
Exploring Therapeutic Avenues in Lung Cancer: The Epigenetic Perspective

 , ,
, ,  ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Overview of Lung Cancer Epigenetics
3. Aberrant DNA Methylation and Gene Modification in Lung Cancer

{kind=link}
{kind=link}
| Gene | Epigenetic Modifications | Reported Histological Type | Mechanism | References |
|---|---|---|---|---|
| RASSF1A | hypermethylation | NSCLC, SCLC | Hypermethylation of the RASSF1A gene promoter region in lung cancer disrupts RASSF1A’s tumor suppressor function, potentially leading to dysregulated cell cycle progression and resistance to Ras-induced apoptosis. | [36] |
| SEMA3B | hypermethylation | NSCLC, SCLC | Hypermethylation of SEMA3B gene promoter region results in decreased SEMA3B expression, which can compromise its tumor-suppressive functions, promote tumor growth, and potentially contribute to metastasis. | [37] |
| DAPK | hypermethylation | NSCLC | Cell cycle progression, apoptosis, cell migration. | [38] |
| P14 | hypermethylation | NSCLC | Hypermethylated P14 gene results in increased activity of CDK4 and CDK6, leading to uncontrolled cell cycle progression and excessive cell growth. | [39] |
| FHIT | hypermethylation | NSCLC | FHIT hypermethylation induces inactivation of FHIT gene, facilitating genomic instability, promoting clonal expansion and enhancing survival under selective pressures. | [40,41] |
| PTEN | hypermethylation | NSCLC | Inhibitor of the AKT/MTOR pathway and cell cycle. | [42] |
| CDKN2A | hypermethylation | NSCLC | Hypermethylation of CDKN2A contributes to tumorigenesis through the inactivation of tumor suppressor genes, specifically p16INK4a and p14ARF. | [43] |
| CDH1 | hypermethylation | NSCLC | Facilitates cellular adhesion while restraining cellular motility, invasion, and metastasis. | [44] |
| CDH13 | hypermethylation | NSCLC | Loss of E-cadherin weakens cell–cell adhesion, promoting cancer cells’ ability to detach from the primary tumor and invade surrounding tissues. | [45] |
| BLU | hypermethylation | NSCLC | Transcription regulation. | [46] |
| TBX-2 | hypermethylation | NSCLC | Hypermethylation of the TBX-2 promoter region creates a repressive chromatin state that inhibits transcription factor binding and RNA polymerase activity, consequently silencing TBX-2 gene expression and disrupting its role in transcription regulation, which contributes to tumorigenesis. | [47] |
| WIF1 | hypermethylation | NSCLC | Hypermethylation of WIF1 gene promoter region silences WIF1 expression by adding methyl groups to CpG islands, impairing its tumor-suppressive function and promoting tumor growth and progression. | [48] |
4. Epigenetic Therapy in Lung Cancer
DNMT Inhibitors
5. Histone Alterations
5.1. HDAC Inhibitors
5.2. PRMT Inhibitors and MTAP Deletion in Lung Cancer
6. Involvement of Epigenetic Modifications in the Development of Lung Cancer Drug Resistance
6.1. Vorinostat
6.2. Panobinostat
6.3. Belinostat
6.4. Trichostatin A (TSA)
6.5. Decitabine and 5-Azacytidine
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Quintanal-Villalonga, Á.; Molina-Pinelo, S. Epigenetics of Lung Cancer: A Translational Perspective. Cell. Oncol. 2019, 42, 739–756. [Google Scholar] [CrossRef]
- Sun, S.; Schiller, J.H.; Gazdar, A.F. Lung Cancer in Never Smokers—A Different Disease. Nat. Rev. Cancer 2007, 7, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Brambilla, E.; Burke, A.P.; Marx, A.; Nicholson, A.G. Introduction to The 2015 World Health Organization Classification of Tumors of the Lung, Pleura, Thymus, and Heart. J. Thorac. Oncol. 2015, 10, 1240–1242. [Google Scholar] [CrossRef] [PubMed]
- Lawson, D.A.; Kessenbrock, K.; Davis, R.T.; Pervolarakis, N.; Werb, Z. Tumour Heterogeneity and Metastasis at Single-Cell Resolution. Nat. Cell Biol. 2018, 20, 1349–1360. [Google Scholar] [CrossRef]
- Brzeziańska, E.; Dutkowska, A.; Antczak, A. The Significance of Epigenetic Alterations in Lung Carcinogenesis. Mol. Biol. Rep. 2013, 40, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Dobersch, S.; Romero-Olmedo, A.J.; Barreto, G. Epigenetics in Lung Cancer Diagnosis and Therapy. Cancer Metastasis Rev. 2015, 34, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Anglim, P.P.; Alonzo, T.A.; Laird-Offringa, I.A. DNA Methylation-Based Biomarkers for Early Detection of Non-Small Cell Lung Cancer: An Update. Mol. Cancer 2008, 7, 81. [Google Scholar] [CrossRef]
- Raos, D.; Ulamec, M.; Katusic Bojanac, A.; Bulic-Jakus, F.; Jezek, D.; Sincic, N. Epigenetically Inactivated RASSF1A as a Tumor Biomarker. Bosn. J. Basic Med. Sci. 2020, 21, 386–397. [Google Scholar] [CrossRef]
- Bajbouj, K.; Al-Ali, A.; Ramakrishnan, R.K.; Saber-Ayad, M.; Hamid, Q. Histone Modification in NSCLC: Molecular Mechanisms and Therapeutic Targets. Int. J. Mol. Sci. 2021, 22, 11701. [Google Scholar] [CrossRef]
- Stresemann, C.; Lyko, F. Modes of Action of the DNA Methyltransferase Inhibitors Azacytidine and Decitabine. Int. J. Cancer 2008, 123, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Fukazawa, T.; Yamatsuji, T.; Matsuoka, J.; Miyachi, H.; Maeda, Y.; Durbin, M.; Naomoto, Y. Anti-Tumor Effect in Human Lung Cancer by a Combination Treatment of Novel Histone Deacetylase Inhibitors: SL142 or SL325 and Retinoic Acids. PLoS ONE 2010, 5, e13834. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.J.; Kim, Y.S.; Kim, M.J.; Jang, S.; Lee, J.-H.; Choi, J.; Ro, S.; Hyun, Y.-L.; Lee, J.S.; Kim, C.-S. A Novel Histone Deacetylase Inhibitor, CG0006, Induces Cell Death through Both Extrinsic and Intrinsic Apoptotic Pathways. Anticancer Drugs 2009, 20, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Reid, T.; Valone, F.; Lipera, W.; Irwin, D.; Paroly, W.; Natale, R.; Sreedharan, S.; Keer, H.; Lum, B.; Scappaticci, F.; et al. Phase II Trial of the Histone Deacetylase Inhibitor Pivaloyloxymethyl Butyrate (Pivanex, AN-9) in Advanced Non-Small Cell Lung Cancer. Lung Cancer 2004, 45, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Traynor, A.M.; Dubey, S.; Eickhoff, J.C.; Kolesar, J.M.; Schell, K.; Huie, M.S.; Groteluschen, D.L.; Marcotte, S.M.; Hallahan, C.M.; Weeks, H.R.; et al. Vorinostat (NSC#701852) in Patients with Relapsed Non-Small Cell Lung Cancer: A Wisconsin Oncology Network Phase II Study. J. Thorac. Oncol. 2009, 4, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination Epigenetic Therapy Has Efficacy in Patients with Refractory Advanced Non–Small Cell Lung Cancer. Cancer Discov. 2011, 1, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining Epigenetic Drugs with Other Therapies for Solid Tumours—Past Lessons and Future Promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in Cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The Fundamental Role of Epigenetic Events in Cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Kaikkonen, M.U.; Lam, M.T.Y.; Glass, C.K. Non-Coding RNAs as Regulators of Gene Expression and Epigenetics. Cardiovasc. Res. 2011, 90, 430–440. [Google Scholar] [CrossRef]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-Cell Lung Cancer. Nat. Rev. Dis. Prim. 2021, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.-J.; Razi, S.; Pourbagheri-Sigaroodi, A.; Bashash, D. The PI3K/Akt/MTOR Pathway in Lung Cancer; Oncogenic Alterations, Therapeutic Opportunities, Challenges, and a Glance at the Application of Nanoparticles. Transl. Oncol. 2022, 18, 101364. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, A.S.; Geraldo, M.V.; Fuziwara, C.S.; Kulcsar, M.A.V.; Friguglietti, C.U.M.; da Costa, R.B.; Baia, G.S.; Kimura, E.T. Notch Pathway Is Activated by MAPK Signaling and Influences Papillary Thyroid Cancer Proliferation. Transl. Oncol. 2013, 6, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Herout, V.; Vossenaar, T. Adenocarcinoma of the Lung 101 Cases Including 32 Cases of Bronchiolar Cell Carcinoma. A Histopathological Study. Sb. Ved. Pr. Lek. Fak. Karlov. Univ. Hradci Kral. 1973, 16, 155–164. [Google Scholar]
- Ma, Y.; Li, M.D. Establishment of a Strong Link Between Smoking and Cancer Pathogenesis through DNA Methylation Analysis. Sci. Rep. 2017, 7, 1811. [Google Scholar] [CrossRef]
- Gkountakos, A.; Sartori, G.; Falcone, I.; Piro, G.; Ciuffreda, L.; Carbone, C.; Tortora, G.; Scarpa, A.; Bria, E.; Milella, M.; et al. PTEN in Lung Cancer: Dealing with the Problem, Building on New Knowledge and Turning the Game Around. Cancers 2019, 11, 1141. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. DNA Hypomethylation in Cancer Cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Schwanbeck, R. The Role of Epigenetic Mechanisms in Notch Signaling during Development. J. Cell. Physiol. 2015, 230, 969–981. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive Genomic Profiles of Small Cell Lung Cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef]
- Li, E.; Zhang, Y. DNA Methylation in Mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef]
- Langevin, S.M.; Kratzke, R.A.; Kelsey, K.T. Epigenetics of Lung Cancer. Transl. Res. 2015, 165, 74–90. [Google Scholar] [CrossRef]
- Hoang, P.H.; Landi, M.T. DNA Methylation in Lung Cancer: Mechanisms and Associations with Histological Subtypes, Molecular Alterations, and Major Epidemiological Factors. Cancers 2022, 14, 961. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, S.A.; Nikula, K.J.; Palmisano, W.A.; Michels, R.; Saccomanno, G.; Gabrielson, E.; Baylin, S.B.; Herman, J.G. Aberrant Methylation of P16 INK4a Is an Early Event in Lung Cancer and a Potential Biomarker for Early Diagnosis. Proc. Natl. Acad. Sci. USA 1998, 95, 11891–11896. [Google Scholar] [CrossRef] [PubMed]
- Qu, F.; Zheng, J.; Gan, W.; Lian, H.; He, H.; Li, W.; Yuan, T.; Yang, Y.; Li, X.; Ji, C.; et al. MiR-199a-3p Suppresses Proliferation and Invasion of Prostate Cancer Cells by Targeting Smad1. Oncotarget 2017, 8, 52465–52473. [Google Scholar] [CrossRef] [PubMed]
- Nuovo, G.J.; Plaia, T.W.; Belinsky, S.A.; Baylin, S.B.; Herman, J.G. In Situ Detection of the Hypermethylation-Induced Inactivation of the P16 Gene as an Early Event in Oncogenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 12754–12759. [Google Scholar] [CrossRef]
- Endoh, H.; Yatabe, Y.; Shimizu, S.; Tajima, K.; Kuwano, H.; Takahashi, T.; Mitsudomi, T. RASSF1A Gene Inactivation in Non-Small Cell Lung Cancer and Its Clinical Implication. Int. J. Cancer 2003, 106, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Loginov, V.I.; Dmitriev, A.A.; Senchenko, V.N.; Pronina, I.V.; Khodyrev, D.S.; Kudryavtseva, A.V.; Krasnov, G.S.; Gerashchenko, G.V.; Chashchina, L.I.; Kazubskaya, T.P.; et al. Tumor Suppressor Function of the SEMA3B Gene in Human Lung and Renal Cancers. PLoS ONE 2015, 10, e0123369. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, M.; Cheng, D.; Ma, X.; Li, Y. Clinical Significance of DAPK Promoter Hypermethylation in Lung Cancer: A Meta-Analysis. Drug Des. Devel. Ther. 2015, 9, 1785. [Google Scholar] [CrossRef]
- Huang, K.; Tang, Y.; He, L.; Dai, Y. MicroRNA-340 Inhibits Prostate Cancer Cell Proliferation and Metastasis by Targeting the MDM2-P53 Pathway. Oncol. Rep. 2016, 35, 887–895. [Google Scholar] [CrossRef]
- Geng, X.; Pu, W.; Tan, Y.; Lu, Z.; Wang, A.; Tan, L.; Chen, S.; Guo, S.; Wang, J.; Chen, X. Quantitative Assessment of the Diagnostic Role of FHIT Promoter Methylation in Non-Small Cell Lung Cancer. Oncotarget 2017, 8, 6845–6856. [Google Scholar] [CrossRef]
- Miuma, S.; Saldivar, J.C.; Karras, J.R.; Waters, C.E.; Paisie, C.A.; Wang, Y.; Jin, V.; Sun, J.; Druck, T.; Zhang, J.; et al. Fhit Deficiency-Induced Global Genome Instability Promotes Mutation and Clonal Expansion. PLoS ONE 2013, 8, e80730. [Google Scholar] [CrossRef] [PubMed]
- Soria, J.-C.; Lee, H.-Y.; Lee, J.I.; Wang, L.; Issa, J.-P.; Kemp, B.L.; Liu, D.D.; Kurie, J.M.; Mao, L.; Khuri, F.R. Lack of PTEN Expression in Non-Small Cell Lung Cancer Could Be Related to Promoter Methylation. Clin. Cancer Res. 2002, 8, 1178–1184. [Google Scholar] [PubMed]
- Zhao, R.; Choi, B.Y.; Lee, M.-H.; Bode, A.M.; Dong, Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (P16 INK4a) in Cancer. EBioMedicine 2016, 8, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Kim, M.J.; Lee, J.Y.; Kim, Y.Z.; Kim, E.J.; Park, J.Y. Aberrant Methylation of E-Cadherin and H-Cadherin Genes in Nonsmall Cell Lung Cancer and Its Relation to Clinicopathologic Features. Cancer 2007, 110, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating MicroRNAs as Stable Blood-Based Markers for Cancer Detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Ito, G.; Kondo, M.; Uchiyama, M.; Fukui, T.; Mori, S.; Yoshioka, H.; Ueda, Y.; Shimokata, K.; Sekido, Y. Frequent Inactivation of RASSF1A, BLU, and SEMA3B on 3p21.3 by Promoter Hypermethylation and Allele Loss in Non-Small Cell Lung Cancer. Cancer Lett. 2005, 225, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Nehme, E.; Rahal, Z.; Sinjab, A.; Khalil, A.; Chami, H.; Nemer, G.; Kadara, H. Epigenetic Suppression of the T-Box Subfamily 2 (TBX2) in Human Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1159. [Google Scholar] [CrossRef]
- Liu, S.; Chen, X.; Chen, R.; Wang, J.; Zhu, G.; Jiang, J.; Wang, H.; Duan, S.; Huang, J. Diagnostic Role of Wnt Pathway Gene Promoter Methylation in Non Small Cell Lung Cancer. Oncotarget 2017, 8, 36354–36367. [Google Scholar] [CrossRef]
- Li, J.; Poi, M.J.; Tsai, M.-D. Regulatory Mechanisms of Tumor Suppressor P16(INK4A) and Their Relevance to Cancer. Biochemistry 2011, 50, 5566–5582. [Google Scholar] [CrossRef]
- Fischer, J.R.; Ohnmacht, U.; Rieger, N.; Zemaitis, M.; Stoffregen, C.; Manegold, C.; Lahm, H. Prognostic Significance of RASSF1A Promoter Methylation on Survival of Non-Small Cell Lung Cancer Patients Treated with Gemcitabine. Lung Cancer 2007, 56, 115–123. [Google Scholar] [CrossRef]
- Cilluffo, D.; Barra, V.; Di Leonardo, A. P14ARF: The Absence That Makes the Difference. Genes 2020, 11, 824. [Google Scholar] [CrossRef] [PubMed]
- Toyooka, S.; Mitsudomi, T.; Soh, J.; Aokage, K.; Yamane, M.; Oto, T.; Kiura, K.; Miyoshi, S. Molecular Oncology of Lung Cancer. Gen. Thorac. Cardiovasc. Surg. 2011, 59, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Kurakawa, E.; Shimamoto, T.; Utsumi, K.; Hirano, T.; Kato, H.; Ohyashiki, K. Hypermethylation of P16INK4a and P15INK4b Genes in Non-Small Cell Lung Cancer. Int. J. Oncol. 2001, 19, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Sarne, V.; Huter, S.; Braunmueller, S.; Rakob, L.; Jacobi, N.; Kitzwögerer, M.; Wiesner, C.; Obrist, P.; Seeboeck, R. Promoter Methylation of Selected Genes in Non-Small-Cell Lung Cancer Patients and Cell Lines. Int. J. Mol. Sci. 2020, 21, 4595. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Chen, X.; Hong, Q.; Deng, Z.; Ma, H.; Xin, Y.; Fang, Y.; Ye, H.; Wang, R.; Zhang, C.; et al. Meta-Analyses of Gene Methylation and Smoking Behavior in Non-Small Cell Lung Cancer Patients. Sci. Rep. 2015, 5, 8897. [Google Scholar] [CrossRef] [PubMed]
- Belinsky, S.A. Unmasking the Lung Cancer Epigenome. Annu. Rev. Physiol. 2015, 77, 453–474. [Google Scholar] [CrossRef] [PubMed]
- Cui, H. Loss of Imprinting of IGF2 as an Epigenetic Marker for the Risk of Human Cancer. Dis. Markers 2007, 23, 105–112. [Google Scholar] [CrossRef]
- Rauch, T.A.; Zhong, X.; Wu, X.; Wang, M.; Kernstine, K.H.; Wang, Z.; Riggs, A.D.; Pfeifer, G.P. High-Resolution Mapping of DNA Hypermethylation and Hypomethylation in Lung Cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 252–257. [Google Scholar] [CrossRef]
- Kim, S.H.; Lee, S.; Lee, C.H.; Lee, M.K.; Kim, Y.D.; Shin, D.H.; Choi, K.U.; Kim, J.Y.; Park, D.Y.; Sol, M.Y. Expression of Cancer-Testis Antigens MAGE-A3/6 and NY-ESO-1 in Non-Small-Cell Lung Carcinomas and Their Relationship with Immune Cell Infiltration. Lung 2009, 187, 401–411. [Google Scholar] [CrossRef]
- Martin-Kleiner, I. BORIS in Human Cancers—A Review. Eur. J. Cancer 2012, 48, 929–935. [Google Scholar] [CrossRef]
- Jang, S.J.; Soria, J.C.; Wang, L.; Hassan, K.A.; Morice, R.C.; Walsh, G.L.; Hong, W.K.; Mao, L. Activation of Melanoma Antigen Tumor Antigens Occurs Early in Lung Carcinogenesis. Cancer Res. 2001, 61, 7959–7963. [Google Scholar] [PubMed]
- Ahuja, N.; Sharma, A.R.; Baylin, S.B. Epigenetic Therapeutics: A New Weapon in the War Against Cancer. Annu. Rev. Med. 2016, 67, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.L.; Pecot, C.V. Targeting Epigenetics in Lung Cancer. Cold Spring Harb. Perspect. Med. 2021, 11, a038000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, C.; Wu, C.; Cui, W.; Wang, L. DNA Methyltransferases in Cancer: Biology, Paradox, Aberrations, and Targeted Therapy. Cancers 2020, 12, 2123. [Google Scholar] [CrossRef] [PubMed]
- Dan, H.; Zhang, S.; Zhou, Y.; Guan, Q. DNA Methyltransferase Inhibitors: Catalysts For Antitumour Immune Responses. OncoTargets Ther. 2019, 12, 10903–10916. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Borghese, C.; Casagrande, N. The CCL5/CCR5 Axis in Cancer Progression. Cancers 2020, 12, 1765. [Google Scholar] [CrossRef] [PubMed]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; DeStefano Shields, C.E.; Niknafs, N.; Yen, R.-W.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300.e21. [Google Scholar] [CrossRef]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA Methyltransferase Inhibitors and Their Emerging Role in Epigenetic Therapy of Cancer. Anticancer Res. 2013, 33, 2989–2996. [Google Scholar]
- Billam, M.; Sobolewski, M.D.; Davidson, N.E. Effects of a Novel DNA Methyltransferase Inhibitor Zebularine on Human Breast Cancer Cells. Breast Cancer Res. Treat. 2010, 120, 581–592. [Google Scholar] [CrossRef]
- Holleran, J.L.; Parise, R.A.; Joseph, E.; Eiseman, J.L.; Covey, J.M.; Glaze, E.R.; Lyubimov, A.V.; Chen, Y.-F.; D’Argenio, D.Z.; Egorin, M.J. Plasma Pharmacokinetics, Oral Bioavailability, and Interspecies Scaling of the DNA Methyltransferase Inhibitor, Zebularine. Clin. Cancer Res. 2005, 11, 3862–3868. [Google Scholar] [CrossRef]
- Bryan, J.N.; Kumar, S.R.; Jia, F.; Balkin, E.R.; Lewis, M.R. Zebularine Significantly Sensitises MEC1 Cells to External Irradiation and Radiopharmaceutical Therapy When Administered Sequentially in Vitro. Cell Biol. Int. 2014, 38, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Fu, Y.; Tian, S.; Huang, S.; Luo, X.; Lin, L.; Zhang, X.; Wang, H.; Lin, Z.; Zhao, H.; et al. Zebularine Elevates STING Expression and Enhances CGAMP Cancer Immunotherapy in Mice. Mol. Ther. 2021, 29, 1758–1771. [Google Scholar] [CrossRef] [PubMed]
- Fang, D.; Han, J. Histone Mutations and Cancer. In Advances in Experimental Medicine and Biology; Springer Singapore: Singapore, 2021; Volume 1283, ISBN 978-981-15-8103-8. [Google Scholar]
- Verza, F.A.; Das, U.; Fachin, A.L.; Dimmock, J.R.; Marins, M. Roles of Histone Deacetylases and Inhibitors in Anticancer Therapy. Cancers 2020, 12, 1664. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Bertrand, P. Inside HDACs with More Selective HDAC Inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef]
- Sanaei, M.; Kavoosi, F. Histone Deacetylase Inhibitors, Intrinsic and Extrinsic Apoptotic Pathways, and Epigenetic Alterations of Histone Deacetylases (HDACs) in Hepatocellular Carcinoma. Iran. J. Pharm. Res. IJPR 2021, 20, 324–336. [Google Scholar] [CrossRef]
- Slonka, G.F.; Leland, S.E. In Vitro Cultivation of Ancylostoma Tubaeforme from Egg to Fourth-Stage Larva. Am. J. Vet. Res. 1970, 31, 1901–1904. [Google Scholar] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Miyanaga, A.; Gemma, A.; Noro, R.; Kataoka, K.; Matsuda, K.; Nara, M.; Okano, T.; Seike, M.; Yoshimura, A.; Kawakami, A.; et al. Antitumor Activity of Histone Deacetylase Inhibitors in Non-Small Cell Lung Cancer Cells: Development of a Molecular Predictive Model. Mol. Cancer Ther. 2008, 7, 1923–1930. [Google Scholar] [CrossRef]
- Li, C.-T.; Hsiao, Y.-M.; Wu, T.-C.; Lin, Y.; Yeh, K.-T.; Ko, J.-L.; Vorinostat, S.A.H.A. Represses Telomerase Activity via Epigenetic Regulation of Telomerase Reverse Transcriptase in Non-Small Cell Lung Cancer Cells. J. Cell. Biochem. 2011, 112, 3044–3053. [Google Scholar] [CrossRef]
- Mammì, G. Clinical and Instrumental Study of Patients with Syndrome X Associated or Unassociated with Excessive Myocardial Oxygen Consumption Low Work Loads. Clin. Ter. 1988, 125, 435–441. [Google Scholar] [PubMed]
- Shieh, J.-M.; Wei, T.-T.; Tang, Y.-A.; Huang, S.-M.; Wen, W.-L.; Chen, M.-Y.; Cheng, H.-C.; Salunke, S.B.; Chen, C.-S.; Lin, P.; et al. Mitochondrial Apoptosis and FAK Signaling Disruption by a Novel Histone Deacetylase Inhibitor, HTPB, in Antitumor and Antimetastatic Mouse Models. PLoS ONE 2012, 7, e30240. [Google Scholar] [CrossRef] [PubMed]
- Imre, G.; Gekeler, V.; Leja, A.; Beckers, T.; Boehm, M. Histone Deacetylase Inhibitors Suppress the Inducibility of Nuclear Factor-ΚB by Tumor Necrosis Factor-α Receptor-1 Down-Regulation. Cancer Res. 2006, 66, 5409–5418. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Y.; Wu, J.-Z.; Cao, H.-X.; Ma, R.; Wu, J.-Q.; Zhong, Y.-J.; Feng, J.-F. Blockade of DNA Methylation Enhances the Therapeutic Effect of Gefitinib in Non-Small Cell Lung Cancer Cells. Oncol. Rep. 2013, 29, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Guccione, E.; Richard, S. The Regulation, Functions and Clinical Relevance of Arginine Methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Hamard, P.-J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef]
- Yang, J.-H.; Chiou, Y.-Y.; Fu, S.-L.; Shih, I.-Y.; Weng, T.-H.; Lin, W.-J.; Lin, C.-H. Arginine Methylation of HnRNPK Negatively Modulates Apoptosis upon DNA Damage through Local Regulation of Phosphorylation. Nucleic Acids Res. 2014, 42, 9908–9924. [Google Scholar] [CrossRef] [PubMed]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP Deletion Confers Enhanced Dependency on the PRMT5 Arginine Methyltransferase in Cancer Cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef]
- Patro, C.P.K.; Biswas, N.; Pingle, S.C.; Lin, F.; Anekoji, M.; Jones, L.D.; Kesari, S.; Wang, F.; Ashili, S. MTAP Loss: A Possible Therapeutic Approach for Glioblastoma. J. Transl. Med. 2022, 20, 620. [Google Scholar] [CrossRef]
- Tang, B.; Lee, H.-O.; Gupta, S.; Wang, L.; Kurimchak, A.M.; Duncan, J.S.; Kruger, W.D. Extracellular 5′-Methylthioadenosine Inhibits Intracellular Symmetric Dimethylarginine Protein Methylation of FUSE-Binding Proteins. J. Biol. Chem. 2022, 298, 102367. [Google Scholar] [CrossRef]
- Schmid, M.; Malicki, D.; Nobori, T.; Rosenbach, M.D.; Campbell, K.; Carson, D.A.; Carrera, C.J. Homozygous Deletions of Methylthioadenosine Phosphorylase (MTAP) Are More Frequent than P16INK4A (CDKN2) Homozygous Deletions in Primary Non-Small Cell Lung Cancers (NSCLC). Oncogene 1998, 17, 2669–2675. [Google Scholar] [CrossRef] [PubMed]
- Eram, M.S.; Shen, Y.; Szewczyk, M.M.; Wu, H.; Senisterra, G.; Li, F.; Butler, K.V.; Kaniskan, H.Ü.; Speed, B.A.; dela Seña, C.; et al. A Potent, Selective, and Cell-Active Inhibitor of Human Type I Protein Arginine Methyltransferases. ACS Chem. Biol. 2016, 11, 772–781. [Google Scholar] [CrossRef] [PubMed]
- Fedoriw, A.; Rajapurkar, S.R.; O’Brien, S.; Gerhart, S.V.; Mitchell, L.H.; Adams, N.D.; Rioux, N.; Lingaraj, T.; Ribich, S.A.; Pappalardi, M.B.; et al. Anti-Tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 2019, 36, 100–114.e25. [Google Scholar] [CrossRef] [PubMed]
- Marjon, K.; Cameron, M.J.; Quang, P.; Clasquin, M.F.; Mandley, E.; Kunii, K.; McVay, M.; Choe, S.; Kernytsky, A.; Gross, S.; et al. MTAP Deletions in Cancer Create Vulnerability to Targeting of the MAT2A/PRMT5/RIOK1 Axis. Cell Rep. 2016, 15, 574–587. [Google Scholar] [CrossRef] [PubMed]
- Alhalabi, O.; Chen, J.; Zhang, Y.; Lu, Y.; Wang, Q.; Ramachandran, S.; Tidwell, R.S.; Han, G.; Yan, X.; Meng, J.; et al. MTAP Deficiency Creates an Exploitable Target for Antifolate Therapy in 9p21-Loss Cancers. Nat. Commun. 2022, 13, 1797. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Zhu, H.; Liu, W.; Zhai, X.; Tian, H.; Yu, J. MTAP-Deficiency Could Predict Better Treatment Response in Advanced Lung Adenocarcinoma Patients Initially Treated with Pemetrexed-Platinum Chemotherapy and Bevacizumab. Sci. Rep. 2020, 10, 843. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Vasan, N.; Baselga, J.; Hyman, D.M. A View on Drug Resistance in Cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef]
- Nishino, M.; Hida, T.; Kravets, S.; Dahlberg, S.E.; Lydon, C.A.; Hatabu, H.; Johnson, B.E.; Awad, M.M. Tumor Volume Dynamics and Tumor Growth Rate in ALK-Rearranged Advanced Non-Small-Cell Lung Cancer Treated with Crizotinib. Eur. J. Radiol. Open 2020, 7, 100210. [Google Scholar] [CrossRef]
- Tellez-Gabriel, M.; Ory, B.; Lamoureux, F.; Heymann, M.-F.; Heymann, D. Tumour Heterogeneity: The Key Advantages of Single-Cell Analysis. Int. J. Mol. Sci. 2016, 17, 2142. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug Resistance and Combating Drug Resistance in Cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Ruley, H.E.; Jacks, T.; Housman, D.E. P53-Dependent Apoptosis Modulates the Cytotoxicity of Anticancer Agents. Cell 1993, 74, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Kawamata, N.; Takeuchi, S.; Yin, D.; Chien, W.; Miller, C.W.; Koeffler, H.P. SAHA, a HDAC Inhibitor, Has Profound Anti-Growth Activity against Non-Small Cell Lung Cancer Cells. Oncol. Rep. 2006, 15, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Hase, T.; Shimizu, S.; Ando, M.; Hata, A.; Murakami, H.; Kawakami, T.; Nagase, K.; Yoshimura, K.; Fujiwara, T.; et al. Phase I Study of Vorinostat with Gefitinib in BIM Deletion Polymorphism/Epidermal Growth Factor Receptor Mutation Double-positive Lung Cancer. Cancer Sci. 2020, 111, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Reguart, N.; Rosell, R.; Cardenal, F.; Cardona, A.F.; Isla, D.; Palmero, R.; Moran, T.; Rolfo, C.; Pallarès, M.C.; Insa, A.; et al. Phase I/II Trial of Vorinostat (SAHA) and Erlotinib for Non-Small Cell Lung Cancer (NSCLC) Patients with Epidermal Growth Factor Receptor (EGFR) Mutations after Erlotinib Progression. Lung Cancer 2014, 84, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Han, J.-Y.; Lee, S.H.; Lee, G.K.; Yun, T.; Lee, Y.J.; Hwang, K.H.; Kim, J.Y.; Kim, H.T. Phase I/II Study of Gefitinib (Iressa®) and Vorinostat (IVORI) in Previously Treated Patients with Advanced Non-Small Cell Lung Cancer. Cancer Chemother. Pharmacol. 2015, 75, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Takhar, H.S.; Singhal, N.; Gowda, R.; Penniment, M.; Takhar, P.; Brown, M.P. Phase I Study Evaluating the Safety and Efficacy of Oral Panobinostat in Combination with Radiotherapy or Chemoradiotherapy in Patients with Inoperable Stage III Non-Small-Cell Lung Cancer. Anticancer Drugs 2015, 26, 1069–1077. [Google Scholar] [CrossRef]
- Wang, L.; Syn, N.L.-X.; Subhash, V.V.; Any, Y.; Thuya, W.L.; Cheow, E.S.H.; Kong, L.; Yu, F.; Peethala, P.C.; Wong, A.L.-A.; et al. Pan-HDAC Inhibition by Panobinostat Mediates Chemosensitization to Carboplatin in Non-Small Cell Lung Cancer via Attenuation of EGFR Signaling. Cancer Lett. 2018, 417, 152–160. [Google Scholar] [CrossRef]
- Sanda, M.G.; Dunn, R.L.; Michalski, J.; Sandler, H.M.; Northouse, L.; Hembroff, L.; Lin, X.; Greenfield, T.K.; Litwin, M.S.; Saigal, C.S.; et al. Quality of Life and Satisfaction with Outcome among Prostate-Cancer Survivors. N. Engl. J. Med. 2008, 358, 1250–1261. [Google Scholar] [CrossRef]
- To, K.K.-W.; Tong, W.-S.; Fu, L. Reversal of Platinum Drug Resistance by the Histone Deacetylase Inhibitor Belinostat. Lung Cancer 2017, 103, 58–65. [Google Scholar] [CrossRef]
- Tang, D.; Yao, R.; Zhao, D.; Zhou, L.; Wu, Y.; Yang, Y.; Sun, Y.; Lu, L.; Gao, W. Trichostatin A Reverses the Chemoresistance of Lung Cancer with High IGFBP2 Expression through Enhancing Autophagy. Sci. Rep. 2018, 8, 3917. [Google Scholar] [CrossRef]
- Shindo, Y.; Arai, W.; Konno, T.; Kohno, T.; Kodera, Y.; Chiba, H.; Miyajima, M.; Sakuma, Y.; Watanabe, A.; Kojima, T. Effects of Histone Deacetylase Inhibitors Tricostatin A and Quisinostat on Tight Junction Proteins of Human Lung Adenocarcinoma A549 Cells and Normal Lung Epithelial Cells. Histochem. Cell Biol. 2021, 155, 637–653. [Google Scholar] [CrossRef]
- Drzewiecka, H.; Gałęcki, B.; Jarmołowska-Jurczyszyn, D.; Kluk, A.; Dyszkiewicz, W.; Jagodziński, P.P. Decreased Expression of Connective Tissue Growth Factor in Non-Small Cell Lung Cancer Is Associated with Clinicopathological Variables and Can Be Restored by Epigenetic Modifiers. J. Cancer Res. Clin. Oncol. 2016, 142, 1927–1946. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.-T.; Tan, H.-Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic Regulation in Human Cancer: The Potential Role of Epi-Drug in Cancer Therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef]
- Sun, W.; Lv, S.; Li, H.; Cui, W.; Wang, L. Enhancing the Anticancer Efficacy of Immunotherapy through Combination with Histone Modification Inhibitors. Genes 2018, 9, 633. [Google Scholar] [CrossRef]

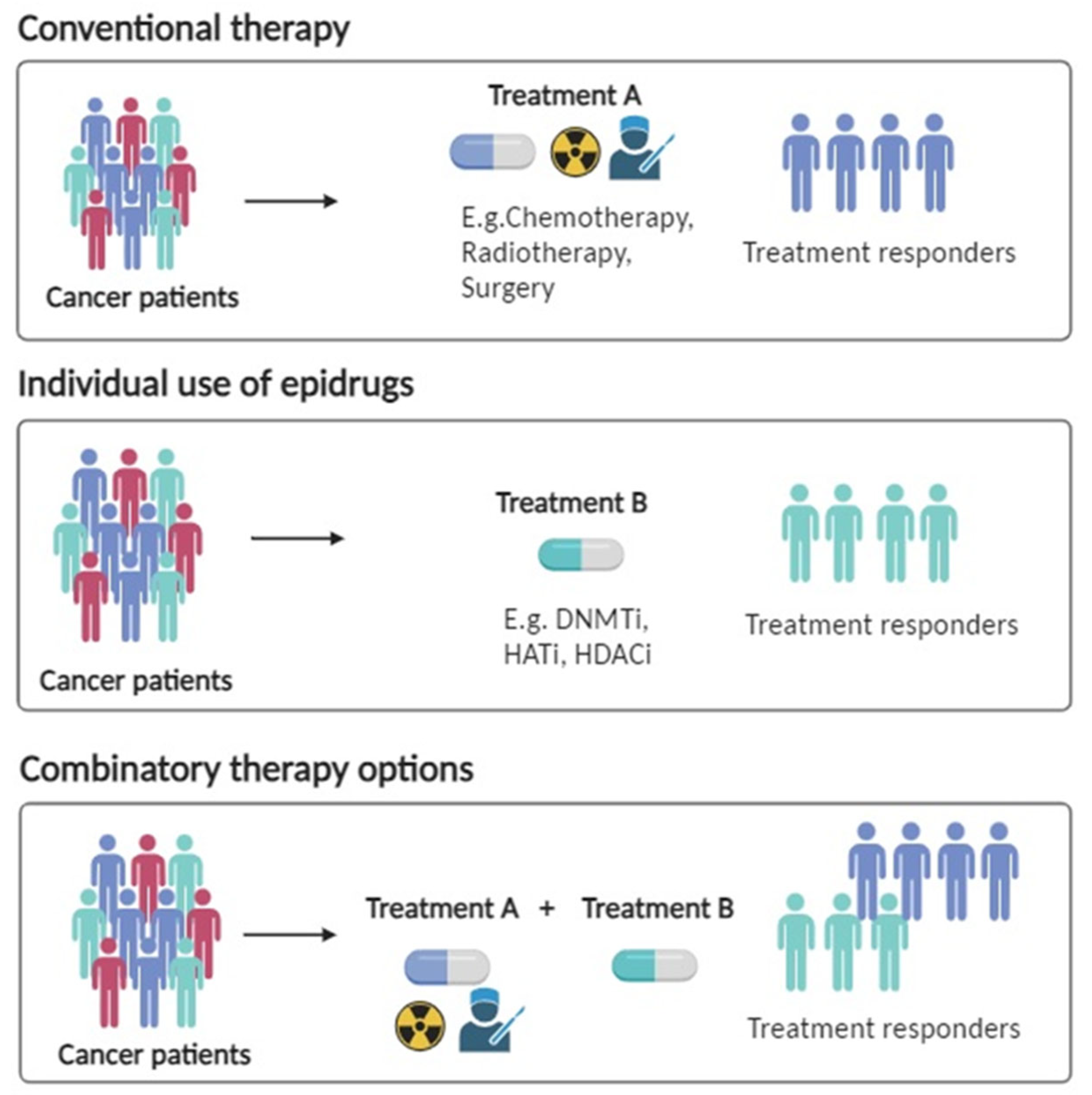
| Study ID, Type and Phase | Objective | Patient Characteristics and Condition | Treatment | Outcome | Status |
|---|---|---|---|---|---|
| ID: NCT00978250 Interventional Phase II Study | Assess the synergy of FdCyd and THU in controlling tumor growth and evaluate their combined safety and tolerability | 25 Patients aged 18 to ≥65 years, Non-Small Cell Cancer | DNMT inhibition with 5-Fluoro-2′-Deoxycytidine (FdCyd) + Tetrahydrouridine (THU) | Median progression-free survival for the group of 25 participants was 2.3 months, with a 95% confidence interval ranging from 1.6 months to 3.9 months | Completed |
| ID: NCT05960773 Interventional Phase II Study | Evaluation of DNA demethylating agent, in individuals with BAP1 Cancer Predisposition Syndrome who have subclinical or early-stage mesothelioma | Estimated number of patients: 15 Age ≥ 18 years. | Decitabine/Cedazuridine | Pre-recruitment phase | Not yet recruiting Patients (estimated study completion 2026) |
| ID: NCT01207726 Interventional Phase II Study | Impact of 5-azacitidine and Entinostat on 3-year progression-free survival in stage I non-small cell lung cancer patients after resection | 13 Patients aged 18 to ≥65 years | 5-Azacitidine and Entinostat | Study terminated prematurely due to low enrollment, making data analysis and conclusions impossible | Terminated |
| ID: NCT02664181 Interventional, Phase II Study | Effectiveness of combining the investigational drug tetrahydrouridine-decitabine (THU-Dec) with nivolumab, compared to nivolumab alone, in patients with Non-Small Cell Lung Cancer (NSCLC) | 13 patients, Age ≥ 18 years | Tetrahydrouridine/Decitabine + Nivolumab | A total of 50% of the participants experience progressive disease. A total of 25% showed stable disease. A total of 25% presented partial remission. | Ongoing |
| ID: NCT00385398 Interventional, Phase II Study | Evaluate the impact of stereotactic radiosurgery (SRS), temozolomide, and erlotinib hydrochloride on cognitive function in NSCLC patients with brain metastases. Determine the frequency of O6-methylguanine-DNA methyltransferase promoter methylation in these patients. | Patients with Age ≥ 18 years | Erlotinib hydrochloride, Temozolomide, Radiation | Specific data or reasons for the study’s withdrawal were not provided | Withdrawn |
| ID: NCT02959437 Interventional Phase I Phase II | The study consists of two parts, with Part 1 dedicated to dose escalation for safety assessment. Following dose determination, Part 2 enrolls subjects with previously treated NSCLC, microsatellite-stable colorectal cancer (CRC), head and neck squamous cell carcinoma, urothelial carcinoma, and melanoma into expansion cohorts. | 70 Patients, Age ≥ 18 years | Azacitidine Pembrolizumab Epacadostat | All participants received a minimum of one dose of the investigational drug and had evaluable baseline and on-treatment biopsies. None of the individuals were enrolled in Part 2 of the study due to the early termination of the research as a strategic decision. | Terminated |
| Study ID, Type and Phase | Objective | Patient Characteristics and Condition | Treatment | Outcome | Status |
|---|---|---|---|---|---|
| ID: NCT00005093 Interventional Phase III | Evaluate the efficacy and safety of gemcitabine, both as a standalone treatment and in combination with CI-994, in patients with advanced non-small cell lung cancer | Age ≥ 18 years No additional data regarding patient accrual is provided | Tacedinaline CI-994 Gemcitabine hydrochloride | No outcome data available provided | Completed |
| ID: NCT00128102 Interventional Phase III | Assessing the oral investigational drug vorinostat’s efficacy and safety versus a placebo in treating advanced malignant pleural mesothelioma after prior chemotherapy, with the primary goal of enhancing overall survival | A total of 661 patients, Age ≥ 18 years with confirmed pleural mesothelioma | Vorinostat Placebo | In the Vorinostat group, the objective response rate (ORR) stands at 0.63% with a 95% Confidence Interval (CI) ranging from 0.08% to 2.25%. Conversely, the placebo group reports an ORR of 0.31% with a 95% CI spanning from 0.01% to 1.71% | Completed |
| ID: NCT00473889 Interventional Phase III | Assess the survival outcomes of advanced non-small cell lung cancer patients treated with vorinostat in combination with paclitaxel and carboplatin | 253 participants, Age ≥ 18 years, confirmed with NSCLC | Vorinostat Paclitaxel Carboplatin Placebo | Out of 125 participants, 28 (22.4%) responded favorably to Vorinostat, Paclitaxel, and Carboplatin treatment, while 97 (77.6%) showed an unfavorable disease response. In the placebo group with Paclitaxel + Carboplatin, 36 (28.8%) exhibited a positive response, with 87 (69.6%) demonstrating an inadequate disease response | Terminated based on the recommendation by the DSMB following a pre-planned protocol interim analysis because the endpoint was not achieved |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Munteanu, R.; Tomuleasa, C.; Iuga, C.-A.; Gulei, D.; Ciuleanu, T.E. Exploring Therapeutic Avenues in Lung Cancer: The Epigenetic Perspective. Cancers 2023, 15, 5394. https://doi.org/10.3390/cancers15225394
Munteanu R, Tomuleasa C, Iuga C-A, Gulei D, Ciuleanu TE. Exploring Therapeutic Avenues in Lung Cancer: The Epigenetic Perspective. Cancers. 2023; 15(22):5394. https://doi.org/10.3390/cancers15225394
Chicago/Turabian StyleMunteanu, Raluca, Ciprian Tomuleasa, Cristina-Adela Iuga, Diana Gulei, and Tudor Eliade Ciuleanu. 2023. "Exploring Therapeutic Avenues in Lung Cancer: The Epigenetic Perspective" Cancers 15, no. 22: 5394. https://doi.org/10.3390/cancers15225394
APA StyleMunteanu, R., Tomuleasa, C., Iuga, C.-A., Gulei, D., & Ciuleanu, T. E. (2023). Exploring Therapeutic Avenues in Lung Cancer: The Epigenetic Perspective. Cancers, 15(22), 5394. https://doi.org/10.3390/cancers15225394