Homologous Recombination Deficiency Detection Algorithms: A Systematic Review

 , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Background
2. Materials and Methods
2.1. Literature Search
2.2. Inclusion and Exclusion Criteria
2.3. Data Extraction
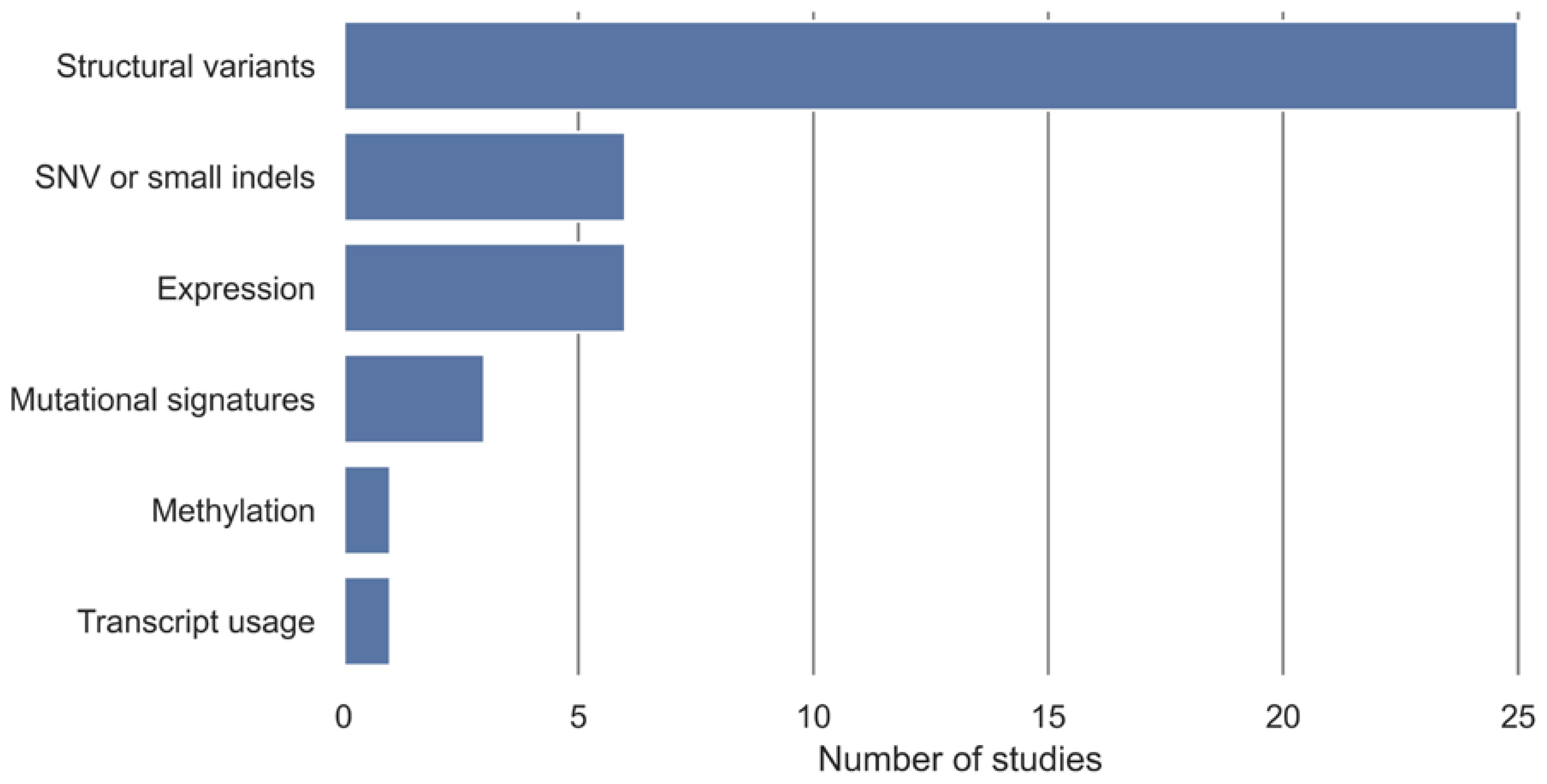
3. Results
3.1. Study Selection
3.2. Study Characteristics
3.3. Definition of HRD
3.4. HRD Detection Algorithms
3.5. HRD Test Performance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
List of Abbreviations
| HRD | homologous recombination deficiency |
| PARPi | poly (ADP-ribose) polymerase inhibitors |
| HR | homolog recombination repair |
| BRCA1 | breast cancer 1 gene |
| BRCA2 | breast cancer 2 gene |
| LOH | loss of heterozygosity |
| LST | large-scale transition |
| TAI | telomeric allelic imbalance |
| PRISMA | Preferred Reported Items for Systematic Reviews and Meta-Analysis |
| ROC | receiver operating characteristic |
| PPV | positive predictive value |
| NPV | negative predictive value |
| TCGA | The Cancer Genome Atlas |
| GEO | Gene Expression Omnibus |
| SNP | single-nucleotide polymorphism |
| arrayCGH | comparative genomic hybridization array |
| FFPE | formalin fixed paraffin embedded |
| NGS | next-generation sequencing |
| WES | whole-exome sequencing |
| WGS | whole-genome sequencing |
| RNA-seq | RNA sequencing |
| SVM | support vector machine |
| LASSO | least absolute shrinkage and selection operator |
| CHORD | Classifier of HOmologous Recombination Deficiency |
| tHRD | transcriptional HRD |
| SigMA | Signature Multivariate Analysis |
| NMF | non-negative matrix factorization |
| LGA | large-scale genomic alterations |
| HRD score | combined homologous recombination deficiency score |
| GSA | genomic scar algorithm |
| SCINS | scores of chromosomal instability scarring |
| TD-score | tandem duplications score |
| HRDS | hypothesized HR-deficiency score (HRDS) |
References
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet. Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-ribose polymerase inhibition: Frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Szostak, J.W.; Orr-Weaver, T.L.; Rothstein, R.J.; Stahl, F.W. The double-strand-break repair model for recombination. Cell 1983, 33, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-Mccune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [PubMed]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-like Breast Carcinomas with BRCA1/2 Inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.-Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric Allelic Imbalance Indicates Defective DNA Repair and Sensitivity to DNA-Damaging Agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Katsumata, N.; Yasuda, M.; Takahashi, F.; Isonishi, S.; Jobo, T.; Aoki, D.; Tsuda, H.; Sugiyama, T.; Kodama, S.; Kimura, E.; et al. Dose-dense paclitaxel once a week in combination with carboplatin every 3 weeks for advanced ovarian cancer: A phase 3, open-label, randomised controlled trial. Lancet 2009, 374, 1331–1338. [Google Scholar] [CrossRef] [PubMed]
- Welcsh, P.L.; King, M.-C. BRCA1 and BRCA2 and the genetics of breast and ovarian cancer. Hum. Mol. Genet. 2001, 10, 705–713. [Google Scholar] [CrossRef]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef] [PubMed]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Hill, S.J.; Decker, B.; Roberts, E.A.; Horowitz, N.S.; Muto, M.G.; Worley, M.J.J.; Feltmate, C.M.; Nucci, M.R.; Swisher, E.M.; Nguyen, H.; et al. Prediction of DNA Repair Inhibitor Response in Short-Term Patient-Derived Ovarian Cancer Organoids. Cancer Discov. 2018, 8, 1404–1421. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.W.; Karlan, B.Y.; Cass, L.; Baldwin, R.L. BRCA1 promoter methylation predicts adverse ovarian cancer prognosis. Gynecol. Oncol. 2006, 101, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, 105906. [Google Scholar] [CrossRef]
- Veritas Health Innovation. Covidence Systematic Review Software [Internet]. Available online: https://support.covidence.org/help/how-can-i-cite-covidence (accessed on 21 September 2022).
- Joosse, S.A.; Van Beers, E.H.; Tielen, I.H.G.; Horlings, H.; Peterse, J.L.; Hoogerbrugge, N.; Ligtenberg, M.J.; Wessels, L.F.A.; Axwijk, P.; Verhoef, S.; et al. Prediction of BRCA1-association in hereditary non-BRCA1/2 breast carcinomas with array-CGH. Breast Cancer Res. Treat. 2009, 116, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Lips, E.H.; Laddach, N.; Savola, S.P.; Vollebergh, M.A.; Oonk, A.M.; Imholz, A.L.T.; Wessels, L.F.A.; Wesseling, J.; Nederlof, P.M.; Rodenhuis, S. Quantitative copy number analysis by Multiplex Ligation-dependent Probe Amplification (MLPA) of BRCA1-associated breast cancer regions identifies BRCAness. Breast Cancer Res. 2011, 13, R107. [Google Scholar] [CrossRef]
- Joosse, S.A.; Brandwijk, K.I.M.; Devilee, P.; Wesseling, J.; Hogervorst, F.B.L.; Verhoef, S.; Nederlof, P.M. Prediction of BRCA2-association in hereditary breast carcinomas using array-CGH. Breast Cancer Res. Treat. 2012, 132, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, D.; Li, C.; Zhou, M.; Hao, D. Correlation between gene expression and mutator phenotype predicts homologous recombination deficiency and outcome in ovarian cancer. J. Mol. Med. 2014, 92, 1159–1168. [Google Scholar] [CrossRef]
- Zhang, S.; Yuan, Y.; Hao, D. A genomic instability score in discriminating nonequivalent outcomes of BRCA1/2 mutations and in predicting outcomes of ovarian cancer treated with platinum-based chemotherapy. PLoS ONE 2014, 9, e113169. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.; Weekes, D.; Shah, V.; Gazinska, P.; Joshi, S.; Sidhu, B.; Gillett, C.; Pinder, S.; Vanoli, F.; Jasin, M.; et al. Genomic complexity profiling reveals that hormad1 overexpression contributes to homologous recombination deficiency in triple-negative breast cancers. Cancer Discov. 2015, 5, 488–505. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Severson, T.M.; Wolf, D.M.; Yau, C.; Peeters, J.; Wehkam, D.; Schouten, P.C.; Chin, S.F.; Majewski, I.J.; Michaut, M.; Bosma, A.; et al. The BRCA1ness signature is associated significantly with response to PARP inhibitor treatment versus control in the I-SPY 2 randomized neoadjuvant setting. Breast Cancer Res. 2017, 19, 99. [Google Scholar] [CrossRef]
- Wang, T.; Wang, G.; Wang, G.; Zhang, X.; Wu, D.; Yang, L.; Hao, D. The expression of miRNAs is associated with tumour genome instability and predicts the outcome of ovarian cancer patients treated with platinum agents. Sci. Rep. 2017, 7, 14736. [Google Scholar] [CrossRef]
- Diossy, M.; Reiniger, L.; Sztupinszki, Z.; Krzystanek, M.; Timms, K.M.; Neff, C.; Solimeno, C.; Pruss, D.; Eklund, A.C.; Tóth, E.; et al. Breast cancer brain metastases show increased levels of genomic aberration-based homologous recombination deficiency scores relative to their corresponding primary tumors. Ann. Oncol. 2018, 29, 1948–1954. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Cafferkey, C.; Loehr, A.; Waddell, T.; Begum, R.; Peckitt, C.; Harding, T.C.; Nguyen, M.; Okines, A.F.; Raponi, M.; et al. Genomic loss of heterozygosity and survival in the REAL3 trial. Oncotarget 2018, 9, 36654–36665. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Y.; Salas, L.A.; Miller, T.W.; Mark, K.; Marotti, J.D.; Kettenbach, A.N.; Cheng, C.; Christensen, B.C. Molecular and epigenetic profiles of BRCA1-like hormone-receptor-positive breast tumors identified with development and application of a copy-number-based classifier. Breast Cancer Res. 2019, 21, 1–13. [Google Scholar] [CrossRef]
- Gulhan, D.C.; Lee, J.J.K.; Melloni, G.E.M.; Cortés-Ciriano, I.; Park, P.J. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet. 2019, 51, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Eeckhoutte, A.; Houy, A.; Manié, E.; Reverdy, M.; Bièche, I.; Marangoni, E.; Goundiam, O.; Vincent-Salomon, A.; Stoppa-Lyonnet, D.; Bidard, F.C.; et al. ShallowHRD: Detection of homologous recombination deficiency from shallow whole genome sequencing. Bioinformatics 2020, 36, 3888–3889. [Google Scholar] [CrossRef] [PubMed]
- Lips, E.H.; Benard-Slagter, A.; Opdam, M.; Scheerman, C.E.; Wesseling, J.; Hogervorst, F.B.L.; Linn, S.C.; Savola, S.; Nederlof, P.M. BRCAness digitalMLPA profiling predicts benefit of intensified platinum-based chemotherapy in triple-negative and luminal-type breast cancer. Breast Cancer Res. 2020, 22, 79. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; W M Martens, J.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef] [PubMed]
- Barenboim, M.; Kovac, M.; Ameline, B.; Jones, D.T.W.; Witt, O.; Bielack, S.; Burdach, S.; Baumhoer, D.; Nathrath, M. DNA methylation-based classifier and gene expression signatures detect BRCAness in osteosarcoma. PLoS Comput. Biol. 2021, 17, e1009562. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Shao, M.; Meng, P.; Wang, C.; Li, Q.; Cai, Y.; Song, C.; Wang, X.; Shi, T. GSA: An independent development algorithm for calling copy number and detecting homologous recombination deficiency (HRD) from target capture sequencing. BMC Bioinform. 2021, 22, 562. [Google Scholar] [CrossRef] [PubMed]
- Schouten, P.C.; Richters, L.K.; Vis, D.J.; Kommoss, S.; van Dyk, E.; Ernst, C.; Kluin, R.J.C.; Marme, F.; Lips, E.H.; Schmidt, S.; et al. Ovarian cancer specific BRCA-like copy number aberration classifiers detect mutations associated with homologous recombination deficiency in the AGO-TR1 trial. Clin. Cancer Res. 2021, 27, 6559–6569. [Google Scholar] [CrossRef]
- Zhuang, S.; Chen, T.; Li, Y.; Wang, Y.; Ai, L.; Geng, Y.; Zou, M.; Liu, K.; Xu, H.; Wang, L.; et al. A transcriptional signature detects homologous recombination deficiency in pancreatic cancer at the individual level. Mol. Ther.—Nucleic Acids 2021, 26, 1014–1026. [Google Scholar] [CrossRef]
- Kang, H.G.; Hwangbo, H.; Kim, M.J.; Kim, S.; Lee, E.J.; Park, M.J.; Kim, J.W.; Kim, B.G.; Cho, E.H.; Chang, S.; et al. Aberrant Transcript Usage Is Associated with Homologous Recombination Deficiency and Predicts Therapeutic Response. Cancer Res. 2022, 82, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, B.D.; Dougherty, B.V.; Bell, J.S.K.; Kapilivsky, J.; Michuda, J.; Sedgewick, A.J.; Munson, W.A.; Chandra, T.A.; Dry, J.R.; Beaubier, N.; et al. Validation of Genomic and Transcriptomic Models of Homologous Recombination Deficiency in a Real-World Pan-Cancer Cohort. MedRxiv 2022, 22, 587. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.; Yang, Y.; Xie, A.; Jiang, Z.; Liao, J.; Yan, M.; Zhou, Y.; Zhu, J.; Hu, J.; Zhang, Y.; et al. Applicability of Anticancer Drugs for the Triple-Negative Breast Cancer Based on Homologous Recombination Repair Deficiency. Front. Cell Dev. Biol. 2022, 10, 457. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Martens, J.W.M.; Hollestelle, A.; Smid, M. Identifying Transcripts with Tandem Duplications from RNA-Sequencing Data to Predict BRCA1-Type Primary Breast Cancer. Cancers 2022, 14, 753. [Google Scholar] [CrossRef]
- National Cancer Institute. The Cancer Genome Atlas Program [Internet]. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 21 September 2022).
- METABRIC. Breast Cancer (METABRIC, Nature 2012 & Nat Commun 2016) [Internet]. 2016. Available online: https://www.cbioportal.org/study/summary?id=brca_metabric (accessed on 12 September 2022).
- NCBI. Gene Expression Omnibus [Internet]. 2022. Available online: https://www.ncbi.nlm.nih.gov/geo/ (accessed on 12 September 2022).
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Telli, M.L.; Jensen, K.C.; Vinayak, S.; Kurian, A.W.; Lipson, J.A.; Flaherty, P.J.; Timms, K.; Abkevich, V.; Schackmann, E.A.; Wapnir, I.L.; et al. Phase II study of gemcitabine, carboplatin, and iniparib as neoadjuvant therapy for triple-negative and BRCA1/2 mutation-associated breast cancer with assessment of a tumor-based measure of genomic instability: PrECOG 0105. J. Clin. Oncol. 2015, 33, 1895–1901. [Google Scholar] [CrossRef]
- Schölkopf, B.; Smola, A.J. Elements of Statistical Learning Theory. In Learning with Kernels; Springer Series in Statistics; Springer: New York, NY, USA, 2018; p. xxii. [Google Scholar]
- Tibshirani, R.; Hastie, T.; Narasimhan, B.; Chu, G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6567–6572. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V.; Saitta, L. Support-Vector Networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Lee, D.D.; Seung, H.S. Learning the parts of objects by non-negative matrix factorization. Nature 1999, 401, 788–791. [Google Scholar] [CrossRef]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.-J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination–Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- van der Wiel, A.M.A.; Schuitmaker, L.; Cong, Y.; Theys, J.; Van Hoeck, A.; Vens, C.; Lambin, P.; Yaromina, A.; Dubois, L.J. Homologous Recombination Deficiency Scar: Mutations and Beyond—Implications for Precision Oncology. Cancers 2022, 14, 4157. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, D.; Stenzinger, A. Homologous recombination repair deficiency (HRD): From biology to clinical exploitation. Genes Chromosom. Cancer 2021, 60, 299–302. [Google Scholar] [CrossRef]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [PubMed]
- Hasty, P.; Montagna, C. Chromosomal rearrangements in cancer: Detection and potential causal mechanisms. Mol. Cell. Oncol. 2014, 1, e29904. [Google Scholar] [CrossRef]
- Patel, K.J.; Yu, V.P.C.C.; Lee, H.; Corcoran, A.; Thistlethwaite, F.C.; Evans, M.J.; Colledge, W.H.; Friedman, L.S.; Ponder, B.A.J.; Venkitaraman, A.R. Involvement of Brca2 in DNA repair. Mol. Cell 1998, 1, 347–357. [Google Scholar] [CrossRef]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for homologous recombination deficiency in cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef]
- Fawcett, T. An introduction to ROC analysis. Pattern Recognit. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author (et al.) | Year | Algorithm | Cancer Type | Cohort | Cohort Size | Tumor Tissue Type | Method | Algorithm Description |
|---|---|---|---|---|---|---|---|---|
| Joosse [22] | 2009 | BRCA1 classifier | Breast | gBRCA1 mutated | 34 T | FFPE | Array-CGH | Shrunken centroid model |
| Sporadic | 48 T | |||||||
| HBOC | 48 V | |||||||
| Lips [23] | 2011 | BRCA1-like MLPA classifier | Breast | NKI-clinical genetics series | 34 T 18 V | FFPE | MLPA | Nearest shrunken centroid model |
| NKI-AVL neoadjuvant chemotherapy | 50 T 8 V | Frozen | ||||||
| Randomized trial series | 46 V | FFPE | ||||||
| Deventer series | 69 A | FFPE | ||||||
| Abkevich [6] | 2012 | HRD-LOH | Ovarian | Gynecology Cancer Banks at MDACC and UCSF | 152 T | Frozen | SNP array | Sum of LOH segment counts |
| Magee-Womens Hospital of UPMC | 53 V | |||||||
| TCGA ovarian cancer | 435 V | |||||||
| Joosse [24] | 2012 | BRCA2 classifier | Breast | gBRCA2 mutated | 28 T 19 V | FFPE | Array-CGH | Shrunken centroid model |
| Sporadic | 28 T 19 V | |||||||
| HBOC | 89 V | |||||||
| gBRCA1 mutated (Joosse et al. 2009) | 34 A | |||||||
| Popova [7] | 2012 | LST | Breast | BLC | 80 T 60 V | Frozen | SNP array | Two-step decision rule. First, segregate tumors based on ploidy and second, segregate according to number of LST counts. |
| Lu [25] | 2014 | Hypothesized HR-deficiency score (HRDS) | Breast Ovarian | TCGA ovarian cancer | 167 T 141 V | Frozen | WES | Score based on gene expression levels |
| TCGA breast cancer | 127 A | Frozen | ||||||
| Bonome dataset | 185 A | Frozen | ||||||
| Yoshihar dataset | 300 A | Frozen | ||||||
| Tothill dataset | 285 A | Frozen | ||||||
| Zhang [26] | 2014 | Genomic instability score | Ovarian | TCGA ovarian cancer | 325 T | Frozen | NGS panel SNP array | Score based on CNC regions and somatic mutations |
| Watkins [27] | 2015 | Scores of chromosomal instability scarring (SCINS) | Breast Ovarian | Guy’s Hospital King’s College London TNBC | 142 A | Frozen | SNP array Gene expression microarray | Four scores based on different types of allele-specific copy-number profiles |
| METABRIC TNBC | 115 A | Frozen | ||||||
| TCGA TNBC | 80 A | Frozen | ||||||
| PrECOG TNBC | 80 A | Frozen | ||||||
| TCGA HGSC | 299 A | Frozen | ||||||
| Telli [16] | 2016 | Combined homologous recombination deficiency score (HRD score) | Breast Ovarian | Breast cancer: TCGA Timms et al. 2014 cohort | 497 T | Frozen | Microarray SNP array WES Capture panel NGS | Numeric sum of LOH, LST, and TAI counts |
| Ovarian cancer: TCGA Hennesy et al. 2010 | 561 T | Frozen | ||||||
| Breast cancer: PrECOG 0105 | 93 A | FFPE Frozen | ||||||
| Breast cancer: Neoadjuvant cisplatin trials | 79 A | FFPE Frozen | ||||||
| Davies [28] | 2017 | HRDetect | Breast Ovarian Pancreatic | Nik-Zainal et al. 2016 cohort | 560 T | Frozen | WGS | LASSO logistic regression model |
| Low coverage simulated Nik-Zainal et al. 2016 cohort | 560 V | N/A | ||||||
| Breast cancer | 80 V | N/A | ||||||
| Pancreatic cancer | 96 V | Frozen | ||||||
| Breast cancer | 3 V | FFPE | ||||||
| Ovarian cancer | 73 V | Frozen | ||||||
| TNBC | 9 A | Needle biopsy | ||||||
| Severson [29] | 2017 | BRCA1ness signature | Breast | RATHER cohort | 128 T | Frozen | Array | Nearest centroid model |
| I-SPY 2 trial | 116 V | |||||||
| Wang [30] | 2017 | 10-miRNA-score | Ovarian | TCGA ovarian cancer | 319 A | Frozen | miRNA microarray miRNA-Seq | Score based on miRNA expression levels |
| TCGA ovarian cancer samples | 136 A | miRNA-Seq | ||||||
| TCGA breast cancer | 657 A | miRSeq | ||||||
| Diossy [31] | 2018 | WES-HRDetect | Breast Brain metastases | Matched primary breast cancer and brain metastasis | 21 T | FFPE Frozen | WES | LASSO logistic regression model |
| 17 V | FFPE | |||||||
| Smyth [32] | 2018 | Genomic LOH | Esophagogastric | REAL3 cohort | 158 T | FFPE | NGS panel | Sum of the lengths of included LOH segments divided by the length of the interrogated genome. |
| Chen [33] | 2019 | BRCA1-like classifier | Breast | GSE9021 GSE9114 | 74 T | FFPE | Array-CGH | Support vector machine |
| GSE18626 | 106 V | FFPE | ||||||
| TCGA breast cancer | 957 A | Frozen | ||||||
| METABRIC breast | 1968 A | Frozen | ||||||
| Gulhan [34] | 2019 | Signature Multivariate Analysis (SigMA) | Breast Osteosarcoma Ovarian Pancreatic Prostate | TCGA Breast cancer | 730 T | Frozen | WGS | Likelihood-based measure combined with clustering using non-negative matrix factorization |
| Down-sampled TCGA breast cancer | 730 T | Simulated | Down-sampled WGS | |||||
| Breast cancer (MSK-IMPACT data) | 878 V | FFPE | Capture panel NGS | |||||
| Nik-Zainal et al. 2016 cohort | 560 V | Frozen | WGS | |||||
| Eeckhoutte [35] | 2020 | ShallowHRD | Breast Ovarian | Primary breast and ovarian cancer | 26 T | Frozen | Shallow WGS | Sum of LGA counts |
| Primary breast and ovarian cancer | 4 T | FFPE | Shallow WGS | |||||
| Patient-derived xenografts | 39 T | Frozen | Shallow WGS | |||||
| TCGA-BRCA | 108 normal T 79 tumor V | N/A | Down-sampled WGS | |||||
| Lips [36] | 2020 | BRCA1-like digitalMLPA classifier BRCA2-like digitalMLPA classifier | Breast | Cohort for BRCA1-like digitalMLPA classifier | 71 T 70 V | FFPE Frozen | digitalMLPA | Shrunken centroid model |
| Cohort for BRCA2-like digitalMLPA classifier | 55 T 56 V | |||||||
| The Dutch high-dose trial | 122 A | |||||||
| Nguyen [37] | 2020 | Classifier of HOmologous Recombination Deficiency (CHORD) | Pan-cancer | Metastatic Pan-cancer (HMF Priestley) | 3824 T | Frozen | WGS | Random-forest-based model |
| Primary pan-cancer (PCAWG) | 1854 V | |||||||
| Nik-Zainal et al. 2016 cohort | 560 V | |||||||
| Barenboim [38] | 2021 | DNA-methylation-based RF classifier | Osteosarcoma | Osteosarcoma | 43 T 20 V | Frozen | RNA-seq | Random forest model |
| Chen [39] | 2021 | Genomic scar algorithm (GSA) | Breast Ovarian | Breast and ovarian cancer | 195 T | FFPE | MGI panel sequencing | Numeric sum of LST, TAI, LOH subtracted by correction coefficient multiplied a ploidy value |
| Schouten [40] | 2021 | Ovarian cancer BRCA1-like classifier Ovarian cancer BRCA2-like classifier | Ovarian | NKI and EMI cohort | 73 T | FFPE | Array-CGH | Shrunken centroids classifier |
| AGO-TR1 | 523 A | FFPE blood | Low-coverage WGS | |||||
| Zhuang [41] | 2021 | 24 gene pairs (24-GPS) | Pancreatic | TCGA | 147 T | Frozen, blood | RNA-seq | LASSO regression model |
| ICGC-AU | 95 V | N/A | Gene expression array | |||||
| GSE17891 | 27 V | FFPE | ||||||
| GSE57495 | 63 V | Frozen | ||||||
| Kang [42] | 2022 | Transcriptional HRD (tHRD) | Breast Ovarian | TCGA-BRCA | 272 T | Frozen | RNA-seq WGS WES | Random-forest- based model |
| 116 V | ||||||||
| TCGA-OV | 130 T | |||||||
| 32 V | ||||||||
| NAC | 27 A | Frozen FFPE | ||||||
| PR | 36 A | |||||||
| OM | 24 A | |||||||
| OS | 33 A | |||||||
| Leibowitz [43] | 2022 | HRD-DNA | Pan-cancer | Breast cancer | 483 T | FFPE Blood | NGS panel | gwLOH |
| 64 V | ||||||||
| 1511 A | ||||||||
| Ovarian cancer | 289 T | |||||||
| 69 V | ||||||||
| 858 A | ||||||||
| HRD-RNA | Pancreatic cancer | 1375 T | RNA-seq panel | Logistic regression model | ||||
| 301 D | ||||||||
| 165 V | ||||||||
| 1927 A | ||||||||
| Prostate cancer | 925 T | |||||||
| 204 D | ||||||||
| 119 V | ||||||||
| 1536 A | ||||||||
| Other | 9921 T | |||||||
| 2125 D | ||||||||
| 1113 V | ||||||||
| 20772 A | ||||||||
| Liao [44] | 2022 | Transcriptomic HRD score | Breast | TCGA | 1084 T | Frozen | WES Gene expression array | LASSO logistic regression model |
| GSE25055 | 114 A | Fine-needle aspiration core biopsy | Gene expression array | |||||
| GSE25065 | 64 A | Fine-needle aspiration core biopsy | Gene expression array | |||||
| GSE41998 | 140 A | Frozen | Gene expression array | |||||
| METABRIC | 299 A | Frozen | Gene expression array | |||||
| Nik-Zainal et al. 2016 cohort | 75 V | Frozen | WGS Gene expression array | |||||
| Qu [45] | 2022 | Tandem duplications score (TD-score) | Breast | Nik-Zainal et al. 2016 cohort | 266 T | Frozen | RNA-seq WGS | Score of TD counts |
| Author (et al.) | Algorithm | Algorithm Input | Study Type a | Validation | Performance | Gold Standard of HRD |
|---|---|---|---|---|---|---|
| Joosse [22] | BRCA1 classifier | Copy number | Predictive | External | Sensitivity: 88% Specificity: 94% PPV: 93% NPV: 88% | BRCA1 germline variants |
| Lips [23] | BRCA1-like MLPA classifier | Copy number | Predictive Explanatory | External | Sensitivity: 85% Specificity: 87% Accuracy: 86% | Algorithm developed by Joosse et al. [22] |
| Abkevich [6] | HRD-LOH | LOH | Explanatory | No validation | N/A | BRCA1/2 methylation, germline, and somatic variants LOH BRCA1 expression |
| Joosse [24] | BRCA2 classifier | Copy number | Predictive | External | Sensitivity: 89% Specificity: 84% PPV: 85% NPV: 89% | BRCA2 germline variants |
| Popova [7] | LST | LST Ploidy | Predictive | External | Validation: Sensitivity: 100% Specificity: 54% | BRCA1/2 germline and somatic variants BRCA1 promoter methylation |
| Lu [25] | HRDS | Gene expression | Descriptive Explanatory | No validation | N/A | BRCA1/2 variants |
| Zhang [26] | Genomic instability score | Copy number Point mutation Indels | Explanatory | No validation | N/A | BRCA1/2 variants BRCA1 methylation |
| Watkins [27] | SCINS | Copy number | Descriptive Explanatory | No validation | N/A | Copy number measure |
| Telli [16] | HRD score | LOH LST TAI | Explanatory | External | PrECOG 0105: Sensitivity: 100% a Specificity: 41.6% a Neoadjuvant cisplatin trials cohort: Sensitivity: 87.5% a Specificity: 51.3% a | BRCA1/2 variants LOH BRCA1 methylation |
| Davies [28] | HRDetect | Mutational signatures LOH Indels | Predictive | External | Breast cancer cohort: Sensitivity: 86% Low-coverage WGS breast cancer cohort: Sensitivity 86% Ovarian and pancreatic cancer cohort: Sensitivity: approaching 100% | BRCA1/2 variants |
| Severson [29] | BRCA1ness signature | Gene expression | Predictive Explanatory | Internal | Sensitivity: 96.7% (T) Specificity: 73.1% (T) | Algorithm developed by Lips et al. [23]. |
| Wang [30] | 10-miRNA-score | miRNA expression | Descriptive Explanatory | No validation | N/A | Expression in HR genes |
| Diossy [31] | WES-HRDetect | Mutational signatures LOH Indels | Predictive/ Descriptive | External | Sensitivity 76.6% AUC: 96% | LOH LST TAI BRCA1/2 variants |
| Smyth [32] | Genomic LOH | Percentage of genomic LOH | Explanatory | No validation | N/A | Genomic LOH |
| Chen [33] | BRCA1-like classifier | Copy number | Predictive | External | AUC: 75% | MLPA assay (MRC-Holland) |
| Gulhan [34] | SigMA | Mutational signatures | Predictive Explanatory | Internal b | Accuracy: 84% Sensitivity: 74% | Mutational Signature 3 |
| Eeckhoutte [35] | ShallowHRD | Large-scale genomic alterations (LGA) | Predictive | External | Sensitivity: 87.5% Specificity: 90.5% | Variants or LOH in BRCA1/2, RAD51C, PALB2 Methylation of BRCA1 and RAD51C |
| Lips [36] | BRCA1-like digitalMLPA classifier BRCA2-like digitalMLPA classifier | Copy number | Predictive | External | BRCA1-like digitalMLPA classifier: Sensitivity: 93% Specificity: 90% Accuracy: 91% BRCA2-like digitalMLPA classifier: Sensitivity: 75% Specificity: 89% Accuracy: 82% | Algorithms developed by Joosse et al. [24] and Joosse et al. [22] |
| Nguyen [37] | CHORD | Single-base substitution Indels Structural variants | Predictive | External | Cohort 1: AUC: 98.7% Cohort 2: AUC: 99.5% | BRCA1/2 complete copy number loss LOH Germline or somatic variants in BRCA1/2 |
| Barenboim [38] | DNA-methylation based RF classifier | Methylation copy number | Predictive | External | Sensitivity: 93% Specificity: 83% AUC: 87% Accuracy: 90% | Percent of genome change (PCG) score based on CNA, TAI, and LOH |
| Chen [39] | GSA | LOH LST TAI Ploidy | Predictive | Internal | Sensitivity: 95.2% (T) Specificity: 78.4% (T) AUC: 88.3 (T) | BRCA1/2 variants LOH BRCA1 methylation |
| Schouten [40] | Ovarian cancer BRCA1-like classifier Ovarian cancer BRCA2-like classifier | Copy number | Predictive | External | Ovarian cancer BRCA1-like classifier: Sensitivity: 96.2% Specificity: 40% Ovarian cancer BRCA2-like classifier: Sensitivity: 77% Specificity: 41% | BRCA1/2 germline and somatic variants BRCA1 methylation |
| Zhuang [41] | 24-GPS | Gene expression | Predictive Explanatory | Internal | AUC: 98% (T) | Gene expression |
| Kang [42] | tHRD | Transcript usage | Predictive Explanatory | External | OC model: Accuracy: 72% BC model: Accuracy: 84% | LOH LST TAI Mutation Signature 3 |
| Leibowitz [43] | HRD-DNA HRD-RNA | LOH Gene expression | Predictive Explanatory | External | HRD-DNA: Breast Sensitivity: 100% Specificity: 96.3% AUC: 100% F1: 98.3% HRD-DNA: Ovarian Sensitivity: 92.1% Specificity: 100% AUC: 99.3% F1: 95.9% HRD-RNA: prostate cancer Sensitivity: 85% Specificity: 98% AUC: 98% F1: 88% HRD-RNA: pancreatic cancer Sensitivity: 53% Specificity: 100% AUC: 98% F1: 69% | Biallelic loss of BRCA 1/2 |
| Liao [44] | Transcriptomic HRD score | Gene expression | Predictive Explanatory | External | AUC: 79% | LOH LST TAI Deleterious BRCA1/2 variants |
| Qu [45] | TD-score | Tandem duplications | Predictive Explanatory | Internal | AUC: 87% (T) Sensitivity: 88.2% (T) Specificity: 64.7% (T) | BRCA1-type HRD phenotype by CHORD [37] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mark, L.R.; Terp, S.K.; Krarup, H.B.; Thomassen, M.; Pedersen, I.S.; Bøgsted, M. Homologous Recombination Deficiency Detection Algorithms: A Systematic Review. Cancers 2023, 15, 5633. https://doi.org/10.3390/cancers15235633
Mark LR, Terp SK, Krarup HB, Thomassen M, Pedersen IS, Bøgsted M. Homologous Recombination Deficiency Detection Algorithms: A Systematic Review. Cancers. 2023; 15(23):5633. https://doi.org/10.3390/cancers15235633
Chicago/Turabian StyleMark, Lasse Ringsted, Simone Karlsson Terp, Henrik Bygum Krarup, Mads Thomassen, Inge Søkilde Pedersen, and Martin Bøgsted. 2023. "Homologous Recombination Deficiency Detection Algorithms: A Systematic Review" Cancers 15, no. 23: 5633. https://doi.org/10.3390/cancers15235633
APA StyleMark, L. R., Terp, S. K., Krarup, H. B., Thomassen, M., Pedersen, I. S., & Bøgsted, M. (2023). Homologous Recombination Deficiency Detection Algorithms: A Systematic Review. Cancers, 15(23), 5633. https://doi.org/10.3390/cancers15235633