
Advances of Protein Palmitoylation in Tumor Cell Deaths


Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
3. Cell Death
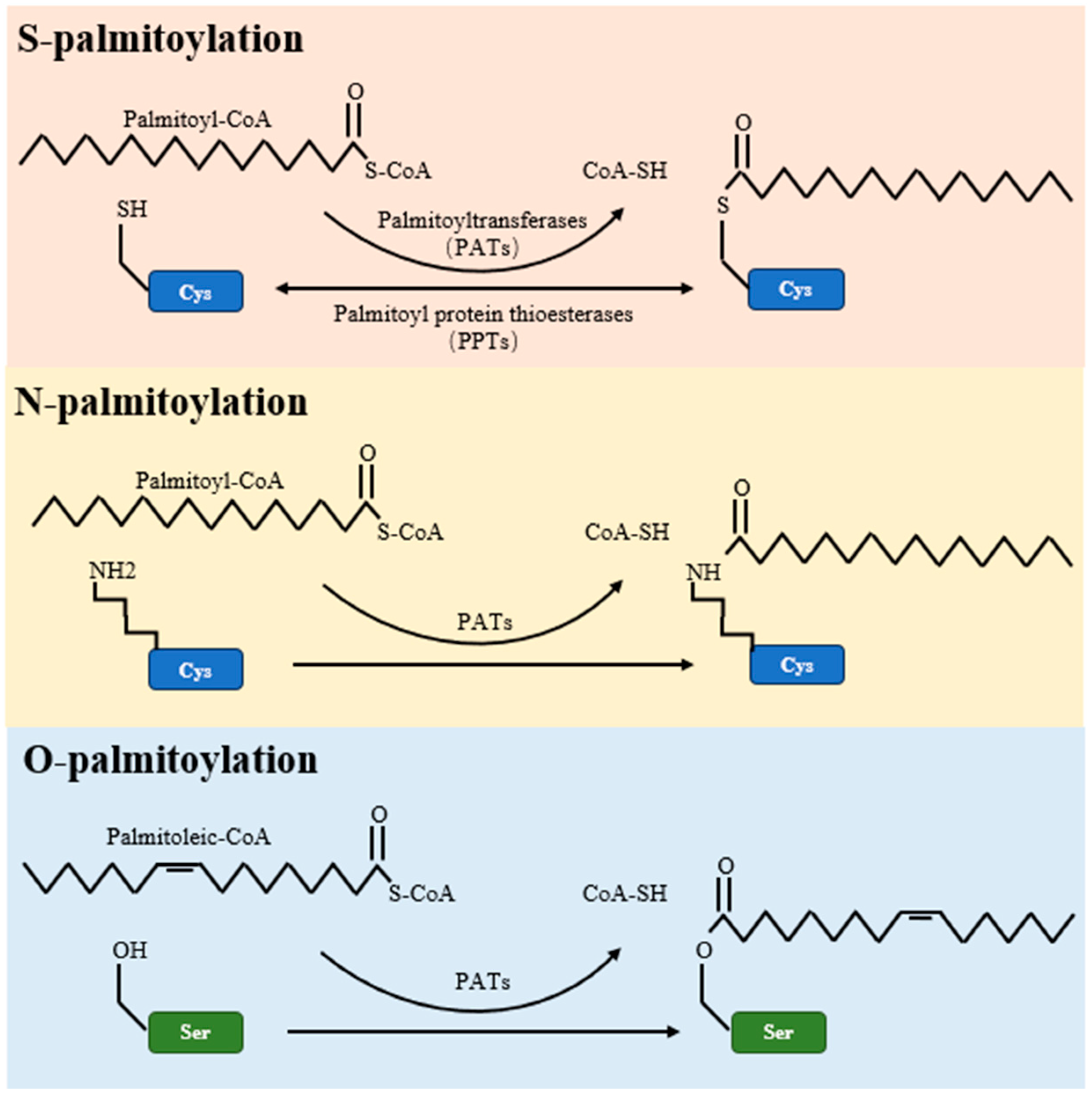
4. Palmitoylation
5. Palmitoylation in Tumor Apoptosis
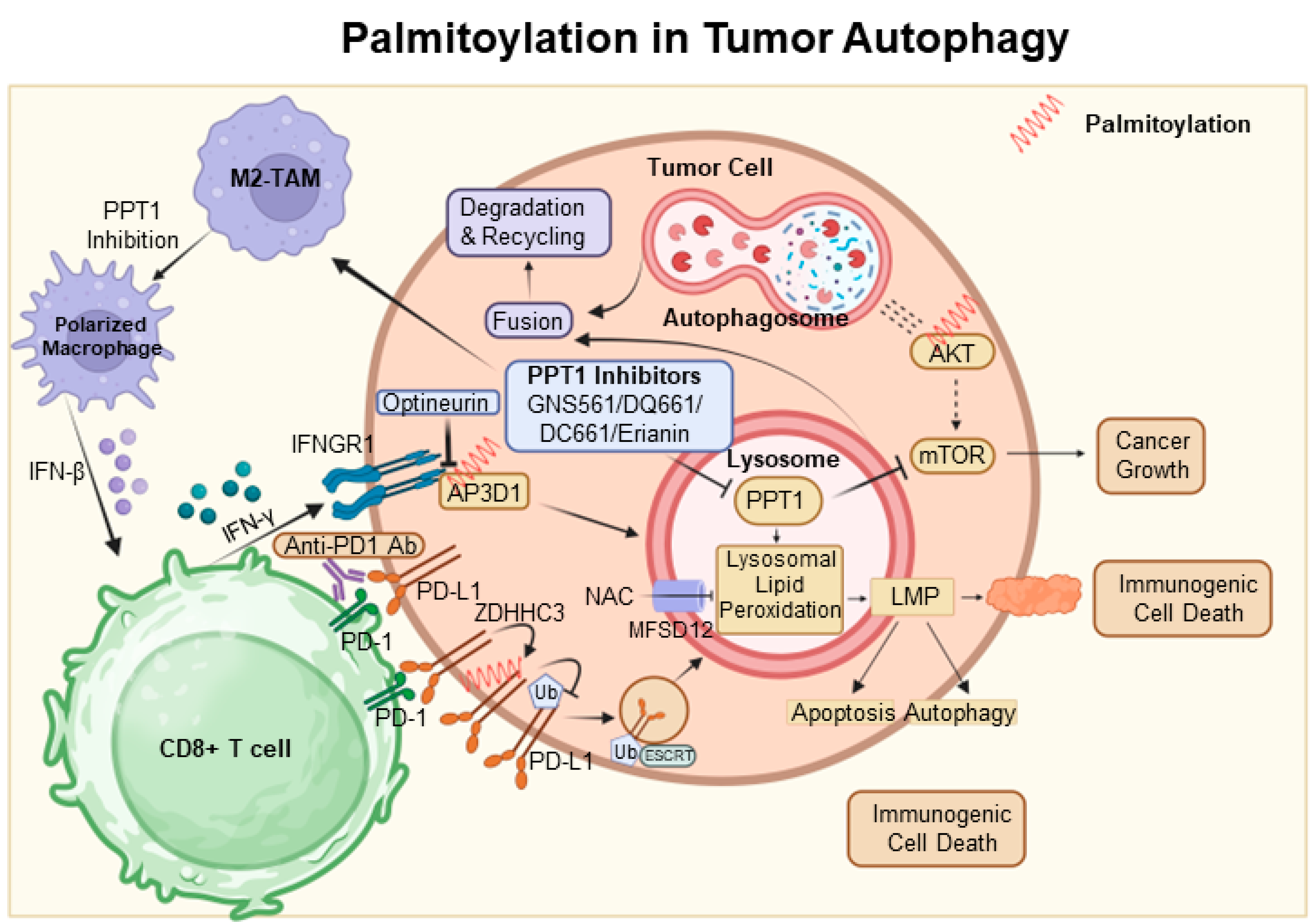
6. Palmitoylation in Tumor Autophagy
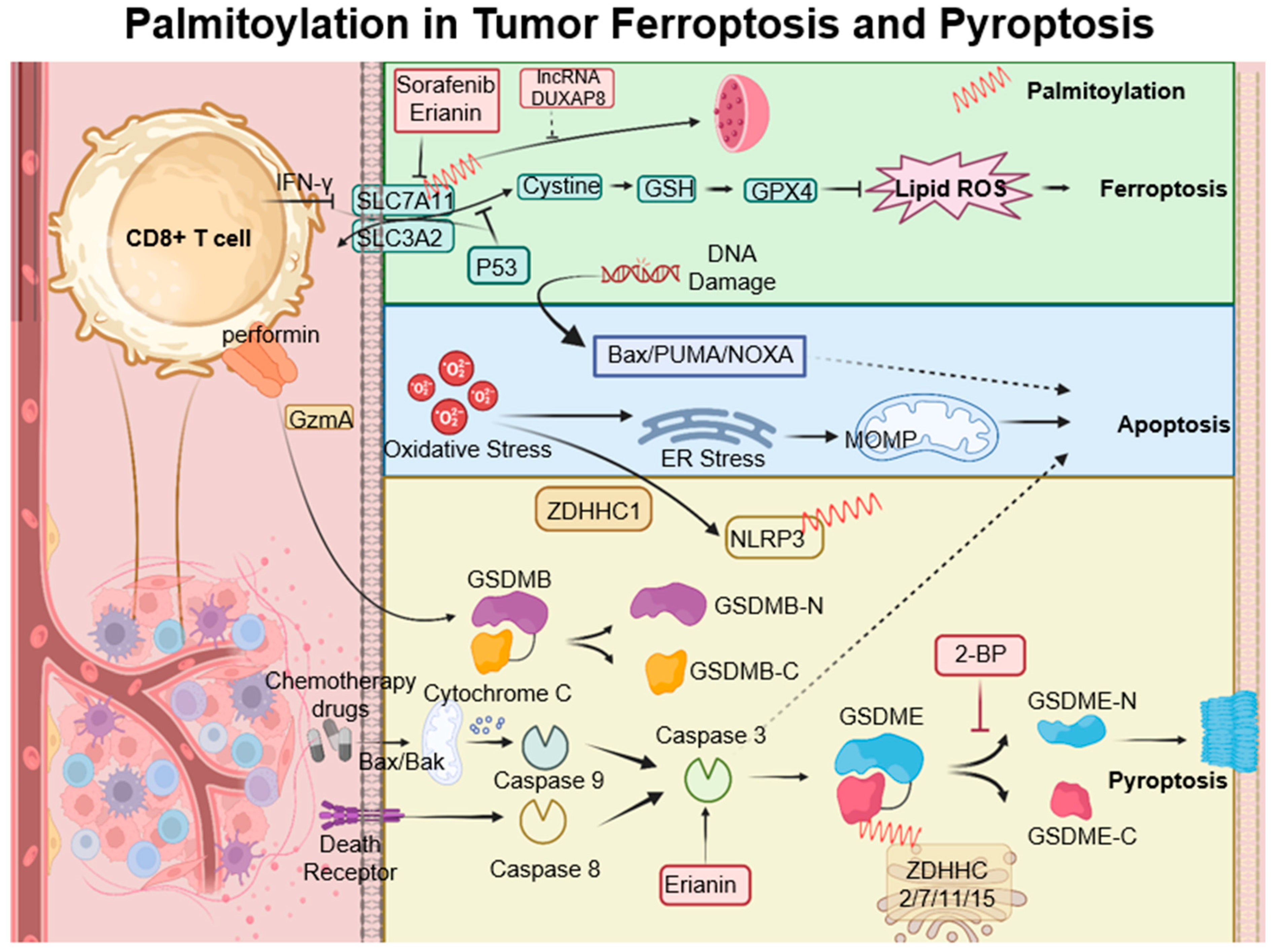
7. Palmitoylation in Ferroptosis and Pyroptosis
8. Palmitoylation in Oncology: A Multifaceted Therapeutic Target

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target/Pathway | Agent/Influence | Clinical Potential and Remarks | Reference |
|---|---|---|---|
| A general inhibitor of protein S-palmitoylation | 2-BP | Inhibit S-palmitoylation: e.g., Inhibit DHHC3 acyltransferase as a promising therapeutic avenue towards enhancing tumor-specific immunity | [102] |
| Pancreatic cancer | 2-BP and PD-1/PD-L1 | Facilitate checkpoint immunotherapy | [111] |
| HCC | GNS561 | Inhibit PPT1 to inhibit autophagy | [98] |
| HCC | DC661 | Inhibit PPT1 to inhibit autophagy | [112] |
| Melanoma | DC661 and anti-PD-1 antibody | Synergistic enhancement of antitumor activity | [99] |
| Melanoma, pancreatic cancer, and colorectal cancer | DQ661 | Inhibit PPT1 to inhibit autophagy and mTORC1 activity by specifically targeting PPT1. | [103] |
| Oral squamous cell carcinoma, osteosarcoma, hepatic cancer, lung cancer, and cervical cancer | Erianin | Promote apoptosis, autophagy, ferroptosis, and pyroptosis and enhance the immunotherapy | [110] |
| Chronic lymphocytic leukemia | ABD957 | Targeted disruption of N-Ras depalmitoylation; Synergy with MEK inhibition | [70,71] |
| Lung Cancer | P1MK5E | Enhance Necroptosis and Anticancer activity | [115] |
| Lung Cancer | Artonin F | c-Met in Competitive inhibition of c-Met palmitoylation | [116] |
9. Future Perspective
9.1. Current Understanding and Challenges in Protein Palmitoylation
9.2. Linking Palmitoylation to Cellular Death in Cancer
9.3. Therapeutic Implications and Future Directions
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Li, W.; Li, F.; Zhang, X.; Lin, H.-K.; Xu, C. Insights into the post-translational modification and its emerging role in shaping the tumor microenvironment. Signal Transduct. Target. Ther. 2021, 6, 422. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, M.; Mo, Y.; Wang, H.; Han, Y.; Zhao, X.; Yang, X.; Liu, Z.; Xu, B. Emerging roles of protein palmitoylation and its modifying enzymes in cancer cell signal transduction and cancer therapy. Int. J. Biol. Sci. 2022, 18, 3447–3457. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Hao, Q.; Liang, Y.; Kong, E. Protein palmitoylation in cancer: Molecular functions and therapeutic potential. Mol. Oncol. 2023, 17, 3–26. [Google Scholar] [CrossRef]
- Fhu, C.W.; Ali, A. Protein Lipidation by Palmitoylation and Myristoylation in Cancer. Front. Cell Dev. Biol. 2021, 9, 673647. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMP s and DAMP s: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef]
- Tong, X.; Tang, R.; Xiao, M.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Targeting cell death pathways for cancer therapy: Recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J. Hematol. Oncol. 2022, 15, 1–32. [Google Scholar] [CrossRef]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, K.R.; Riaz, T.A.; Kim, H.-R.; Chae, H.-J. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp. Mol. Med. 2021, 53, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef]
- Obeng, E. Apoptosis (programmed cell death) and its signals—A review. Braz. J. Biol. 2021, 81, 1133–1143. [Google Scholar] [CrossRef]
- Morana, O.; Wood, W.; Gregory, C.D. The apoptosis paradox in cancer. Int. J. Mol. Sci. 2022, 23, 1328. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 2020, 104, 575–587. [Google Scholar] [CrossRef]
- Chen, Q.; Kang, J.; Fu, C. The independence of and associations among apoptosis, autophagy, and necrosis. Signal Transduct. Target. Ther. 2018, 3, 18. [Google Scholar] [CrossRef]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Fernández, Á.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.-C. Disruption of the beclin 1–BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.-l.; Huang, Z.-j.; Lin, Z.-t.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Hassannia, B.; Vandenabeele, P.; Berghe, T.V. Targeting ferroptosis to iron out cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Tan, Y.; Chen, Q.; Li, X.; Zeng, Z.; Xiong, W.; Li, G.; Li, X.; Yang, J.; Xiang, B.; Yi, M. Pyroptosis: A new paradigm of cell death for fighting against cancer. J. Exp. Clin. Cancer Res. 2021, 40, 153. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Van Gorp, H.; Lamkanfi, M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019, 20, e47575. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Ruan, J.; Wu, H. Monitoring gasdermin pore formation in vitro. Methods Enzym. 2019, 625, 95–107. [Google Scholar] [CrossRef]
- Moujalled, D.; Strasser, A.; Liddell, J.R. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021, 28, 2029–2044. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.R.; Wang, C.; Adibekian, A.; Tully, S.E.; Cravatt, B.F. Global profiling of dynamic protein palmitoylation. Nat. Methods 2012, 9, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Resh, M.D. Chapter 13—Lipid modification of proteins. In Biochemistry of Lipids, Lipoproteins and Membranes (Seventh Edition); Ridgway, N.D., McLeod, R.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2021; pp. 429–456. [Google Scholar] [CrossRef]
- Ko, P.J.; Dixon, S.J. Protein palmitoylation and cancer. EMBO Rep. 2018, 19, e46666. [Google Scholar] [CrossRef]
- Le, X.; Mu, J.; Peng, W.; Tang, J.; Xiang, Q.; Tian, S.; Feng, Y.; He, S.; Qiu, Z.; Ren, G.; et al. DNA methylation downregulated ZDHHC1 suppresses tumor growth by altering cellular metabolism and inducing oxidative/ER stress-mediated apoptosis and pyroptosis. Theranostics 2020, 10, 9495–9511. [Google Scholar] [CrossRef]
- Tang, J.; Peng, W.; Feng, Y.; Le, X.; Wang, K.; Xiang, Q.; Li, L.; Wang, Y.; Xu, C.; Mu, J.; et al. Cancer cells escape p53’s tumor suppression through ablation of ZDHHC1-mediated p53 palmitoylation. Oncogene 2021, 40, 5416–5426. [Google Scholar] [CrossRef]
- Sharma, C.; Wang, H.X.; Li, Q.; Knoblich, K.; Reisenbichler, E.S.; Richardson, A.L.; Hemler, M.E. Protein Acyltransferase DHHC3 Regulates Breast Tumor Growth, Oxidative Stress, and Senescence. Cancer Res. 2017, 77, 6880–6890. [Google Scholar] [CrossRef]
- Sharma, C.; Hemler, M.E. Antioxidant and Anticancer Functions of Protein Acyltransferase DHHC3. Antioxidants 2022, 11, 960. [Google Scholar] [CrossRef]
- Wu, Z.; Tan, R.; Zhu, L.; Yao, P.; Hu, Q. Protein S-Palmitoylation and Lung Diseases. Adv. Exp. Med. Biol. 2021, 1304, 165–186. [Google Scholar] [CrossRef]
- Chen, X.; Ma, H.; Wang, Z.; Zhang, S.; Yang, H.; Fang, Z. EZH2 Palmitoylation Mediated by ZDHHC5 in p53-Mutant Glioma Drives Malignant Development and Progression. Cancer Res. 2017, 77, 4998–5010. [Google Scholar] [CrossRef]
- Chong, X.; Zhu, L.; Yu, D.; Chen, S.; Wang, G.; Yu, Q.; Ma, X.; Xu, J.; Chen, H.; An, H. ZDHHC9 promotes colon tumor growth by inhibiting effector T cells. Oncol. Lett. 2023, 25, 5. [Google Scholar] [CrossRef]
- Dzikiewicz-Krawczyk, A.; Kok, K.; Slezak-Prochazka, I.; Robertus, J.L.; Bruining, J.; Tayari, M.M.; Rutgers, B.; de Jong, D.; Koerts, J.; Seitz, A.; et al. ZDHHC11 and ZDHHC11B are critical novel components of the oncogenic MYC-miR-150-MYB network in Burkitt lymphoma. Leukemia 2017, 31, 1470–1473. [Google Scholar] [CrossRef]
- Dai, H.; Wu, R.; Zhang, J.; Dou, R.; Xu, M.; Wang, J.; Wang, J.; Su, F.; Zhang, T. ZDHHC11B is decreased in lung adenocarcinoma and inhibits tumorigenesis via regulating epithelial-mesenchymal transition. Cancer Med. 2023, 12, 17212–17222. [Google Scholar] [CrossRef]
- Lu, F.; Shen, S.H.; Wu, S.; Zheng, P.; Lin, K.; Liao, J.; Jiang, X.; Zeng, G.; Wei, D. Hypomethylation-induced prognostic marker zinc finger DHHC-type palmitoyltransferase 12 contributes to glioblastoma progression. Ann. Transl. Med. 2022, 10, 334. [Google Scholar] [CrossRef]
- Yeste-Velasco, M.; Mao, X.; Grose, R.; Kudahetti, S.C.; Lin, D.; Marzec, J.; Vasiljević, N.; Chaplin, T.; Xue, L.; Xu, M.; et al. Identification of ZDHHC14 as a novel human tumour suppressor gene. J. Pathol. 2014, 232, 566–577. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Yang, H.; Zhao, C.; Hu, L.; Delong, W.; Wang, R.; Fang, Z.; Chen, X. Local anesthetics impair the growth and self-renewal of glioblastoma stem cells by inhibiting ZDHHC15-mediated GP130 palmitoylation. Stem Cell Res. Ther. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, C.; Xiao, M.; Han, Y.; Zhang, S.; Xu, B. Bioinformatics Analysis of the Prognostic and Biological Significance of ZDHHC-Protein Acyltransferases in Kidney Renal Clear Cell Carcinoma. Front. Oncol. 2020, 10, 565414. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Sun, S.; Yang, H.; Ma, H.; Zhao, C.; Niu, W.; Fan, J.; Fang, Z.; Chen, X. SETD2 Palmitoylation Mediated by ZDHHC16 in Epidermal Growth Factor Receptor-Mutated Glioblastoma Promotes Ionizing Radiation-Induced DNA Damage. Int. J. Radiat. Oncol. Biol. Phys. 2022, 113, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Cao, N.; Li, J.K.; Rao, Y.Q.; Liu, H.; Wu, J.; Li, B.; Zhao, P.; Zeng, L.; Li, J. A potential role for protein palmitoylation and zDHHC16 in DNA damage response. BMC Mol. Biol. 2016, 17, 12. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Choi, M.; Ahn, Y.; Jang, D.; Pak, Y. Flotillin-1 palmitoylation turnover by APT-1 and ZDHHC-19 promotes cervical cancer progression by suppressing IGF-1 receptor desensitization and proteostasis. Cancer Gene Ther. 2022, 30, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Draper, J.M.; Smith, C.D. DHHC20: A human palmitoyl acyltransferase that causes cellular transformation. Mol. Membr. Biol. 2010, 27, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, J.; Tang, J.; Wu, Y.; Dai, F.; Yi, Z.; Wang, Y.; Li, Y.; Wu, Y.; Ren, G.; et al. ZDHHC22-mediated mTOR palmitoylation restrains breast cancer growth and endocrine therapy resistance. Int. J. Biol. Sci. 2022, 18, 2833–2850. [Google Scholar] [CrossRef] [PubMed]
- Edwards, H.; Rubenstein, M.; Dombkowski, A.A.; Caldwell, J.T.; Chu, R.; Xavier, A.C.; Thummel, R.; Neely, M.; Matherly, L.H.; Ge, Y.; et al. Gene Signature of High White Blood Cell Count in B-Precursor Acute Lymphoblastic Leukemia. PLoS ONE 2016, 11, e0161539. [Google Scholar] [CrossRef] [PubMed]
- Won, S.J.; Cheung See Kit, M.; Martin, B.R. Protein depalmitoylases. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 83–98. [Google Scholar] [CrossRef]
- Koster, K.P.; Yoshii, A. Depalmitoylation by Palmitoyl-Protein Thioesterase 1 in Neuronal Health and Degeneration. Front. Synaptic. Neurosci. 2019, 11, 25. [Google Scholar] [CrossRef]
- Rebecca, V.W.; Nicastri, M.C.; Fennelly, C.; Chude, C.I.; Barber-Rotenberg, J.S.; Ronghe, A.; McAfee, Q.; McLaughlin, N.P.; Zhang, G.; Goldman, A.R.; et al. PPT1 Promotes Tumor Growth and Is the Molecular Target of Chloroquine Derivatives in Cancer. Cancer Discov. 2019, 9, 220–229. [Google Scholar] [CrossRef]
- Shen, Z.C.; Xia, Z.X.; Liu, J.M.; Zheng, J.Y.; Luo, Y.F.; Yang, H.; Li, M.D.; Cao, T.; Liu, H.P.; Jin, G.L.; et al. APT1-Mediated Depalmitoylation Regulates Hippocampal Synaptic Plasticity. J. Neurosci. 2022, 42, 2662–2677. [Google Scholar] [CrossRef]
- Lin, D.T.; Conibear, E. ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. Elife 2015, 4, e11306. [Google Scholar] [CrossRef]
- Remsberg, J.R.; Suciu, R.M.; Zambetti, N.A.; Hanigan, T.W.; Firestone, A.J.; Inguva, A.; Long, A.; Ngo, N.; Lum, K.M.; Henry, C.L.; et al. ABHD17 regulation of plasma membrane palmitoylation and N-Ras-dependent cancer growth. Nat. Chem. Biol. 2021, 17, 856–864. [Google Scholar] [CrossRef]
- Veit, M.; Ponimaskin, E.; Schmidt, M.F. Analysis of S-acylation of proteins. Methods Mol. Biol. 2008, 446, 163–182. [Google Scholar] [PubMed]
- Martin, B.R.; Cravatt, B.F. Large-scale profiling of protein palmitoylation in mammalian cells. Nat. Methods 2009, 6, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Yount, J.S.; Moltedo, B.; Yang, Y.-Y.; Charron, G.; Moran, T.M.; López, C.B.; Hang, H.C. Palmitoylome profiling reveals S-palmitoylation–dependent antiviral activity of IFITM3. Nat. Chem. Biol. 2010, 6, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Sanders, S.; Orban, P.; Cijsouw, T.; Arstikaitis, P.; Yanai, A.; Hayden, M.R.; El-Husseini, A. Neuronal palmitoyl acyl transferases exhibit distinct substrate specificity. FASEB J. 2009, 23, 2605. [Google Scholar] [CrossRef]
- Forrester, M.T.; Hess, D.T.; Thompson, J.W.; Hultman, R.; Moseley, M.A.; Stamler, J.S.; Casey, P.J. Site-specific analysis of protein S-acylation by resin-assisted capture [S]. J. Lipid Res. 2011, 52, 393–398. [Google Scholar] [CrossRef]
- Martin, B.R. Nonradioactive analysis of dynamic protein palmitoylation. Curr. Protoc. Protein Sci. 2013, 73, 14.15.1–14.15.9. [Google Scholar] [CrossRef]
- Gao, X.; Hannoush, R.N. Single-cell in situ imaging of palmitoylation in fatty-acylated proteins. Nat. Protoc. 2014, 9, 2607–2623. [Google Scholar] [CrossRef]
- Gao, X.; Hannoush, R.N. Method for cellular imaging of palmitoylated proteins with clickable probes and proximity ligation applied to Hedgehog, tubulin, and Ras. J. Am. Chem. Soc. 2014, 136, 4544–4550. [Google Scholar] [CrossRef]
- Yang, W.; Di Vizio, D.; Kirchner, M.; Steen, H.; Freeman, M.R. Proteome scale characterization of human S-acylated proteins in lipid raft-enriched and non-raft membranes. Mol. Cell. Proteom. 2010, 9, 54–70. [Google Scholar] [CrossRef]
- Dowal, L.; Yang, W.; Freeman, M.R.; Steen, H.; Flaumenhaft, R. Proteomic analysis of palmitoylated platelet proteins. Blood J. Am. Soc. Hematol. 2011, 118, e62–e73. [Google Scholar] [CrossRef]
- Kastenhuber, E.R.; Lowe, S.W. Putting p53 in context. Cell 2017, 170, 1062–1078. [Google Scholar] [CrossRef]
- Gong, M.; Fan, X.; Yu, H.; Niu, W.; Sun, S.; Wang, H.; Chen, X. Loss of p53 Concurrent with RAS and TERT Activation Induces Glioma Formation. Mol. Neurobiol. 2023, 60, 3452–3463. [Google Scholar] [CrossRef]
- Chen, X.; Li, H.; Fan, X.; Zhao, C.; Ye, K.; Zhao, Z.; Hu, L.; Ma, H.; Wang, H.; Fang, Z. Protein palmitoylation regulates cell survival by modulating XBP1 activity in glioblastoma multiforme. Mol. Ther. -Oncolytics 2020, 17, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, M.; Dejanovic, B.; Kashkar, H.; Schwarz, G.; Nussberger, S. S-palmitoylation represents a novel mechanism regulating the mitochondrial targeting of BAX and initiation of apoptosis. Cell Death Dis. 2014, 5, e1057. [Google Scholar] [CrossRef] [PubMed]
- Seyrek, K.; Lavrik, I.N. Modulation of CD95-mediated signaling by post-translational modifications: Towards understanding CD95 signaling networks. Apoptosis 2019, 24, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Gajate, C.; Mollinedo, F. Lipid rafts and raft-mediated supramolecular entities in the regulation of CD95 death receptor apoptotic signaling. Apoptosis 2015, 20, 584–606. [Google Scholar] [CrossRef]
- Berg, V.; Rusch, M.; Vartak, N.; Jüngst, C.; Schauss, A.; Waldmann, H.; Hedberg, C.; Pallasch, C.P.; Bastiaens, P.I.; Hallek, M.; et al. miRs-138 and -424 control palmitoylation-dependent CD95-mediated cell death by targeting acyl protein thioesterases 1 and 2 in CLL. Blood 2015, 125, 2948–2957. [Google Scholar] [CrossRef] [PubMed]
- Zingler, P.; Särchen, V.; Glatter, T.; Caning, L.; Saggau, C.; Kathayat, R.S.; Dickinson, B.C.; Adam, D.; Schneider-Brachert, W.; Schütze, S. Palmitoylation is required for TNF-R1 signaling. Cell Commun. Signal. 2019, 17, 1–16. [Google Scholar] [CrossRef]
- Hsu, T.-H.; Chang, T.-C. RARRES3 regulates signal transduction through post-translational protein modifications. Mol. Cell. Oncol. 2015, 2, e999512. [Google Scholar] [CrossRef]
- Hsu, T.H.; Jiang, S.Y.; Chang, W.L.; Eckert, R.L.; Scharadin, T.M.; Chang, T.C. Involvement of RARRES3 in the regulation of Wnt proteins acylation and signaling activities in human breast cancer cells. Cell Death Differ. 2015, 22, 801–814. [Google Scholar] [CrossRef]
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.H. AKT as a Therapeutic Target for Cancer. Cancer Res. 2019, 79, 1019–1031. [Google Scholar] [CrossRef] [PubMed]
- Blaustein, M.; Piegari, E.; Martínez Calejman, C.; Vila, A.; Amante, A.; Manese, M.V.; Zeida, A.; Abrami, L.; Veggetti, M.; Guertin, D.A.; et al. Akt Is S-Palmitoylated: A New Layer of Regulation for Akt. Front. Cell Dev. Biol. 2021, 9, 626404. [Google Scholar] [CrossRef]
- Tian, W.; Li, C.; Ren, J.; Li, P.; Zhao, J.; Li, S.; Dong, D. Identification of PPT1 as a lysosomal core gene with prognostic value in hepatocellular carcinoma. Biosci. Rep. 2023, 43, BSR20230067. [Google Scholar] [CrossRef]
- Phadatare, P.; Debnath, J. Lysosomal lipid peroxidation mediates immunogenic cell death. J. Clin. Investig. 2023, 133, e169240. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, M.; Lee, J.J.; Versace, A.M.; Harper, S.L.; Goldman, A.R.; Crissey, M.A.S.; Jain, V.; Singh, M.P.; Vernon, M.; Aplin, A.E. Lysosomal lipid peroxidation regulates tumor immunity. J. Clin. Investig. 2023, 133. [Google Scholar] [CrossRef]
- Bestion, E.; Raymond, E.; Mezouar, S.; Halfon, P. Update on Autophagy Inhibitors in Cancer: Opening up to a Therapeutic Combination with Immune Checkpoint Inhibitors. Cells 2023, 12, 1702. [Google Scholar] [CrossRef]
- Brun, S.; Bestion, E.; Raymond, E.; Bassissi, F.; Jilkova, Z.M.; Mezouar, S.; Rachid, M.; Novello, M.; Tracz, J.; Hamaï, A.; et al. GNS561, a clinical-stage PPT1 inhibitor, is efficient against hepatocellular carcinoma via modulation of lysosomal functions. Autophagy 2022, 18, 678–694. [Google Scholar] [CrossRef]
- Sharma, G.; Ojha, R.; Noguera-Ortega, E.; Rebecca, V.W.; Attanasio, J.; Liu, S.; Piao, S.; Lee, J.J.; Nicastri, M.C.; Harper, S.L.; et al. PPT1 inhibition enhances the antitumor activity of anti-PD-1 antibody in melanoma. JCI Insight 2020, 5, e133225. [Google Scholar] [CrossRef]
- Du, W.; Hua, F.; Li, X.; Zhang, J.; Li, S.; Wang, W.; Zhou, J.; Wang, W.; Liao, P.; Yan, Y. Loss of optineurin drives cancer immune evasion via palmitoylation-dependent IFNGR1 lysosomal sorting and degradation. Cancer Discov. 2021, 11, 1826–1843. [Google Scholar] [CrossRef]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Lan, J.; Li, C.; Shi, H.; Brosseau, J.-P.; Wang, H.; Lu, H.; Fang, C.; Zhang, Y.; Liang, L. Inhibiting PD-L1 palmitoylation enhances T-cell immune responses against tumours. Nat. Biomed. Eng. 2019, 3, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Nicastri, M.C.; Rebecca, V.W.; Amaravadi, R.K.; Winkler, J.D. Dimeric quinacrines as chemical tools to identify PPT1, a new regulator of autophagy in cancer cells. Mol. Cell Oncol. 2018, 5, e1395504. [Google Scholar] [CrossRef] [PubMed]
- Rebecca, V.W.; Nicastri, M.C.; McLaughlin, N.; Fennelly, C.; McAfee, Q.; Ronghe, A.; Nofal, M.; Lim, C.Y.; Witze, E.; Chude, C.I.; et al. A Unified Approach to Targeting the Lysosome’s Degradative and Growth Signaling Roles. Cancer Discov. 2017, 7, 1266–1283. [Google Scholar] [CrossRef] [PubMed]
- Millson, S.H.; Piper, P.W. Insights from yeast into whether the inhibition of heat shock transcription factor (Hsf1) by rapamycin can prevent the Hsf1 activation that results from treatment with an Hsp90 inhibitor. Oncotarget 2014, 5, 5054–5064. [Google Scholar] [CrossRef] [PubMed]
- Dawson, G.; Dawson, S.A.; Marinzi, C.; Dawson, P.E. Anti-tumor promoting effects of palmitoyl: Protein thioesterase inhibitors against a human neurotumor cell line. Cancer Lett. 2002, 187, 163–168. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, J.; Wu, J.; He, D.; Zhang, C.; Duan, C.; Li, B. Ferroptosis, a new form of cell death: Opportunities and challenges in cancer. J. Hematol. Oncol. 2019, 12, 1–16. [Google Scholar] [CrossRef]
- Shi, Z.; Li, Z.; Jin, B.; Ye, W.; Wang, L.; Zhang, S.; Zheng, J.; Lin, Z.; Chen, B.; Liu, F. Loss of LncRNA DUXAP8 synergistically enhanced sorafenib induced ferroptosis in hepatocellular carcinoma via SLC7A11 de-palmitoylation. Clin. Transl. Med. 2023, 13, e1300. [Google Scholar] [CrossRef]
- Hu, L.; Chen, M.; Chen, X.; Zhao, C.; Fang, Z.; Wang, H.; Dai, H. Chemotherapy-induced pyroptosis is mediated by BAK/BAX-caspase-3-GSDME pathway and inhibited by 2-bromopalmitate. Cell Death Dis. 2020, 11, 281. [Google Scholar] [CrossRef]
- Luo, Q.; Li, X.; Gan, G.; Yang, M.; Chen, X.; Chen, F. PPT1 Reduction Contributes to Erianin-Induced Growth Inhibition in Oral Squamous Carcinoma Cells. Front. Cell Dev. Biol. 2021, 9, 764263. [Google Scholar] [CrossRef]
- Lin, Z.; Lv, Z.; Liu, X.; Huang, K. Palmitoyl transferases act as novel drug targets for pancreatic cancer. J. Transl. Med. 2023, 21, 249. [Google Scholar] [CrossRef]
- Xu, J.; Su, Z.; Cheng, X.; Hu, S.; Wang, W.; Zou, T.; Zhou, X.; Song, Z.; Xia, Y.; Gao, Y.; et al. High PPT1 expression predicts poor clinical outcome and PPT1 inhibitor DC661 enhances sorafenib sensitivity in hepatocellular carcinoma. Cancer Cell Int. 2022, 22, 115. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, T.; Sun, W.; Wang, Z.; Zuo, D.; Zhou, Z.; Li, S.; Xu, J.; Yin, F.; Hua, Y.; et al. Erianin induces G2/M-phase arrest, apoptosis, and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells in vitro and in vivo. Cell Death Dis. 2016, 7, e2247. [Google Scholar] [CrossRef]
- Chen, P.; Wu, Q.; Feng, J.; Yan, L.; Sun, Y.; Liu, S.; Xiang, Y.; Zhang, M.; Pan, T.; Chen, X.; et al. Erianin, a novel dibenzyl compound in Dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct. Target. Ther. 2020, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Behzadi, M.; Arasteh, S.; Bagheri, M. Palmitoylation of Membrane-Penetrating Magainin Derivatives Reinforces Necroptosis in A549 Cells Dependent on Peptide Conformational Propensities. ACS Appl. Mater. Interfaces 2020, 12, 56815–56829. [Google Scholar] [CrossRef]
- Soonnarong, R.; Putra, I.D.; Sriratanasak, N.; Sritularak, B.; Chanvorachote, P. Artonin F Induces the Ubiquitin-Proteasomal Degradation of c-Met and Decreases Akt-mTOR Signaling. Pharmaceuticals 2022, 15, 633. [Google Scholar] [CrossRef] [PubMed]
- Malgapo, M.I.P.; Linder, M.E. Substrate recruitment by zDHHC protein acyltransferases. Open Biol. 2021, 11, 210026. [Google Scholar] [CrossRef] [PubMed]
- Salaun, C.; Takizawa, H.; Galindo, A.; Munro, K.R.; McLellan, J.; Sugimoto, I.; Okino, T.; Tomkinson, N.C.; Chamberlain, L.H. Development of a novel high-throughput screen for the identification of new inhibitors of protein S-acylation. J. Biol. Chem. 2022, 298, 102469. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shen, L.; Xu, Z.; Liu, W.; Li, A.; Xu, J. Protein palmitoylation modification during viral infection and detection methods of palmitoylated proteins. Front. Cell. Infect. Microbiol. 2022, 12, 821596. [Google Scholar] [CrossRef]
- Solis, G.P.; Kazemzadeh, A.; Abrami, L.; Valnohova, J.; Alvarez, C.; van der Goot, F.G.; Katanaev, V.L. Local and substrate-specific S-palmitoylation determines subcellular localization of Gαo. Nat. Commun. 2022, 13, 2072. [Google Scholar] [CrossRef]
- Li, M.; Zhang, L.; Chen, C.-W. Diverse Roles of Protein Palmitoylation in Cancer Progression, Immunity, Stemness, and Beyond. Cells 2023, 12, 2209. [Google Scholar] [CrossRef]
- Shahid, M.; Kim, M.; Jin, P.; Zhou, B.; Wang, Y.; Yang, W.; You, S.; Kim, J. S-Palmitoylation as a Functional Regulator of Proteins Associated with Cisplatin Resistance in Bladder Cancer. Int. J. Biol. Sci. 2020, 16, 2490–2505. [Google Scholar] [CrossRef] [PubMed]
- Rocks, O.; Gerauer, M.; Vartak, N.; Koch, S.; Huang, Z.-P.; Pechlivanis, M.; Kuhlmann, J.; Brunsveld, L.; Chandra, A.; Ellinger, B.; et al. The Palmitoylation Machinery Is a Spatially Organizing System for Peripheral Membrane Proteins. Cell 2010, 141, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Ao, B.; Lu, X.; Yang, S.; Bao, P.; Wang, H.; Li, R.; Huang, Y. Global research trends on precision oncology: A systematic review, bibliometrics, and visualized study. Medicine 2022, 101, e31380. [Google Scholar] [CrossRef] [PubMed]

| Palmitoylation-Related Cell Death | Extrinsic Apoptosis | Intrinsic Apoptosis | Autophagy | Ferroptosis | Pyroptosis | |
|---|---|---|---|---|---|---|
| Morphological features | Cellular contraction, nuclear fragmentation, and chromatin condensation. | Double-membrane autophagosome merging with lysosome | Reduction in mitochondrial volume, increase in mitochondrial membrane density, disappearance of mitochondrial cristae | Cells continue to swell until the cell membrane ruptures, leading to a massive release of cell contents and pro-inflammatory factors | ||
| Activation Signals | Initiated when death ligands bind to cell surface receptors | DNA damage, oxidative stress | ER stress from misfolded proteins or calcium imbalance | Cellular stress | Oxidative stress | Microbial infections and non-infectious stimuli |
| Key Proteins and Molecules | Death-inducing signaling complex (DISC) and Initiator caspases | p53 protein | CHOP/GADD153 | Akt, mTOR and PI3K | GPX4 and system xc- | Gasdermin and caspase |
| Palmitoylation Type | S-Palmitoylation | O-Palmitoylation | N-Palmitoylation |
|---|---|---|---|
| Amino Acid Residue Involved | Cysteine (Cys) | Serine (Ser) or Threonine (Thr) | N-terminal amino group |
| Bond Type | Thioester | Ester | Amide |
| Reversibility | Reversible | Typically irreversible | Typically irreversible |
| Enzymes Involved | Palmitoylltransferases (PATs) for addition, Acyl-protein thioesterases (APTs) or Palmitoyl-protein thioesterase (PPTs) for removal | Not well-defined | N-myristoyltransferase (NMT), though this is for myristoylation which is more common at the N-terminus |
| Function | Regulation of protein-membrane association, protein–protein interactions, and protein stability | Rare, less studied; potential role in regulation of protein stability and function | Rare; N-terminal myristoylation is more common, plays role in protein-membrane association and stability |
| Examples of Proteins | Ras proteins, G-protein α subunits, PSD-95 | Not well characterized due to its rarity | Rare, more common is N-myristoylation (e.g., Src kinase) |
| Pathways/Processes Involved | Signal transduction, apoptosis, synaptic plasticity | Not well-defined due to rarity | Not well-defined due to rarity, but N-myristoylation is involved in signal transduction |
| Disease Associations | Neurodegenerative diseases, cancers, viral infections | Not well-characterized due to rarity | Not well-characterized due to rarity |
| ZDHHC Family | Alteration | Cancer | Reference |
|---|---|---|---|
| ZDHHC1 | Downregulated | Breast, prostate, and gastric cancers | [47,48] |
| Upregulated | Endometrial, renal, and pancreatic cancers | [46] | |
| ZDHHC2 | Downregulated | Pancreatic cancer | [3] |
| Upregulated | Renal cancer | [46] | |
| ZDHHC3 | Upregulated | Breast, prostate, renal, and colorectal cancers | [49,50] |
| ZDHHC4 | Upregulated | Renal cancer | [46] |
| ZDHHC5 | Upregulated | Lung adenocarcinoma, glioma, and breast cancer, | [51,52] |
| ZDHHC7 | Downregulated | Colorectal Cancer | [3] |
| ZDHHC9 | Upregulated | Breast, colorectal, myeloma, glioblastoma, and prostate cancer | [53] |
| ZDHHC11 | Upregulated | Burkitt lymphoma | [54] |
| ZDHHC11B | Downregulated | Lung adenocarcinoma | [55] |
| ZDHHC12 | Upregulated | Glioma and ovarian cancer | [56] |
| ZDHHC13 | Downregulated | Melanoma | [3] |
| ZDHHC14 | Downregulated | Prostate and testicular germ cell tumor | [57] |
| Upregulated | Pancreatic cancer | [46] | |
| ZDHHC15 | Downregulated | Glioblastoma, kidney renal clear cell carcinoma | [58,59] |
| ZDHHC16 | Downregulated | Glioblastoma | [60,61] |
| ZDHHC18 | Upregulated | Ovarian cancer | [3] |
| ZDHHC19 | Upregulated | Glioblastoma, cervical cancer, kidney renal clear cell carcinoma | [59,62] |
| ZDHHC20 | Upregulated | Ovarian, breast, kidney, colon, and prostate cancer | [63] |
| ZDHHC21 | Upregulated | Urothelial, renal, and non-small cell lung cancer | [3] |
| ZDHHC22 | Downregulated | Estrogen receptor negative breast cancer | [64] |
| ZDHHC23 | Upregulated | B-precursor acute lymphoblastic leukemia and renal cancer | [65] |
| Enzyme | Function | Relevance in Tumors | Notes |
|---|---|---|---|
| PPT1 | Responsible for removing palmitoyl groups from proteins | Associated with neurodegenerative diseases, but direct role in tumors remains less clear | PPT1’s primary function is to maintain stability of membrane proteins |
| APT | Catalyzes depalmitoylation processes, such as depalmitoylation of PSD-95 | Linked to synaptic plasticity and memory formation, potentially affecting tumor-associated signaling and proliferation | Inhibition of APT1 is considered to enhance synaptic function, but its exact role in tumors requires further investigation |
| ABHD17 | Involved in the depalmitoylation process | Viewed as a therapeutic target for NRAS mutant tumors due to its influence on the palmitoylation cycle of N-Ras | Inhibition of ABHD17 may have therapeutic potential in NRAS-driven tumors |
| Decade | Method Name | Brief Description |
|---|---|---|
| 1970s | Radioactive Labeling with [3H]-Palmitate | Proteins labeled with radioactive palmitate, detected via autoradiography post SDS-PAGE. |
| 1980s–1990s | [125I]-Iodopalmitate Metabolic Labeling | Enhanced specificity through metabolic labeling with radioactive iodopalmitate. |
| 1990s–present | Mass Spectrometry | Accurate identification of palmitoylated proteins and specific modification sites. |
| 2000s | Acyl-Biotin Exchange | Palmitate cleavage by hydroxylamine, with biotin tagging of revealed cysteines. |
| 2010s | Acyl-Resin Assisted Capture | Direct capture of de-palmitoylated proteins using thiol-reactive resin. |
| 2010s | Click Chemistry | Bioorthogonal reactions with specialized fatty acids to affix reporter molecules. |
| 2010s | Proximity Ligation Assay | In situ detection through paired antibodies, leading to oligonucleotide ligation and amplified signal. |
| 2010s | PalmPISC | Integration of metabolic labeling, click chemistry, and mass spectrometry for comprehensive analysis. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, X.; Shi, Y.; Zhan, Y.; Xing, Y.; Li, Y.; Zhou, Z.; Chen, G. Advances of Protein Palmitoylation in Tumor Cell Deaths. Cancers 2023, 15, 5503. https://doi.org/10.3390/cancers15235503
Lin X, Shi Y, Zhan Y, Xing Y, Li Y, Zhou Z, Chen G. Advances of Protein Palmitoylation in Tumor Cell Deaths. Cancers. 2023; 15(23):5503. https://doi.org/10.3390/cancers15235503
Chicago/Turabian StyleLin, Xiangyi, Yuxuan Shi, Yuxin Zhan, Yuying Xing, Yu Li, Zhiqing Zhou, and Guoan Chen. 2023. "Advances of Protein Palmitoylation in Tumor Cell Deaths" Cancers 15, no. 23: 5503. https://doi.org/10.3390/cancers15235503
APA StyleLin, X., Shi, Y., Zhan, Y., Xing, Y., Li, Y., Zhou, Z., & Chen, G. (2023). Advances of Protein Palmitoylation in Tumor Cell Deaths. Cancers, 15(23), 5503. https://doi.org/10.3390/cancers15235503