340. [Google Scholar] [CrossRef]
- Barclay, A.N. Signal regulatory protein alpha (SIRPα)/CD47 interaction and function. Curr. Opin. Immunol. 2009, 21, 47–52. [Google Scholar] [CrossRef]
- Richert, I.; Gomez-Brouchet, A.; Bouvier, C.; Pinieux, G.D.B.D.; Karanian, M.; Blay, J.-Y.; Dutour, A. The immune landscape of chondrosarcoma—Potential for therapeutic targeting of CSFR1+ macrophages. J. Bone Oncol. 2020, 20, 100271. [Google Scholar] [CrossRef]
- Tsai, R.K.; Discher, D.E. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 2008, 180, 989–1003. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Dhupkar, P.; Gordon, N.; Stewart, J.; Kleinerman, E.S. Anti-PD-1 therapy redirects macrophages from an M2 to an M1 phenotype inducing regression of OS lung metastases. Cancer Med. 2018, 7, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Cersosimo, F.; Lonardi, S.; Bernardini, G.; Telfer, B.; Mandelli, G.E.; Santucci, A.; Vermi, W.; Giurisato, E. Tumor-Associated Macrophages in Osteosarcoma: From Mechanisms to Therapy. Int. J. Mol. Sci. 2020, 21, 5207. [Google Scholar] [CrossRef]
- Fujiwara, T.; Yakoub, M.A.; Chandler, A.; Christ, A.B.; Yang, G.; Ouerfelli, O.; Rajasekhar, V.K.; Yoshida, A.; Kondo, H.; Hata, T.; et al. CSF1/CSF1R Signaling Inhibitor Pexidartinib (PLX3397) Reprograms Tumor-Associated Macrophages and Stimulates T-cell Infiltration in the Sarcoma Microenvironment. Mol. Cancer Ther. 2021, 20, 1388–1399. [Google Scholar] [CrossRef]
- Ségaliny, A.I.; Mohamadi, A.; Dizier, B.; Lokajczyk, A.; Brion, R.; Lanel, R.; Amiaud, J.; Charrier, C.; Boisson-Vidal, C.; Heymann, D. Interleukin-34 promotes tumor progression and metastatic process in osteosarcoma through induction of angiogenesis and macrophage recruitment. Int. J. Cancer 2015, 137, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Tessaro, F.H.G.; Ko, E.Y.; De Simone, M.; Piras, R.; Broz, M.T.; Goodridge, H.S.; Balzer, B.; Shiao, S.L.; Guarnerio, J. Single-cell RNA-seq of a soft-tissue sarcoma model reveals the critical role of tumor-expressed MIF in shaping macrophage heterogeneity. Cell Rep. 2022, 39, 110977. [Google Scholar] [CrossRef] [PubMed]
- Brandacher, G.; Perathoner, A.; Ladurner, R.; Schneeberger, S.; Obrist, P.; Winkler, C.; Werner, E.R.; Werner-Felmayer, G.; Weiss, H.G.; G√∂bel, G.; et al. Prognostic value of indoleamine 2,3-dioxygenase expression in colorectal cancer: Effect on tumor-infiltrating T cells. Clin. Cancer Res. 2006, 12, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Nafia, I.; Toulmonde, M.; Bortolotto, D.; Chaibi, A.; Bodet, D.; Rey, C.; Velasco, V.; Larmonier, C.B.; Cerf, L.; Adam, J.; et al. IDO Targeting in Sarcoma: Biological and Clinical Implications. Front. Immunol. 2020, 11, 274. [Google Scholar] [CrossRef]
- Koo, J.; Hayashi, M.; Verneris, M.R.; Lee-Sherick, A.B. Targeting Tumor-Associated Macrophages in the Pediatric Sarcoma Tumor Microenvironment. Front. Oncol. 2020, 10, 581107. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Kerkelä, E.; Böhling, T.; Herva, R.; Uria, J.; Saarialho-Kere, U. Human macrophage metalloelastase (MMP-12) expression is induced in chondrocytes during fetal development and malignant transformation. Bone 2001, 29, 487–493. [Google Scholar] [CrossRef]
- Yoon, S.W.; Chun, J.S.; Sung, M.H.; Kim, J.Y.; Poo, H. α-MSH inhibits TNF-α-induced matrix metalloproteinase-13 expression by modulating p38 kinase and nuclear factor κB signaling in human chondrosarcoma HTB-94 cells. Osteoarthr. Cartil. 2008, 16, 115–124. [Google Scholar] [CrossRef]
- David, E.; Blanchard, F.; Heymann, M.F.; De Pinieux, G.; Gouin, F.; Rédini, F.; Heymann, D. The Bone Niche of Chondrosarcoma: A Sanctuary for Drug Resistance, Tumour Growth and also a Source of New Therapeutic Targets. Sarcoma 2011, 2011, 932451. [Google Scholar] [CrossRef]
- Gomez-Brouchet, A.; Illac, C.; Gilhodes, J.; Bouvier, C.; Aubert, S.; Guinebretiere, J.-M.; Marie, B.; Larousserie, F.; Entz-Werlé, N.; de Pinieux, G.; et al. CD163-positive tumor-associated macrophages and CD8-positive cytotoxic lymphocytes are powerful diagnostic markers for the therapeutic stratification of osteosarcoma patients: An immunohistochemical analysis of the biopsies fromthe French OS2006 phase 3 t. Oncoimmunology 2017, 6, e1331193. [Google Scholar] [CrossRef]
- Handl, M.; Hermanova, M.; Hotarkova, S.; Jarkovsky, J.; Mudry, P.; Shatokhina, T.; Vesela, M.; Sterba, J.; Zambo, I. Clinicopathological correlation of tumor-associated macrophages in Ewing sarcoma. Biomed. Pap. 2018, 162, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.; Wilding, C.P.; Krasny, L.; Zhu, X.; Chadha, M.; Tam, Y.B.; Ps, H.; Mahalingam, A.H.; Lee, A.T.J.; Arthur, A.; et al. The proteomic landscape of soft tissue sarcomas. Nat. Commun. 2023, 14, 3834. [Google Scholar] [CrossRef]
- Jiang, G.; Chen, J.; Li, Y.; Zhou, J.; Wang, W.; Wu, G.; Zhang, Y. Association of SMC4 with prognosis and immune infiltration of sarcoma. Aging 2023, 15, 567–582. [Google Scholar] [CrossRef]
- Fan, J.; Qin, X.; He, R.; Ma, J.; Wei, Q. Gene expression profiles for an immunoscore model in bone and soft tissue sarcoma. Aging 2021, 13, 13708–13725. [Google Scholar] [CrossRef]
- Dufresne, A.; Lesluyes, T.; Ménétrier-Caux, C.; Brahmi, M.; Darbo, E.; Toulmonde, M.; Italiano, A.; Mir, O.; Le Cesne, A.; Le Guellec, S.; et al. Specific immune landscapes and immune checkpoint expressions in histotypes and molecular subtypes of sarcoma. Oncoimmunology 2020, 9, 1792036. [Google Scholar] [CrossRef]
- Kostine, M.; Cleven, A.H.; de Miranda, N.F.C.C.; Italiano, A.; Cleton-Jansen, A.-M.; Bovée, J.V.M.G. Analysis of PD-L1, T-cell infiltrate and HLA expression in chondrosarcoma indicates potential for response to immunotherapy specifically in the dedifferentiated subtype. Mod. Pathol. 2016, 29, 1028–1037. [Google Scholar] [CrossRef]
- Simard, F.A.; Richert, I.; Vandermoeten, A.; Decouvelaere, A.-V.; Michot, J.-P.; Caux, C.; Blay, J.-Y.; Dutour, A. Description of the immune microenvironment of chondrosarcoma and contribution to progression. Oncoimmunology 2017, 6, e1265716. [Google Scholar] [CrossRef]
- Buddingh, E.P.; Kuijjer, M.L.; Duim, R.A.J.; Bürger, H.; Agelopoulos, K.; Myklebost, O.; Serra, M.; Mertens, F.; Hogendoorn, P.C.W.; Lankester, A.C.; et al. Tumor-Infiltrating Macrophages Are Associated with Metastasis Suppression in High-Grade Osteosarcoma: A Rationale for Treatment with Macrophage Activating Agents. Clin. Cancer Res. 2011, 17, 2110–2119. [Google Scholar] [CrossRef] [PubMed]
- Nyström, H.; Jönsson, M.; Nilbert, M.; Carneiro, A. Immune-cell infiltration in high-grade soft tissue sarcomas; prognostic implications of tumor-associated macrophages and B-cells. Acta Oncol. 2023, 62, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Toulmonde, M.; Penel, N.; Adam, J.; Chevreau, C.; Blay, J.-Y.; Le Cesne, A.; Bompas, E.; Piperno-Neumann, S.; Cousin, S.; Grellety, T.; et al. Use of PD-1 Targeting, Macrophage Infiltration, and IDO Pathway Activation in Sarcomas. JAMA Oncol. 2018, 4, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Smolle, M.A.; Herbsthofer, L.; Goda, M.; Granegger, B.; Brcic, I.; Bergovec, M.; Scheipl, S.; Prietl, B.; El-Heliebi, A.; Pichler, M.; et al. Influence of tumor-infiltrating immune cells on local control rate, distant metastasis, and survival in patients with soft tissue sarcoma. Oncoimmunology 2021, 10, 1896658. [Google Scholar] [CrossRef]
- Han, Q.; Shi, H.; Liu, F. CD163 + M2-type tumor-associated macrophage support the suppression of tumor-infiltrating T cells in osteosarcoma. Int. Immunopharmacol. 2016, 34, 101–106. [Google Scholar] [CrossRef]
- Inagaki, Y.; Hookway, E.; Williams, K.A.; Hassan, A.B.; Oppermann, U.; Tanaka, Y.; Soilleux, E.; Athanasou, N.A. Dendritic and mast cell involvement in the inflammatory response to primary malignant bone tumours. Clin. Sarcoma Res. 2016, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Takeya, H.; Wakigami, N.; Kusada, N.; Bekki, H.; Ishihara, S.; Takeya, M.; Nakashima, Y.; Oda, Y. Positive correlation between the density of macrophages and T-cells in undifferentiated sarcoma. Med. Mol. Morphol. 2019, 52, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lu, Y.-F.; Wang, C.-S.; Xie, Y.-X.; Zhao, Y.-Q.; Qian, Y.-C.; Liu, W.-T.; Wang, M.; Jiang, B.-H. HB-EGF Activates the EGFR/HIF-1α Pathway to Induce Proliferation of Arsenic-Transformed Cells and Tumor Growth. Front. Oncol. 2020, 10, 1019. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-H.; Espinosa, I.; Vrijaldenhoven, S.; Subramanian, S.; Montgomery, K.D.; Zhu, S.; Marinelli, R.J.; Peterse, J.L.; Poulin, N.; Nielsen, T.O.; et al. Prognostic Significance of Macrophage Infiltration in Leiomyosarcomas. Clin. Cancer Res. 2008, 14, 1423–1430. [Google Scholar] [CrossRef]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J. Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef]
- Nabeshima, A.; Matsumoto, Y.; Fukushi, J.; Iura, K.; Matsunobu, T.; Endo, M.; Fujiwara, T.; Iida, K.; Fujiwara, Y.; Hatano, M.; et al. Tumour-associated macrophages correlate with poor prognosis in myxoid liposarcoma and promote cell motility and invasion via the HB-EGF-EGFR-PI3K/Akt pathways. Br. J. Cancer 2015, 112, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.A.; LaFranzo, N.A.; LaFleur, B.J.; Gittelman, R.M.; Vignali, M.; Zhang, S.; Flanagan, K.C.; Rytlewski, J.; Riolobos, L.; Schulte, B.C.; et al. CD4+ T cell and M2 macrophage infiltration predict dedifferentiated liposarcoma patient outcomes. J. Immunother. Cancer 2021, 9, e002812. [Google Scholar] [CrossRef]
- Shiraishi, D.; Fujiwara, Y.; Horlad, H.; Saito, Y.; Iriki, T.; Tsuboki, J.; Cheng, P.; Nakagata, N.; Mizuta, H.; Bekki, H.; et al. CD163 Is Required for Protumoral Activation of Macrophages in Human and Murine Sarcoma. Cancer Res. 2018, 78, 3255–3266. [Google Scholar] [CrossRef]
- Oike, N.; Kawashima, H.; Ogose, A.; Hotta, T.; Hatano, H.; Ariizumi, T.; Sasaki, T.; Yamagishi, T.; Umezu, H.; Endo, N. Prognostic impact of the tumor immune microenvironment in synovial sarcoma. Cancer Sci. 2018, 109, 3043–3054. [Google Scholar] [CrossRef]
- Schroeder, B.A.; Zhang, Y.; Smythe, K.S.; Desai, P.; Thomas, A.; Viveiros, P.; Alexiev, B.A.; Obeidin, F.; Chen, E.Y.; Cranmer, L.D.; et al. Immunologic Gene Signature Analysis Correlates Myeloid Cells and M2 Macrophages with Time to Trabectedin Failure in Sarcoma Patients. Cancers 2022, 14, 1290. [Google Scholar] [CrossRef] [PubMed]
- Umakoshi, M.; Nakamura, A.; Tsuchie, H.; Li, Z.; Kudo-Asabe, Y.; Miyabe, K.; Ito, Y.; Yoshida, M.; Nagasawa, H.; Okada, K.; et al. Macrophage numbers in the marginal area of sarcomas predict clinical prognosis. Sci. Rep. 2023, 13, 1290. [Google Scholar] [CrossRef]
- Dumars, C.; Ngyuen, J.-M.; Gaultier, A.; Lanel, R.; Corradini, N.; Gouin, F.; Heymann, D.; Heymann, M.-F. Dysregulation of macrophage polarization is associated with the metastatic process in osteosarcoma. Oncotarget 2016, 7, 78343–78354. [Google Scholar] [CrossRef]
- Heymann, M.-F.; Lézot, F.; Heymann, D. The contribution of immune infiltrates and the local microenvironment in the pathogenesis of osteosarcoma. Cell Immunol. 2019, 343, 103711. [Google Scholar] [CrossRef] [PubMed]
- Kleinerman, E.S.; Raymond, A.K.; Bucana, C.D.; Jaffe, N.; Harris, M.B.; Krakoff, I.H.; Benjamin, R.; Fidler, I.J. Unique histological changes in lung metastases of osteosarcoma patients following therapy with liposomal muramyl tripeptide (CGP 19835A lipid). Cancer Immunol. Immunother. 1992, 34, 211–220. [Google Scholar] [CrossRef]
- Chou, A.J.; Kleinerman, E.S.; Krailo, M.D.; Chen, Z.; Betcher, D.L.; Healey, J.H.; Conrad, E.U.; Nieder, M.L.; Weiner, M.A.; Wells, R.J.; et al. Addition of muramyl tripeptide to chemotherapy for patients with newly diagnosed metastatic osteosarcoma. Cancer 2009, 115, 5339–5348. [Google Scholar] [CrossRef]
- Punzo, F.; Bellini, G.; Tortora, C.; Di Pinto, D.; Argenziano, M.; Pota, E.; Di Paola, A.; Di Martino, M.; Rossi, F. Mifamurtide and TAM-like macrophages: Effect on proliferation, migration and differentiation of osteosarcoma cells. Oncotarget 2020, 11, 687–698. [Google Scholar] [CrossRef]
- Biteau, K.; Guiho, R.; Chatelais, M.; Taurelle, J.; Chesneau, J.; Corradini, N.; Heymann, D.; Redini, F. L-MTP-PE and zoledronic acid combination in osteosarcoma: Preclinical evidence of positive therapeutic combination for clinical transfer. Am. J. Cancer Res. 2016, 6, 677–689. [Google Scholar]
- Anand, N.; Peh, K.H.; Kolesar, J.M. Macrophage Repolarization as a Therapeutic Strategy for Osteosarcoma. Int. J. Mol. Sci. 2023, 24, 2858. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Manji, G.A.; Van Tine, B.A.; Lee, S.M.; Raufi, A.G.; Pellicciotta, I.; Hirbe, A.C.; Pradhan, J.; Chen, A.; Rabadan, R.; Schwartz, G.K. A Phase I Study of the Combination of Pexidartinib and Sirolimus to Target Tumor-Associated Macrophages in Unresectable Sarcoma and Malignant Peripheral Nerve Sheath Tumors. Clin. Cancer Res. 2021, 27, 5519–5527. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterom, A.T.; Judson, I.R.; Verweij, J.; Stroobants, S.; Dumez, H.; Donato di Paola, E.; Sciot, R.; Van Glabbeke, M.; Dimitrijevic, S.; Nielsen, O.S. Update of phase I study of imatinib (STI571) in advanced soft tissue sarcomas and gastrointestinal stromal tumors: A report of the EORTC Soft Tissue and Bone Sarcoma Group. Eur. J. Cancer 2002, 38, S83–S87. [Google Scholar] [CrossRef] [PubMed]
- Cavnar, M.J.; DeMatteo, R.P. Sarcoma response to targeted therapy dynamically polarizes tumor-associated macrophages. Oncoimmunology 2014, 3, e28463. [Google Scholar] [CrossRef]
- Chugh, R.; Thomas, D.; Wathen, K.; Thall, P.F.; Benjamin, R.S.; Maki, R.S.; Samuels, B.L.; Keohan, M.L.; Priebat, D.A.; Baker, L.H. Imatinib mesylate in soft tissue and bone sarcomas: Interim results of a Sarcoma Alliance for Research thru Collaboration (SARC) phase II trial. J. Clin. Oncol. 2004, 22, 9001. [Google Scholar] [CrossRef]
- Ou, D.-L.; Chen, C.-W.; Hsu, C.-L.; Chung, C.-H.; Feng, Z.-R.; Lee, B.-S.; Cheng, A.-L.; Yang, M.-H.; Hsu, C. Regorafenib enhances antitumor immunity via inhibition of p38 kinase/Creb1/Klf4 axis in tumor-associated macrophages. J. Immunother. Cancer 2021, 9, e001657. [Google Scholar] [CrossRef]
- Davis, L.E.; Bolejack, V.; Ryan, C.W.; Ganjoo, K.N.; Loggers, E.T.; Chawla, S.; Agulnik, M.; Livingston, M.B.; Reed, D.; Keedy, V.; et al. Randomized Double-Blind Phase II Study of Regorafenib in Patients with Metastatic Osteosarcoma. J. Clin. Oncol. 2019, 37, 1424–1431. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Liguori, M.; Buracchi, C.; Pasqualini, F.; Bergomas, F.; Pesce, S.; Sironi, M.; Grizzi, F.; Mantovani, A.; Belgiovine, C.; Allavena, P. Functional TRAIL receptors in monocytes and tumor-associated macrophages: A possible targeting pathway in the tumor microenvironment. Oncotarget 2016, 7, 41662–41676. [Google Scholar] [CrossRef]
- Kontny, H.U.; Hämmerle, K.; Klein, R.; Shayan, P.; Mackall, C.L.; Niemeyer, C.M. Sensitivity of Ewing’s sarcoma to TRAIL-induced apoptosis. Cell Death Differ. 2001, 8, 506–514. [Google Scholar] [CrossRef][Green Version]
- Germano, G.; Frapolli, R.; Simone, M.; Tavecchio, M.; Erba, E.; Pesce, S.; Pasqualini, F.; Grosso, F.; Sanfilippo, R.; Casali, P.G.; et al. Antitumor and Anti-inflammatory Effects of Trabectedin on Human Myxoid Liposarcoma Cells. Cancer Res. 2010, 70, 2235–2244. [Google Scholar] [CrossRef]
- Edris, B.; Weiskopf, K.; Volkmer, A.K.; Volkmer, J.-P.; Willingham, S.B.; Contreras-Trujillo, H.; Liu, J.; Majeti, R.; West, R.B.; Fletcher, J.A.; et al. Antibody therapy targeting the CD47 protein is effective in a model of aggressive metastatic leiomyosarcoma. Proc. Natl. Acad. Sci. USA 2012, 109, 6656–6661. [Google Scholar] [CrossRef] [PubMed]


| Clinical Trial Number | Status | Conditions | Interventions | Phases |
|---|---|---|---|---|
| NCT02584647 | Recruiting | Sarcoma and malignant peripheral nerve sheath tumors | Drug: PLX3397 (Receptor Tyrosine Kinase Inhibitor), Sirolimus | Phase I/II |
| NCT02502786 | Recruiting | Recurrent osteosarcoma | Biological: Humanized anti-GD2 antibody Drug: GM-CSF | Phase II |
| NCT03217266 | Active, not recruiting | Soft tissue sarcoma | Drug: MDM2 Inhibitor KRT-232 Radiation: Radiation Therapy | Phase I |
| NCT01050296 | Recruiting | Pediatric solid tumors | Not applicable | Not applicable |
| NCT04465643 | Recruiting | Nerve sheath tumors | Drug: Nivolumab, Ipilimumab | Phase I |
| NCT03866525 | Recruiting | Solid tumor, gastrointestinal cancer | Biological: OH2 (anti-PD-1 antibody) injection, with or without irinotecan or HX008 | Phase I/II |
| NCT02389244 | Recruiting | Ewing sarcomas, chondrosarcomas, osteosarcomas, chondroma, CIC-rearranged sarcoma | Drug: Regorafenib, placebo | Phase II |
| NCT05427461 | Recruiting | Leiomyosarcoma | Other: Blood samples will be collected at different times | Not applicable |
| NCT02390752 | Recruiting | Neurofibroma, plexiform, precursor cell, lymphoblastic leukemia-lymphoma, leukemia, promyelocytic, acute, sarcoma | Drug: Turalio (CSF1R inhibitor) | Phase I |
| NCT04900519 | Recruiting | Solid tumor relapsed solid neoplasm refractory tumor | Biological: STI-6643 Anti-CD47 human monoclonal antibody | Phase I |
| NCT03990233 | Recruiting | Solid tumor, adult | Drug: BI 765063 (antagonist to SIRPα receptor), BI 754091 (antagonist to PD-1 receptor) | Phase I |
| NCT04660929 | Recruiting | All HER2 overexpressing solid tumors | Biological: CT-0508, Pembrolizumab | Phase I |
| NCT04168528 | Recruiting | Malignant solid tumor, breast cancer, head and neck cancer, melanoma | Drug: 68GaNOTA-Anti-MMR-VHH2 | Phase I/II |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zając, A.E.; Czarnecka, A.M.; Rutkowski, P. The Role of Macrophages in Sarcoma Tumor Microenvironment and Treatment. Cancers 2023, 15, 5294. https://doi.org/10.3390/cancers15215294
Zając AE, Czarnecka AM, Rutkowski P. The Role of Macrophages in Sarcoma Tumor Microenvironment and Treatment. Cancers. 2023; 15(21):5294. https://doi.org/10.3390/cancers15215294
Chicago/Turabian StyleZając, Agnieszka E., Anna M. Czarnecka, and Piotr Rutkowski. 2023. "The Role of Macrophages in Sarcoma Tumor Microenvironment and Treatment" Cancers 15, no. 21: 5294. https://doi.org/10.3390/cancers15215294
APA StyleZając, A. E., Czarnecka, A. M., & Rutkowski, P. (2023). The Role of Macrophages in Sarcoma Tumor Microenvironment and Treatment. Cancers, 15(21), 5294. https://doi.org/10.3390/cancers15215294