Simple Summary
Homologous recombination deficiency (HRD) originates from genomic mutations or alterations in the homologous recombination repair pathway. Various promising tests have been developed to detect HRD. Some of these tests have shown good ability to predict response to Poly (ADP-ribose) polymerase inhibitors in cancer patients. However, a standardized way to define HRD has yet to be established. In this systematic review an overview of available HRD tests is provided. Important factors to consider are highlighted when planning clinical trials and studies involving HRD tests.
Abstract
Homologous recombination deficiency (HRD) can arise from germline or somatic pathogenic variants as well as other genomic damage and epigenetic alterations in the HR repair pathway. Patients with tumors presenting with an HRD phenotype can show sensitivity to Poly (ADP-ribose) polymerase inhibitors (PARPis). Several promising tests to detect HRD have been developed based on different HRD definitions, biomarkers, and algorithms. However, no consensus on a gold standard HRD test has been established. In this systematic review, a comprehensive list of tests for the detection of HRD was identified and compared regarding HRD definition, biomarkers, and algorithms. PubMed’s Medline and Elsevier’s Embase were systematically searched, resulting in 27 eligible articles meeting the inclusion criteria. The primary challenge when comparing HRD tests lies in the lack of a consensus definition of HRD, as the HRD definition influences the proportion of samples being classified as HRD and impacts the classification performance. This systematic review provides an overview of available HRD tests that can inspire other researchers in searching for a gold standard HRD definition and highlights the importance of the factors that should be considered when choosing an HRD definition and tests for future planning of clinical trials and studies.
1. Background
Genomic profiling of tumors can be useful for understanding defects in DNA damage repair mechanisms and identifying patients who are candidates for targeted treatment [1,2,3,4]. Homolog recombination repair (HR) is a DNA damage repair mechanism that facilitates the repair of double-stranded breaks in DNA using a sister chromatid as template, thereby mediating an almost error-free repair of the double-stranded break [5]. Deficiency of the homologous recombination repair mechanism has been reported as a promoter of tumorigenesis as cells with HRD utilize more error-prone DNA repair mechanisms and accumulate mutations leading to genome instability [6,7,8,9]. HRD can be a result of germline or somatic pathogenic variants in genes involved in the HR repair pathway, primarily in the two key genes, Breast cancer 1 (BRCA1) and Breast cancer 2 (BRCA2) [10]. In addition, tumors can present with an HRD phenotype without identifiable germline or somatic HR variants. This HRD phenotype has yet to be fully characterized since HRD represents a broader phenomenon caused by abnormalities in the HR repair pathway, epigenetic alterations, or instability of the genome [11,12].
Patients with tumors presenting with an HRD phenotype show sensitivity to Poly (ADP-ribose) polymerase inhibitors (PARPis), which are targeted treatments inhibiting single-strand break repair, causing the phenomenon called synthetic lethality [2,3,4,13].
HRD-related genomic damage, often referred to as genomic scars, consists of different genomic aberrations which have been used in HRD tests as circumstantial evidence for HRD. The three most described genomic scars are loss of heterozygosity (LOH) [6], large-scale transition (LST) [7], and telomeric allelic imbalance (TAI) [8]. LOH is a genetic event where one of the alleles is missing [6], LST is chromosomal breaks between genomic regions [7], and TAI provides a measure for telomeric allelic imbalance [8]. Other measures of genomic scars providing an HRD phenotype are mutational signatures, originally described by Alexandrov et al. [14]. Mutational signatures are extracted by unsupervised clustering of point substitutions while considering adjacent sequence bases. BRCA1 and BRCA2 mutations have been described to be strongly associated with Signature 3 [14]. In addition, some tumors have shown a large contribution of Signature 3 without harboring BRCA1 and BRCA2 mutations, which might indicate that other genes with abnormalities might trigger similar mutational profiles [14].
Methylation of genes or pathogenic variants in genes in the HR repair pathway have also been used as biomarkers for HRD, as well as functional assays such as estimations of nuclear RAD51 foci [11,15,16,17,18].
Several promising tests to detect HRD have been developed based on different biomarkers and algorithms. Some HRD tests have been used in clinical trials to better define which cancers are most likely to have HRD. In the SOLO1 clinical trial, patients recently diagnosed with ovarian cancer showed benefits from PARPis harboring pathogenic BRCA variants [18]. The PRIMA and VELIA clinical trials have shown that ovarian cancer patients with HRD based on the HRD test myChoice from Myriad Genetics could benefit from a treatment combining platinum chemotherapy and PARPis [12,19]. However, one of the main challenges is the lack of consensus and a clear definition of HRD. This makes a direct comparison between HRD tests challenging as they are based on various definitions of HRD, biomarkers, and algorithms [11].
To our knowledge, a systematic review of tests for the detection of HRD has not yet been conducted. This systematic review assessed studies in which an HRD test was developed. The review was limited to HRD tests based on genomic/genetic data, including RNA profiling, but excluding HRD detection by functional assays and tests based solely on pathogenic variants, such as BRCA1/2 variants. The aim of the review was to compare and evaluate the current HRD tests used for stratifying patients into HR groups while also addressing HRD definition and biomarkers used.
2. Materials and Methods
A systematic literature review was conducted following the Preferred Reported Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines [20].
2.1. Literature Search
PubMed’s Medline and Elsevier’s Embase databases were systematically searched for eligible articles. The full search strings for PubMed and Embase are presented in Supplementary Table S1. The search strings included three search groups with multiple search terms that represented the search group topic. The three search group topics were (1) homologous recombination deficiency, (2) HRD test, and (3) data type/method. The initial search was conducted on 13 October 2021, and a second search was conducted on 11 May 2022. The second search included seven search terms (see in Supplementary Table S1) identified as missing during the initial search as well as a relocation of a misplaced search term into its correct search group. Moving the misplaced search term into the correct search group did not add any relevant records compared to the initial search. Both searches were conducted with no limitation on the date of publication. Additional studies were identified by assessment of reviews and the bibliography of included articles. Two authors (LRM and SKT), independently and blinded to each other, screened titles and abstracts as well as full texts for assessment of eligibility using Covidence systematic review software (https://support.covidence.org/help/how-can-i-cite-covidence (accessed on 21 September 2022)) [21].
2.2. Inclusion and Exclusion Criteria
Studies were included in the review if fulfilling the following inclusion criteria: (1) concerning homologous recombination deficiency or BRCAness, (2) developing or training an algorithm/classifier for stratifying patients into HRD groups based on a threshold, (3) analyzing patient samples, (4) articles published in English, and (5) original research articles.
Articles were excluded if they used functional assays to stratify patients into HRD groups. Any discrepancies regarding article suitability were solved by consulting two other authors (MB and ISP).
2.3. Data Extraction
Two authors (LRM and SKT) critically reviewed included articles and independently extracted data manually into an Excel spreadsheet. Data concerning study type, disease, cohort size, sample material, HRD definition, algorithm description, and algorithm input were extracted. Studies were grouped according to their algorithm or classifier type. Key findings were also retrieved when available, including any available sensitivities, specificities, positive and negative predictive values (PPV and NPV), accuracies, or areas under the receiver operating characteristic (ROC) curve.
3. Results
3.1. Study Selection
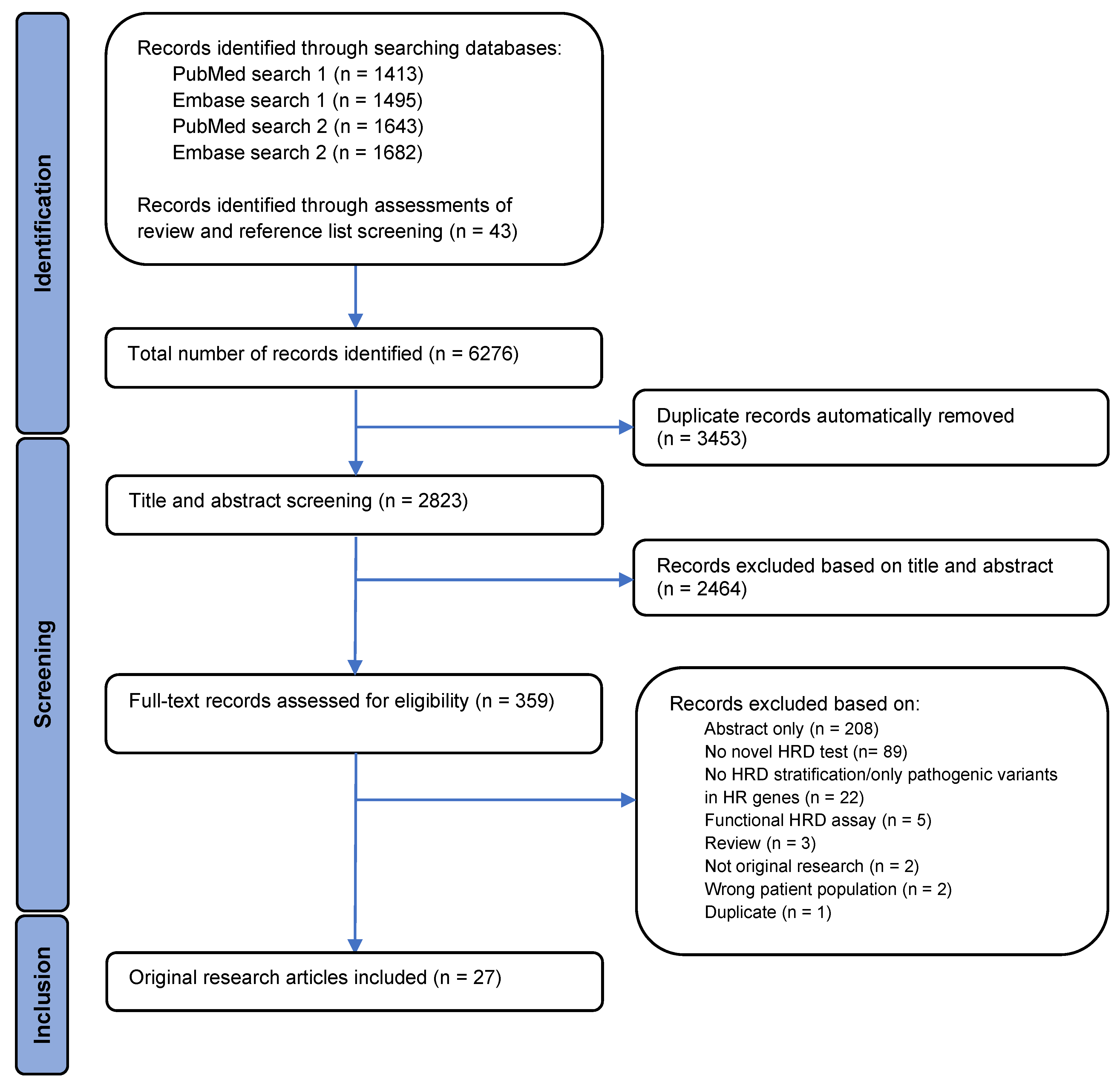
An overview of the study selection process is illustrated by a PRISMA flowchart in Figure 1. A total of 6276 records were identified, with 6233 records identified through searches in PubMed and Embase and 43 by assessment of reviews and the bibliography of the included articles. Automatic removal of 3453 duplicates was conducted using Covidence systematic review software, resulting in 2823 records left for the title and abstract screen. In the title and abstract screening, 2464 records were excluded, leaving 359 records available for full-text assessment of eligibility. Full-text screening of records resulted in 27 articles meeting the inclusion criteria.
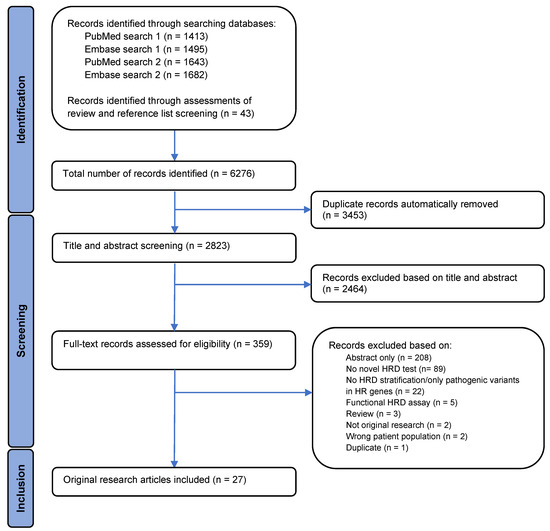
Figure 1.
A PRISMA flowchart displaying the identification, screening, and inclusion process. The flowchart illustrates the filtration of identified records to the final number of articles included in the analysis, as well as exclusion reasons.
The 332 full-text records which failed to meet the inclusion criteria were excluded for several reasons. Eighty-nine articles did not present a novel HRD test, and twenty-two records did not stratify patients into HR groups or only examined variants in HR-related genes. Five records used functional assays for HRD assessment, three records were identified as reviews, and two records were not original research. In addition, two records did not analyze patient samples and one record was identified as a duplicate that the automatic removal process in the Covidence systematic review software had not removed. The remaining records excluded were abstracts and, therefore, not eligible for full-text assessment.
3.2. Study Characteristics
The studies that are included in this review are based on different premises. These are things such as data origin, pre-analytical methods, and biological tissue types. This section will outline these various aspects. Table 1 displays the characteristics of the 27 included studies published between 1 January 2009 and 11 May 2022. Several types of cancers have been studied across the 27 studies, with the majority focusing on breast or ovarian cancer (Table 1).

Table 1.
Characteristics of the eligible studies, including cancer type, cohort size, tissue sample type, methods, and a description of the developed algorithms.
Almost all studies included a training and a validation cohort, except for five studies which only included a training or analysis cohort. The number of validation cohorts included in each study ranged from one to four. The size of the training cohorts varied substantially from 21 to several thousand patients. Similar variation in cohort size was observed in the validation cohorts (for details see Table 1).
The studies included several cohorts, of which data accessibility differs, with publicly accessible cohorts and cohorts needing approved permission for access. Twelve studies included cohorts from The Cancer Genome Atlas (TCGA) database [46], three used METABRICS data [47], and nine used data from Gene Expression Omnibus (GEO) [48]. Five studies used data from the Nik-Zainal et al. [49] cohort that analyzed 560 breast cancer patients, and two used data from the PrECOG cohort [50]. In addition, some studies included internal cohorts, which are not directly available but only used and described in the given study (Table 1).
Data were obtained using different sample materials, and the analysis platforms differed substantially between the studies (Table 1). Fifteen studies included data obtained from arrays. Of these, nine studies included single-nucleotide polymorphism (SNP) arrays conducted on frozen tumor tissue. Four studies included a comparative genomic hybridization array (arrayCGH) conducted on formalin-fixed paraffin-embedded (FFPE) tumor tissue. A gene expression array was used by four studies conducted on frozen tumor tissue, with one study also using FFPE tumor tissue (Table 1). Five studies used microarrays without elaborating on the array type, with analyses conducted on frozen tumor tissue.
Nineteen studies used a next-generation sequencing (NGS) approach to obtain data for their algorithm input (Table 1). Six studies conducted whole-exome sequencing (WES) using frozen or FFPE tumor tissue. Whole-genome sequencing (WGS) was conducted by six studies, of which three used frozen tumor tissue and three used frozen and FFPE tumor tissue. Low coverage/shallow WGS was performed on frozen tumor tissue in two studies, with one of the studies also using FFPE tumor tissue. Seven studies used panel sequencing, of which five used FFPE tumor tissue and three used frozen tumor tissue. RNA sequencing (RNA-seq) was used in five studies, and microRNA sequencing (miRseq) was performed in one study, with all studies using frozen tumor tissue and two studies also using FFPE (Table 1)
Other platforms were also used for obtaining data, with one study using digital multiplex ligation-dependent probe amplification (digitalMLPA) on FFPE tumor tissue and another using MLPA on FFPE and frozen tumor tissue (Table 1).
3.3. Definition of HRD
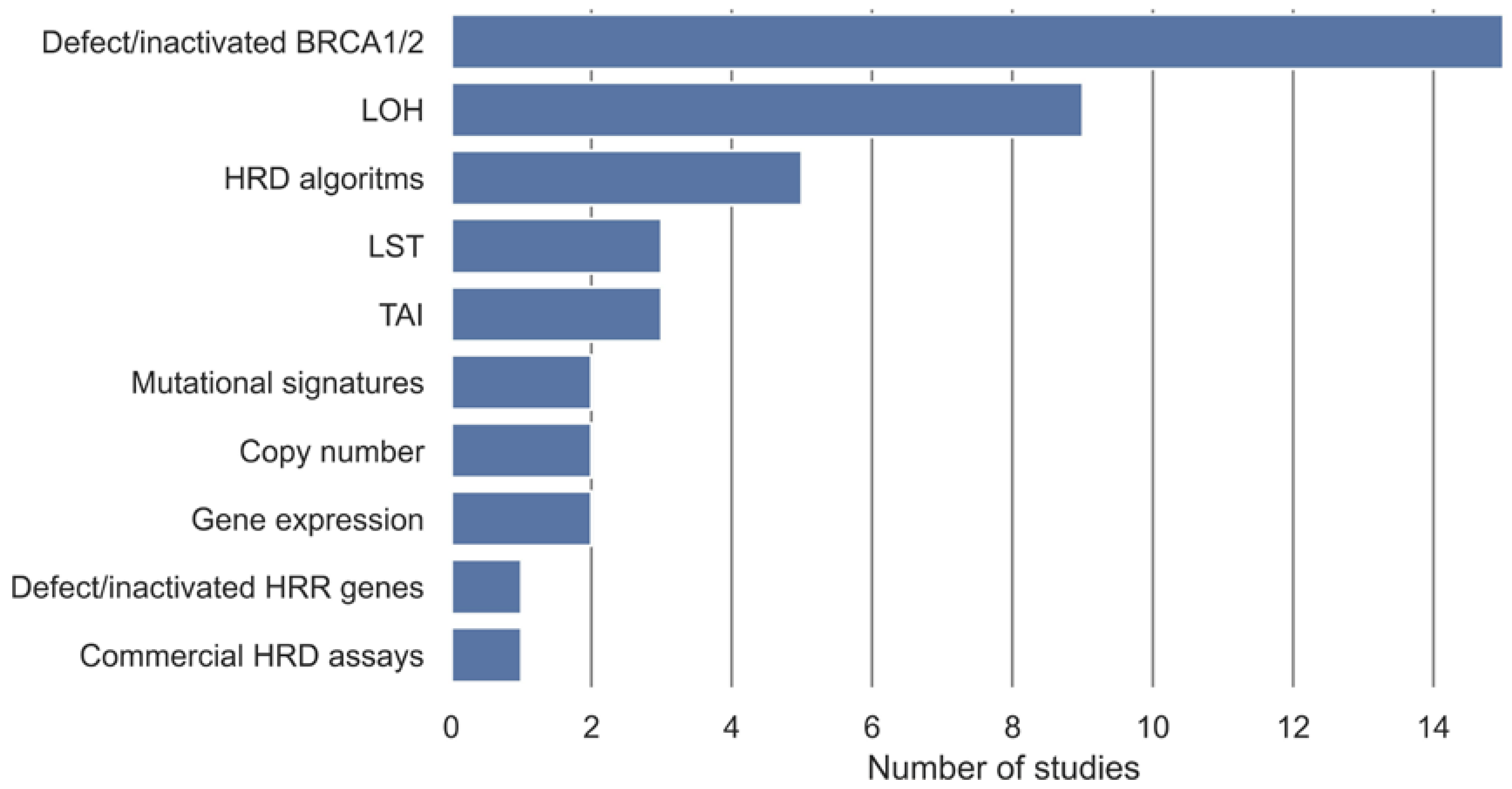
In the included studies, different measures, either alone or in combination, have been used to define a gold standard for samples being either non-HRD or HRD (Table 2). These gold standards are used as class labels in the development of the HRD tests. These measures have been categorized into ten categories (Figure 2). Methylation, somatic, and germline variants in BRCA1 and BRCA2 were the most used gold standard measures of HRD and have been used in 15 studies. Nine studies used a measure of LOH as the definition of HRD, and five used an already-established HRD algorithm as the gold standard. LST and TAI were used as the gold standard in three studies, and a commercial HRD assay was used in one study. Gene expression, copy number, and mutational signatures were each used in two studies as the gold standard for HRD, and methylation, somatic, or germline variants in HR genes were used in one study.
Table 2.
An overview of algorithm input, study type, performance measures, and HRD gold standard. Internal validation is defined as validation primarily conducted on training data. This also includes cross-validation if no external data have been used. External validation is defined as validation conducted on external data or if a part of the dataset has been held out from training with the main purpose to use as a validation set.
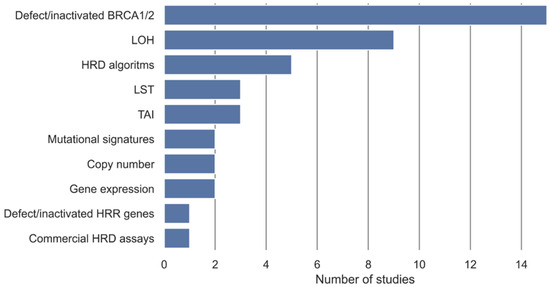
Figure 2.
Overview of gold standards for definition of HRD, either alone or in combination, and their frequencies in the included studies. Defect/inactivated BRCA1/2 covers biallelic or monoallelic methylation, somatic, and germline variants in BRCA1 and BRCA2. Defect/inactivated homologous recombination repair (HRR) genes cover biallelic or monoallelic methylation, somatic, and germline variants in genes involved in the homologous recombination repair pathway in addition to BRCA1 and BRCA2. Loss of heterozygosity (LOH) covers measures of LOH, ranging from LOH in individual genes to genome wide LOH. HRD algorithms cover HRD defined from an HRD algorithm developed in another study.
3.4. HRD Detection Algorithms
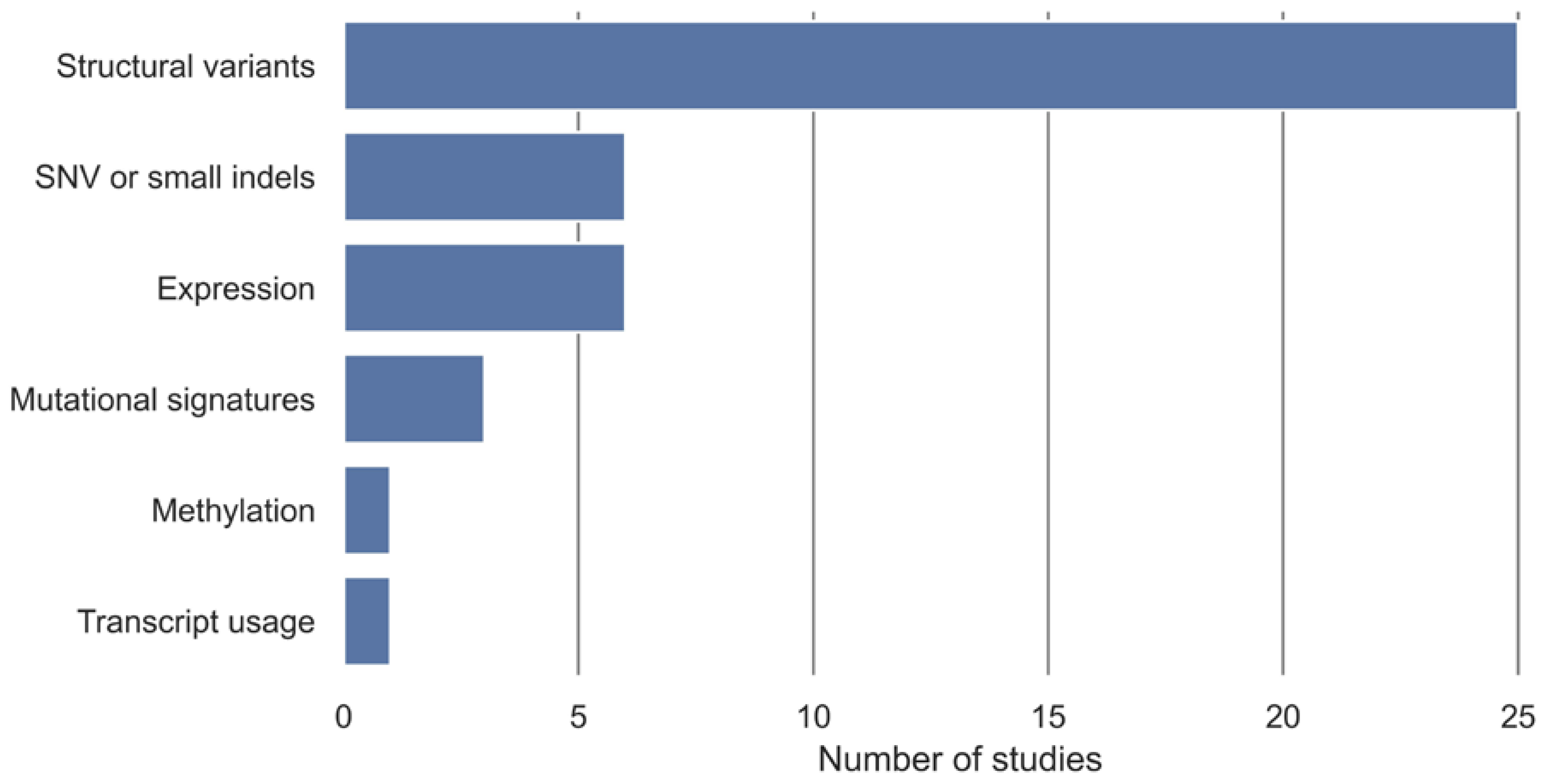
In 16 studies, the development of HRD tests has been based on existing classification algorithms for classifying samples into non-HRD and HRD groups with an HRD definition used as a class label. The additional 11 studies have developed a novel classification algorithm to classify samples into non-HRD and HRD groups (Table 2). The algorithm input data have been stratified into categories, displayed in Figure 3, with structural variants being the most utilized algorithm input, followed by SNV or small indels and expression.
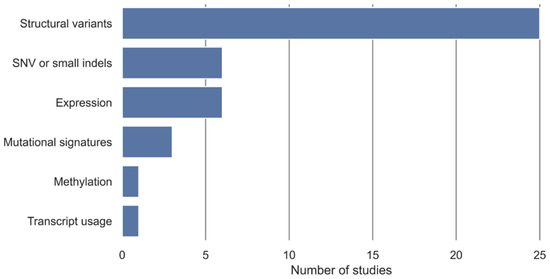
Figure 3.
Overview of algorithmic input and number of studies that are included. Structural variants include measures such as copy number, loss of heterozygosity (LOH), large-scale transitions (LST), ploidy, percentage of genomic LOH, large-scale genomic alterations (LGA), structural variants, and methylation copy number. Expression includes gene expression and miRNA expression. SNV and small indels include point mutations, single-base substitution, and smaller indels. Some HRD tests have been based on more than one algorithm input.
Two studies used a nearest centroid classifier for discriminating non-HRD and HRD groups, with Lips et al. 2011 [23] reporting the BRCA1-like MLPA classifier to classify BRCA1-like breast cancers based on copy number input. Severson et al. 2017 [29] presented the BRCA1ness signature of breast cancer based on gene expression. The nearest centroid classifier finds the centroid for all predictors per class, being the mean value of each predictor, and new samples are then assigned a class based on the closest centroid [51].
Four studies based their HRD test on a shrunken centroid model, which is based on the same concept as a nearest centroid but includes an additional step, shifting class-based centroids towards the centroid of all features. If a feature is shrunken down to the centroid of all features, it can be excluded as the feature does not add any discriminating information. This additional step in the shrunken centroid model acts as a feature selection for the model [52]. Of the four studies using a shrunken centroid model, Joosse et al. 2009 [22] reported the BRCA1 classifier, which was developed to classify BRCA1-like breast cancers. In the study by Joosse et al. 2012 [24], a similar classifier was developed, the BRCA2 classifier, for the classification of BRCA2-like breast cancers. Lips et al. 2020 extended the two algorithms from Joosse et al. [22,24] to a new platform and presented the BRCA1-like digitalMLPA classifier and BRCA2-like digitalMLPA classifier also based on copy number. Schouten et al. [40] applied the same methodological approach as Joosse et al. [22,24] to develop two HRD tests that were specific for ovarian cancer, the Ovarian cancer BRCA1-like classifier and Ovarian cancer BRCA2-like classifier, which both are based on copy numbers as input.
Chen et al. [33] reported the BRCA1-like classifier that was developed to classify BRCA1-like breast cancers based on a support vector machine (SVM) classifier using copy number as input. An SVM classifier finds a hyperplane in the feature space, which can be used to separate the classes of data points. The optimum hyperplane is the plane with the maximum margin between points from separate classes. The classifier can then be applied to new samples and assign a class label [53].
Leibowitz et al. [43] reported the HRD-RNA for pan-cancer using a logistic regression model. Logistic regression models the probability of classifying a sample into possible outcomes with a number of dependent variables [51]. Leibowitz et al. [43] also included the HRD-DNA based on genome-wide LOH.
Four studies based their HRD test on a least absolute shrinkage and selection operator (LASSO) regression model, a regression analysis used as a technique to reduce model complexity. LASSO selects and shrinks the model to use the optimum number of features based on regularization [54]. Of the four studies using LASSO, Zhuang et al. [41] reported the 24 gene pair (24-GPS) classifier that provided an HRD signature for pancreatic cancer based on gene expression input. Davies et al. [28] developed HRDetect to classify BRCA1/BRCA2-deficient breast, ovarian, and pancreatic cancer based on a LASSO logistic regression model that included mutational signatures, LOH, and indels from WGS as input. Diossy et al. [31] extended the HRDetect from Davies et al. [28] to use mutational signatures, LOH, and indels from WES data of breast cancer and brain metastases. They presented the retrained WES-HRDetect based on a LASSO logistic regression model. The study by Liao et al. [44] also used a LASSO logistic regression model for their Transcriptomic HRD score for breast cancer, which was based on gene expression input.
Three studies used a random forest (RF) model, which is an ensemble method constructed by a large number of independently trained decision trees where features for each decision tree are selected randomly [55]. Of the three studies using RF-based models, Nguyen et al. [37] reported the pan-cancer Classifier of HOmologous Recombination Deficiency (CHORD)I based on single-base substitutions, indels, and structural variants. Barenboim et al. [38] reported the DNA-methylation-based RF classifier providing a BRCAness signature for osteosarcoma using methylation copy numbers from array data. Kang et al. [42] reported transcriptional HRD (tHRD) based on transcript usage.
Gulhan et al. [34] developed Signature Multivariate Analysis (SigMA), which uses an approach based on mutational signatures extracted by non-negative matrix factorization (NMF). NMF is an unsupervised machine learning algorithm that factorizes the original dataset into a feature set and a coefficient set. Each feature set has an associated weight in the coefficient set. The feature and coefficient set can be used to select, reduce, or analyze individual features of data [56].
Most of the 11 studies with a novel classification algorithm to classify samples into non-HRD and HRD groups were based on genomic scar measures.
Popova et al. [7] reported LST as a classifier of HRD in breast cancer. The LST test consists of a two-step decision rule with segregation of tumors based on ploidy followed by segregation according to the number of LST counts. Large-scale genomic alterations (LGA), which are reported to correspond to LST, were used by Eeckhoutte et al. [35], who developed ShallowHRD for breast and ovarian cancer based on the sum of LGA counts from WGS at low coverage (~1X coverage).
Abkevich et al. [6] reported HRD-LOH as a classifier of HRD in ovarian cancer based on the sum of LOH segment counts. Smyth et al. [32] also included a measure of LOH, the genomic LOH, for HRD classification in esophagogastric cancer. The genomic LOH test was based on the sum of the lengths of included LOH segments divided by the length of the interrogated genome.
Telli et al. [16] reported the combined homologous recombination deficiency score (HRD score) for breast and ovarian cancer, which consists of the numeric sum of LOH, LST, and TAI counts. Similar to the HRD score, Chen et al. [39] reported a genomic scar algorithm (GSA) to provide a measure of HRD for breast and ovarian cancer. The GSA consists of the numeric sum of LST, TAI, and LOH, which then is subtracted by a correction coefficient multiplied by a ploidy value.
The study by Watkins et al. [27] reported scores of chromosomal instability scarring (SCINS) for breast and ovarian cancer, consisting of four scores based on different types of allele-specific copy-number profiles.
Zhang et al. [26] developed a genomic instability score for ovarian cancer based on the sum of the number of copy number changes and somatic mutations multiplied by a constant.
Tandem duplications as a genomic scar that provided a measure of HRD in BRCA1-type breast cancers were reported by Qu et al. [45], who developed the tandem duplications score (TD-score) based on the counts of small (<10 kb) tandem duplications.
Two studies based their HRD test on other measurements of HRD than genomic scars, with Lu et al. [25] reporting the hypothesized HR-deficiency score (HRDS) classifying breast and ovarian cancer based on gene expression levels and Wang et al. [30] developing the 10-miRNA-score for HRD prediction of ovarian cancer based on the expression levels of miRNA.
3.5. HRD Test Performance
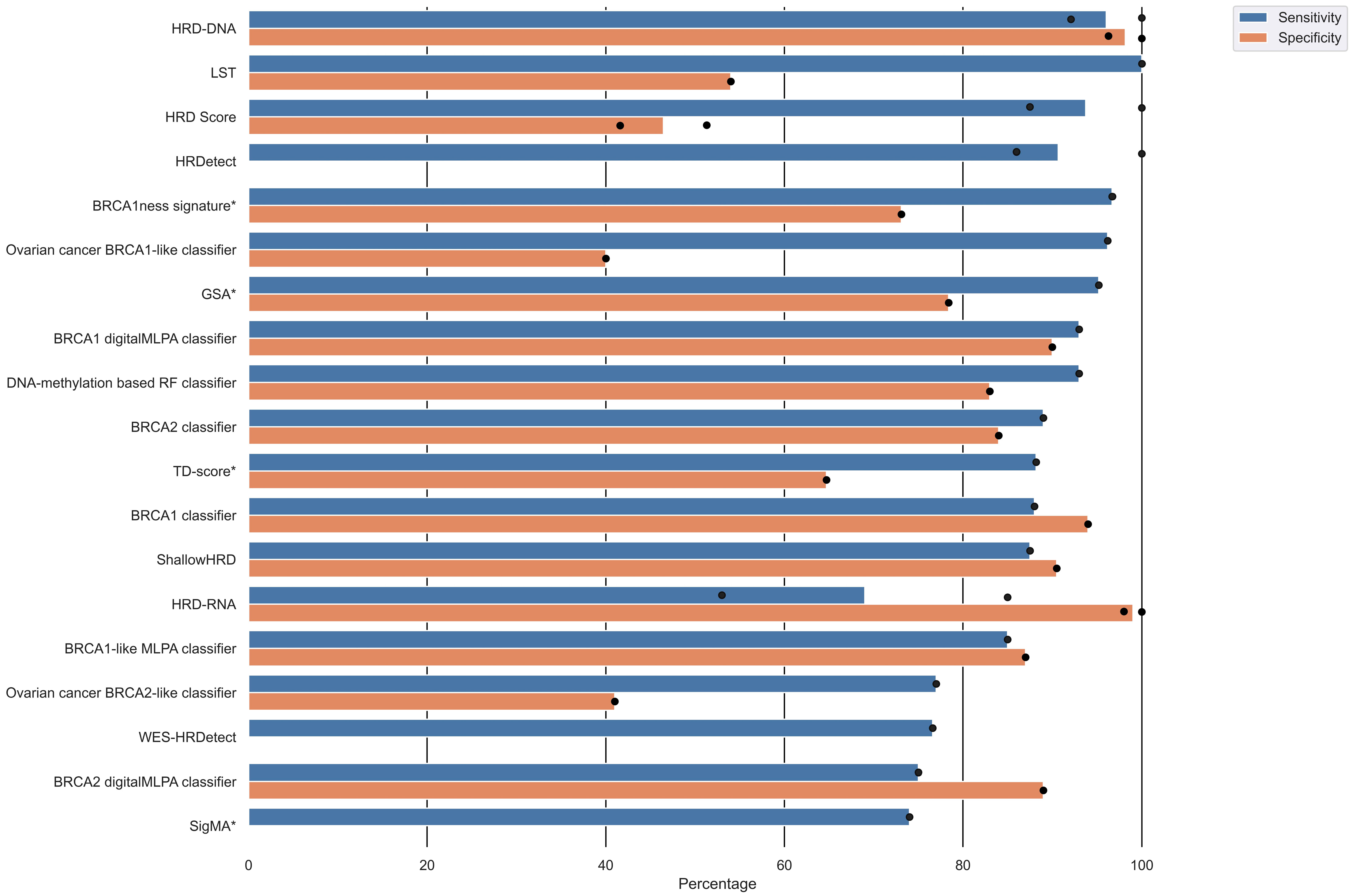
The included studies provided a variety of performance measures and validation results (Table 2) based on a study-individual gold standard of HRD. Sixteen studies reported the sensitivity of their HRD test, of which five studies reported more than one sensitivity due to multiple validations. Corresponding specificities were only reported in 13 studies, of which four provided more than one specificity. An overview of the sensitivity and specificity of the different HRD tests is displayed in Figure 4 and Table 2. The sensitivity ranged from 53% to 100%, and the specificity ranged from 40% to 100%. The HRD test with the highest sensitivity and corresponding specificity was HRD-DNA [43], followed by BRCA1-like digitalMLPA classifier [36], BRCA1 classifier [22], ShallowHRD [35], BRCA1-like MLPA classifier [23], DNA-methylation-based RF classifier [38], and BRCA2 classifier [24]. The HRD score [16], LST [7], BRCA1ness signature [29], and Ovarian cancer BRCA1-like classifier [40] had high sensitivity, but the corresponding specificities were relatively low (Figure 4). HRDetect [28] also had a high sensitivity but did not report a corresponding specificity.
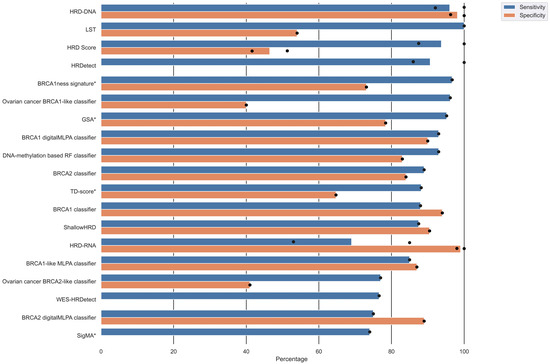
Figure 4.
Overview of sensitivity and specificity of the HRD tests. Several studies reported multiple validation results. Dots represents individual reported validation results, and bars represents mean sensitivity/specificity. Note that not all studies reported both sensitivity and specificity. Algorithms marked with an asterisk (*) next to the algorithm alias have only been internally validated.
Nine studies provided an area under the curve (AUC) ranging from 75% to 100% (Table 2), with AUC being highest for HRD-DNA [43], HRD-RNA [43], CHORD [37], 24-GPS [41], and WES-HRDetect [31].
The accuracy was reported in five studies ranging from 72 to 91% (Table 2), with accuracy being highest for the BRCA1-like digitalMLPA classifier [36].
4. Discussion
This systematic literature review identified 27 studies in which an HRD test was developed or trained to stratify patients into HR groups, with all HRD tests being based on genomic or RNA profiling.
In the included studies, the definition of HRD was rather heterogeneous and lacked consensus between the studies. The definition of HRD was based on multiple measures used either alone or in combination (Figure 2), with defect BRCA1/2 and LOH being the most frequently used measures of HRD.
Most of the HRD tests included in this review were developed to predict HRD in breast and ovarian cancer, followed by prostate and pancreatic cancers (Table 1). The rationale for developing HRD tests for these cancer types could be that more than 15% of breast, ovarian, and pancreatic cancers and 14% of prostate cancer have mutations in HR-related genes [57]. In addition, mutations in BRCA1/2 are associated with an increased lifetime risk of developing breast, ovarian, prostate, and pancreatic cancers [58]. Furthermore, early studies of PARPis showed promising results in BRCA1/2-deficient cells, which built the foundation for clinical trials investigating PARPi response in ovarian cancer. Later, clinical trials with PARPis were expanded to breast, prostate, and pancreatic cancers [59]. As the majority of HRD tests included in this review are developed for HRD detection in ovarian and breast cancer, it is important to recognize that other cancer types might include different HRD patterns. For instance, Diossy et al. [31] found that brain metastases from breast cancer tend to have a higher HRD score than primary breast cancer, which should be considered in a clinical context. The studies included in this review have used a variety of different tissue types in the development of the HRD test. It is important to recognize that HRD measures generated from various tissue types might produce different results and should be validated accordingly. Furthermore, there might be several important considerations when implementing an HRD test in clinical practice, such as the stability of the material used, cost of running the analysis, and the turnaround time.
The lack of consensus on the HRD definition and a gold standard measure of HRD provides a growing problem when developing HRD tests. This became evident during the PRIMA and VELIA clinical trials, where patients with HRD-positive tumors, defined by the myChoice HRD test based on LOH, LST, and TAI, responded to combination treatment with PARPis. However, the response to PARPis was not at the same magnitude as for patients with HRD defined by somatic or germline pathogenic variants in BRCA1/2 [12,19]. This highlights that defects in BRCA1/2 are one of the most robust measures of HRD, although it does not cover all phenotypes of HRD. In addition, secondary or reversion mutations in BRCA1/2 have been found to restore the functionality of the HR mechanism [60,61]. Hence, genomic scars, such as LOH, LST, and TAI, provide an imperfect measure of the HR function, as these measures are results of prior HRD exposure [62,63]. HRD tests based on functional assays can assess the HR mechanism’s functionality, potentially providing a more precise and clinically relevant measure of HRD. Unfortunately, such functional assays are in the early stages of development and are prone to a similar lack of consensus on HRD definition as other HRD tests, making clinical implementation difficult [64].
Most HRD tests were trained with HRD defined as various measures specific to BRCA1 and/or BRCA2. There is, however, evidence that HRD can arise based on variants in a wider set of genes related to the HR pathway [10,11]. Interestingly, only ShallowHRD was developed based on an HRD definition, including more HR-related genes than BRCA1 and BRCA2 [35]. HRD definitions mainly based on variants in BRCA1/2 or genes related to the HR pathway have been referred to be the etiology or origin of HRD, as these variants are the main reason that a given gene is inactivated or defective [11]. HRD tests such as tHRD, SigMA, SCINS, WES-HRDetect, genomic LOH, DNA-methylation-based RF classifier, and transcriptomic HRD score defined HRD as various measures of structural variants across the genome, which have been referred to as genomic scars or prior HRD exposure. HRD tests based on genomic scars aim to detect a genomic pattern resulting from prior HRD exposure without detecting the underlying reason [11].
The performance of the included HRD tests varied substantially (Figure 4 and Table 2), with the HRD tests HRD-DNA [43], BRCA1 digitalMLPA classifier [36], and BRCA1 classifier [22] having average sensitivities and specificities above 90%. In common for these HRD tests is the use of copy number as an algorithmic input and a definition of HRD as biallelic loss or variants in BRCA1 and/or BRCA2. The BRCA1 digitalMLPA classifier, however, indirectly defines HRD as variants in BRCA1 by using the BRCA1 classifier as the HRD definition, which makes interpreting this algorithm somewhat difficult. Although these HRD tests perform well when predicting samples with biallelic loss or variants in BRCA1 and/or BRCA2, their utility might be limited by their HRD definition.
Some studies evaluated the HRD tests’ performance by AUC, with HRD-DNA, HRD-RNA, CHORD, 24-GPS, and WES-HRDetect all having AUCs above 96%. However, the 24-GPS was not evaluated in an external dataset, which is why this test needs further evaluation to validate the performance. Interestingly, for these five HRD tests, their individual input biomarkers were also included in their individual HRD class label definition, contrary to most of the other studies using various biomarkers to predict an HRD class label defined as, for instance, BRCA mutations.
Prediction models are usually validated using sensitivity and specificity [65]. A high sensitivity describes a model’s ability to predict true positives, and specificity describes the model’s ability to predict true negatives. Hence, a model having both high sensitivity and specificity minimizes false positives and false negatives [65]. However, many of the included studies used a non-classical approach to assess performance in which they suggested that false-positive samples, impacting the specificity, might not be misclassified samples but instead samples which harbor similar patterns as HRD-positive samples. Thereby, the false-positive samples are suggested to be true HRD samples that do not comprise the measures used as the HRD definition. This complicates the comparison of performance for the HRD tests even further. For instance, Davies et al. [28] found that one-third of tumors with a high HRDetect score could not be verified as BRCA mutated, but they argued that these tumors seem biologically comparable to BRCA-mutated tumors and might respond similar to PARP inhibitors.
A way to empirically compare the performance of the HRD tests could be based on drugs targeting HRD, such as PARPis and platinum chemotherapy, with drug response being used as a surrogate marker for HRD. Although the response to PARPis might be affected by other mechanisms, the approach could be useful for the comparison of various HRD tests and support the clinical utility of the tests [11,64].
In a clinical context, it is important to be aware of the HRD definition and how it influences the proportion of patients selected as HRD-positive. For instance, in the VELIA clinical trial, the percentage of patients eligible for PARPi treatment when considering HRD as BRCA mutations were 26% compared to 55% when considering HRD as either BRCA mutations or a measure of genomic scars defined by the myChoice HRD test [19]. This further highlights the importance of the HRD definition as it can highly influence the proportion of patients with HRD-positive tumors and, thereby, patients eligible for treatment. When summarizing the issues concerning HRD definition and the non-classical approach to specificity, it is intuitive to believe that there will be some potential limitations in identifying the group of patients who have functional HR repair and, thereby, likely not to benefit from treatments targeting HRD. This issue has been raised elsewhere in several studies [11,64].
The main limitation of this review was the limited opportunity to compare HRD tests based on their performance measures, as many HRD tests lacked information regarding performance and/or did not conduct an external validation of the HRD test, as well as the lack of consensus of HRD definitions. Furthermore, the studies were reported in a rather heterogeneous manner, which hindered a clear interpretation of the effects of, e.g., analysis platform, data input, or disease on the performance of the HRD tests. However, this systematic review provides a detailed summary of the numerous parameters included in the HRD detection algorithms and addresses the challenges of choosing a suitable HRD test due to the heterogeneity of the parameters. Although we conducted the systematic literature search using two widely used databases and assessed reviews of relevant topics and the bibliographies of the included articles, we cannot exclude having missed relevant articles. Studies published in languages other than English and those without available full text were not included in the review, so we cannot exclude a publication bias. In addition, the review was limited to including HRD tests based on genomic and RNA profiling and excluding HRD detection by functional assays and HRD tests based solely on HR-related pathogenic variants. The decision to exclude HRD tests based on functional assays and studies based solely on HR-related pathogenic variants was based on a large number of studies eligible for full-text review when including these HRD tests too. Furthermore, we limited inclusion to studies that developed or trained a novel HRD test. Studies that evaluated an HRD test in additional cohorts without training or modifying the HRD test were excluded due to the significant addition of studies eligible for full-text screening. Therefore, we cannot exclude that this limitation on the HRD tests included can bias our evaluation of the HRD tests’ performance.
5. Conclusions
This systematic review provided an overview of the HRD tests that have been developed and summarized the variety of different biomarkers, algorithms, and HRD definitions used. The primary challenge for the comparison of HRD tests lies in the definition of HRD. The performance of the included HRD test varied with some performing better than others. However, this review also highlights that the HRD definition influences the proportion of samples classified as HRD and impacts the classification performance.
With this systematic review comparing HRD tests, we have provided an overview that can inspire other researchers in searching for a gold standard HRD definition, as this field requires one such to most suitably classify tumors as HRD or non-HRD. In addition, we have highlighted the importance of the factors that should be considered when choosing an HRD definition and HRD test for future planning of clinical trials and studies, as a consensus definition of HRD is truly needed.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/cancers15235633/s1, Table S1: Search terms in Pubmed and Embase for the first and second search.
Author Contributions
Conceptualization, L.R.M., S.K.T., H.B.K., M.T., I.S.P. and M.B.; Design of the study, L.R.M., S.K.T., H.B.K., M.T., I.S.P. and M.B.; Analysis and data extraction, L.R.M. and S.K.T.; Interpretation of data, L.R.M. and S.K.T.; Writing the manuscript, L.R.M. and S.K.T.; Critical revising, H.B.K., M.T., I.S.P. and M.B.; L.R.M. and S.K.T. contributed equally to this paper and share first authorship. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors would like to thank the Medical Library at Aalborg University Hospital, Denmark, for assistance in the search process.
Conflicts of Interest
The authors declare no conflict of interest.
List of Abbreviations
| HRD | homologous recombination deficiency |
| PARPi | poly (ADP-ribose) polymerase inhibitors |
| HR | homolog recombination repair |
| BRCA1 | breast cancer 1 gene |
| BRCA2 | breast cancer 2 gene |
| LOH | loss of heterozygosity |
| LST | large-scale transition |
| TAI | telomeric allelic imbalance |
| PRISMA | Preferred Reported Items for Systematic Reviews and Meta-Analysis |
| ROC | receiver operating characteristic |
| PPV | positive predictive value |
| NPV | negative predictive value |
| TCGA | The Cancer Genome Atlas |
| GEO | Gene Expression Omnibus |
| SNP | single-nucleotide polymorphism |
| arrayCGH | comparative genomic hybridization array |
| FFPE | formalin fixed paraffin embedded |
| NGS | next-generation sequencing |
| WES | whole-exome sequencing |
| WGS | whole-genome sequencing |
| RNA-seq | RNA sequencing |
| SVM | support vector machine |
| LASSO | least absolute shrinkage and selection operator |
| CHORD | Classifier of HOmologous Recombination Deficiency |
| tHRD | transcriptional HRD |
| SigMA | Signature Multivariate Analysis |
| NMF | non-negative matrix factorization |
| LGA | large-scale genomic alterations |
| HRD score | combined homologous recombination deficiency score |
| GSA | genomic scar algorithm |
| SCINS | scores of chromosomal instability scarring |
| TD-score | tandem duplications score |
| HRDS | hypothesized HR-deficiency score (HRDS) |
References
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet. Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Szostak, J.W.; Orr-Weaver, T.L.; Rothstein, R.J.; Stahl, F.W. The double-strand-break repair model for recombination. Cell 1983, 33, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-Mccune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-like Breast Carcinomas with BRCA1/2 Inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.-Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric Allelic Imbalance Indicates Defective DNA Repair and Sensitivity to DNA-Damaging Agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, N.; Yasuda, M.; Takahashi, F.; Isonishi, S.; Jobo, T.; Aoki, D.; Tsuda, H.; Sugiyama, T.; Kodama, S.; Kimura, E.; et al. Dose-dense paclitaxel once a week in combination with carboplatin every 3 weeks for advanced ovarian cancer: A phase 3, open-label, randomised controlled trial. Lancet 2009, 374, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Welcsh, P.L.; King, M.-C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol. Genet. 2001, 10, 705–713. [Google Scholar] [CrossRef]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J.J.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.W.; Karlan, B.Y.; Cass, L.; Baldwin, R.L. BRCA1 promoter methylation predicts adverse ovarian cancer prognosis. Gynecol. Oncol. 2006, 101, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, 105906. [Google Scholar] [CrossRef]
- Veritas Health Innovation. Covidence Systematic Review Software [Internet]. Available online: https://support.covidence.org/help/how-can-i-cite-covidence (accessed on 21 September 2022).
- Joosse, S.A.; Van Beers, E.H.; Tielen, I.H.G.; Horlings, H.; Peterse, J.L.; Hoogerbrugge, N.; Ligtenberg, M.J.; Wessels, L.F.A.; Axwijk, P.; Verhoef, S.; et al. Prediction of BRCA1-association in hereditary non-BRCA1/2 breast carcinomas with array-CGH. Breast Cancer Res. Treat. 2009, 116, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Lips, E.H.; Laddach, N.; Savola, S.P.; Vollebergh, M.A.; Oonk, A.M.; Imholz, A.L.T.; Wessels, L.F.A.; Wesseling, J.; Nederlof, P.M.; Rodenhuis, S. Quantitative copy number analysis by Multiplex Ligation-dependent Probe Amplification (MLPA) of BRCA1-associated breast cancer regions identifies BRCAness. Breast Cancer Res. 2011, 13, R107. [Google Scholar] [CrossRef]
- Joosse, S.A.; Brandwijk, K.I.M.; Devilee, P.; Wesseling, J.; Hogervorst, F.B.L.; Verhoef, S.; Nederlof, P.M. Prediction of BRCA2-association in hereditary breast carcinomas using array-CGH. Breast Cancer Res. Treat. 2012, 132, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, D.; Li, C.; Zhou, M.; Hao, D. Correlation between gene expression and mutator phenotype predicts homologous recombination deficiency and outcome in ovarian cancer. J. Mol. Med. 2014, 92, 1159–1168. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, Y.; Hao, D. A genomic instability score in discriminating nonequivalent outcomes of BRCA1/2 mutations and in predicting outcomes of ovarian cancer treated with platinum-based chemotherapy. PLoS ONE 2014, 9, e113169. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.; Weekes, D.; Shah, V.; Gazinska, P.; Joshi, S.; Sidhu, B.; Gillett, C.; Pinder, S.; Vanoli, F.; Jasin, M.; et al. Genomic complexity profiling reveals that hormad1 overexpression contributes to homologous recombination deficiency in triple-negative breast cancers. Cancer Discov. 2015, 5, 488–505. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Severson, T.M.; Wolf, D.M.; Yau, C.; Peeters, J.; Wehkam, D.; Schouten, P.C.; Chin, S.F.; Majewski, I.J.; Michaut, M.; Bosma, A.; et al. The BRCA1ness signature is associated significantly with response to PARP inhibitor treatment versus control in the I-SPY 2 randomized neoadjuvant setting. Breast Cancer Res. 2017, 19, 99. [Google Scholar] [CrossRef]
- Wang, T.; Wang, G.; Wang, G.; Zhang, X.; Wu, D.; Yang, L.; Hao, D. The expression of miRNAs is associated with tumour genome instability and predicts the outcome of ovarian cancer patients treated with platinum agents. Sci. Rep. 2017, 7, 14736. [Google Scholar] [CrossRef]
- Diossy, M.; Reiniger, L.; Sztupinszki, Z.; Krzystanek, M.; Timms, K.M.; Neff, C.; Solimeno, C.; Pruss, D.; Eklund, A.C.; Tóth, E.; et al. Breast cancer brain metastases show increased levels of genomic aberration-based homologous recombination deficiency scores relative to their corresponding primary tumors. Ann. Oncol. 2018, 29, 1948–1954. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Cafferkey, C.; Loehr, A.; Waddell, T.; Begum, R.; Peckitt, C.; Harding, T.C.; Nguyen, M.; Okines, A.F.; Raponi, M.; et al. Genomic loss of heterozygosity and survival in the REAL3 trial. Oncotarget 2018, 9, 36654–36665. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Salas, L.A.; Miller, T.W.; Mark, K.; Marotti, J.D.; Kettenbach, A.N.; Cheng, C.; Christensen, B.C. Molecular and epigenetic profiles of BRCA1-like hormone-receptor-positive breast tumors identified with development and application of a copy-number-based classifier. Breast Cancer Res. 2019, 21, 1–13. [Google Scholar] [CrossRef]
- Gulhan, D.C.; Lee, J.J.K.; Melloni, G.E.M.; Cortés-Ciriano, I.; Park, P.J. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet. 2019, 51, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Eeckhoutte, A.; Houy, A.; Manié, E.; Reverdy, M.; Bièche, I.; Marangoni, E.; Goundiam, O.; Vincent-Salomon, A.; Stoppa-Lyonnet, D.; Bidard, F.C.; et al. ShallowHRD: Detection of homologous recombination deficiency from shallow whole genome sequencing. Bioinformatics 2020, 36, 3888–3889. [Google Scholar] [CrossRef] [PubMed]
- Lips, E.H.; Benard-Slagter, A.; Opdam, M.; Scheerman, C.E.; Wesseling, J.; Hogervorst, F.B.L.; Linn, S.C.; Savola, S.; Nederlof, P.M. BRCAness digitalMLPA profiling predicts benefit of intensified platinum-based chemotherapy in triple-negative and luminal-type breast cancer. Breast Cancer Res. 2020, 22, 79. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; W M Martens, J.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef] [PubMed]
- Barenboim, M.; Kovac, M.; Ameline, B.; Jones, D.T.W.; Witt, O.; Bielack, S.; Burdach, S.; Baumhoer, D.; Nathrath, M. DNA methylation-based classifier and gene expression signatures detect BRCAness in osteosarcoma. PLoS Comput. Biol. 2021, 17, e1009562. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shao, M.; Meng, P.; Wang, C.; Li, Q.; Cai, Y.; Song, C.; Wang, X.; Shi, T. GSA: An independent development algorithm for calling copy number and detecting homologous recombination deficiency (HRD) from target capture sequencing. BMC Bioinform. 2021, 22, 562. [Google Scholar] [CrossRef] [PubMed]
- Schouten, P.C.; Richters, L.K.; Vis, D.J.; Kommoss, S.; van Dyk, E.; Ernst, C.; Kluin, R.J.C.; Marme, F.; Lips, E.H.; Schmidt, S.; et al. Ovarian cancer specific BRCA-like copy number aberration classifiers detect mutations associated with homologous recombination deficiency in the AGO-TR1 trial. Clin. Cancer Res. 2021, 27, 6559–6569. [Google Scholar] [CrossRef]
- Zhuang, S.; Chen, T.; Li, Y.; Wang, Y.; Ai, L.; Geng, Y.; Zou, M.; Liu, K.; Xu, H.; Wang, L.; et al. A transcriptional signature detects homologous recombination deficiency in pancreatic cancer at the individual level. Mol. Ther.—Nucleic Acids 2021, 26, 1014–1026. [Google Scholar] [CrossRef]
- Kang, H.G.; Hwangbo, H.; Kim, M.J.; Kim, S.; Lee, E.J.; Park, M.J.; Kim, J.W.; Kim, B.G.; Cho, E.H.; Chang, S.; et al. Aberrant Transcript Usage Is Associated with Homologous Recombination Deficiency and Predicts Therapeutic Response. Cancer Res. 2022, 82, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, B.D.; Dougherty, B.V.; Bell, J.S.K.; Kapilivsky, J.; Michuda, J.; Sedgewick, A.J.; Munson, W.A.; Chandra, T.A.; Dry, J.R.; Beaubier, N.; et al. Validation of Genomic and Transcriptomic Models of Homologous Recombination Deficiency in a Real-World Pan-Cancer Cohort. MedRxiv 2022, 22, 587. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Yang, Y.; Xie, A.; Jiang, Z.; Liao, J.; Yan, M.; Zhou, Y.; Zhu, J.; Hu, J.; Zhang, Y.; et al. Applicability of Anticancer Drugs for the Triple-Negative Breast Cancer Based on Homologous Recombination Repair Deficiency. Front. Cell Dev. Biol. 2022, 10, 457. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Martens, J.W.M.; Hollestelle, A.; Smid, M. Identifying Transcripts with Tandem Duplications from RNA-Sequencing Data to Predict BRCA1-Type Primary Breast Cancer. Cancers 2022, 14, 753. [Google Scholar] [CrossRef]
- National Cancer Institute. The Cancer Genome Atlas Program [Internet]. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 21 September 2022).
- METABRIC. Breast Cancer (METABRIC, Nature 2012 & Nat Commun 2016) [Internet]. 2016. Available online: https://www.cbioportal.org/study/summary?id=brca_metabric (accessed on 12 September 2022).
- NCBI. Gene Expression Omnibus [Internet]. 2022. Available online: https://www.ncbi.nlm.nih.gov/geo/ (accessed on 12 September 2022).
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Telli, M.L.; Jensen, K.C.; Vinayak, S.; Kurian, A.W.; Lipson, J.A.; Flaherty, P.J.; Timms, K.; Abkevich, V.; Schackmann, E.A.; Wapnir, I.L.; et al. Phase II study of gemcitabine, carboplatin, and iniparib as neoadjuvant therapy for triple-negative and BRCA1/2 mutation-associated breast cancer with assessment of a tumor-based measure of genomic instability: PrECOG 0105. J. Clin. Oncol. 2015, 33, 1895–1901. [Google Scholar] [CrossRef]
- Schölkopf, B.; Smola, A.J. Elements of Statistical Learning Theory. In Learning with Kernels; Springer Series in Statistics; Springer: New York, NY, USA, 2018; p. xxii. [Google Scholar]
- Tibshirani, R.; Hastie, T.; Narasimhan, B.; Chu, G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6567–6572. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V.; Saitta, L. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Lee, D.D.; Seung, H.S. Learning the parts of objects by non-negative matrix factorization. Nature 1999, 401, 788–791. [Google Scholar] [CrossRef]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- van der Wiel, A.M.A.; Schuitmaker, L.; Cong, Y.; Theys, J.; Van Hoeck, A.; Vens, C.; Lambin, P.; Yaromina, A.; Dubois, L.J. Homologous Recombination Deficiency Scar: Mutations and Beyond—Implications for Precision Oncology. Cancers 2022, 14, 4157. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.; Stenzinger, A. Homologous recombination repair deficiency (HRD): From biology to clinical exploitation. Genes Chromosom. Cancer 2021, 60, 299–302. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Montagna, C. Chromosomal rearrangements in cancer: Detection and potential causal mechanisms. Mol. Cell. Oncol. 2014, 1, e29904. [Google Scholar] [CrossRef]
- Patel, K.J.; Yu, V.P.C.C.; Lee, H.; Corcoran, A.; Thistlethwaite, F.C.; Evans, M.J.; Colledge, W.H.; Friedman, L.S.; Ponder, B.A.J.; Venkitaraman, A.R. Involvement of Brca2 in DNA repair. Mol. Cell 1998, 1, 347–357. [Google Scholar] [CrossRef]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for homologous recombination deficiency in cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).