
Tumour Growth Models of Breast Cancer for Evaluating Early Detection—A Summary and a Simulation Study

Abstract
:Simple Summary
Abstract
1. Introduction
2. Inference Methods
2.1. Components of Models of Breast Cancer Tumour Progression
- Onset of the primary tumour—onset is defined as the point at which a tumour reaches a diameter of 0.5 mm (from which it can reasonably be assumed to have deterministic growth), and its distribution is defined according to the Moolgavkar-Venson-Knudson carcinogenesis model [24]. Tumours are assumed to be spherical.
- Growth of the primary tumour—tumour volume is assumed to follow an exponential growth function where the inverse growth rate is a gamma random effect.
- Lymph node spread—a nonhomogeneous Poisson process with rate of spread assumed to be proportional to the number of cell divisions (raised to a power) and the rate of growth of the primary tumour.
- Symptomatic detection—the hazard rate of symptomatic detection is proportional to the (latent) tumour volume.
- Detection via mammography—screening test sensitivity follows a logistic function of the (latent) tumour diameter.
2.2. Likelihood Inference for Incident Cases and Cohort Designs
2.2.1. Continuous Growth Models for Collection of Incident Cases
- Pfree—a disease-free state (prior to breast cancer tumour onset).
- Ptumour—a breast cancer state (preclinical/as yet undetected).
- Pafter—a post-symptomatic detection state.
- The rate of births in the population is constant across calendar time.
- The distribution of age at tumour onset is constant across calendar time.
- The distribution of time to symptomatic detection is constant across calendar time.
2.2.2. Continuous Growth Models for Screening Cohorts
2.3. Simulation-Based and Inference-Based Evaluations of Early Detection
3. A Simulation Study—Extending the Age of Screening Participation
3.1. Parameter Values Used in the Simulation
3.2. Description of the Simulation Approach
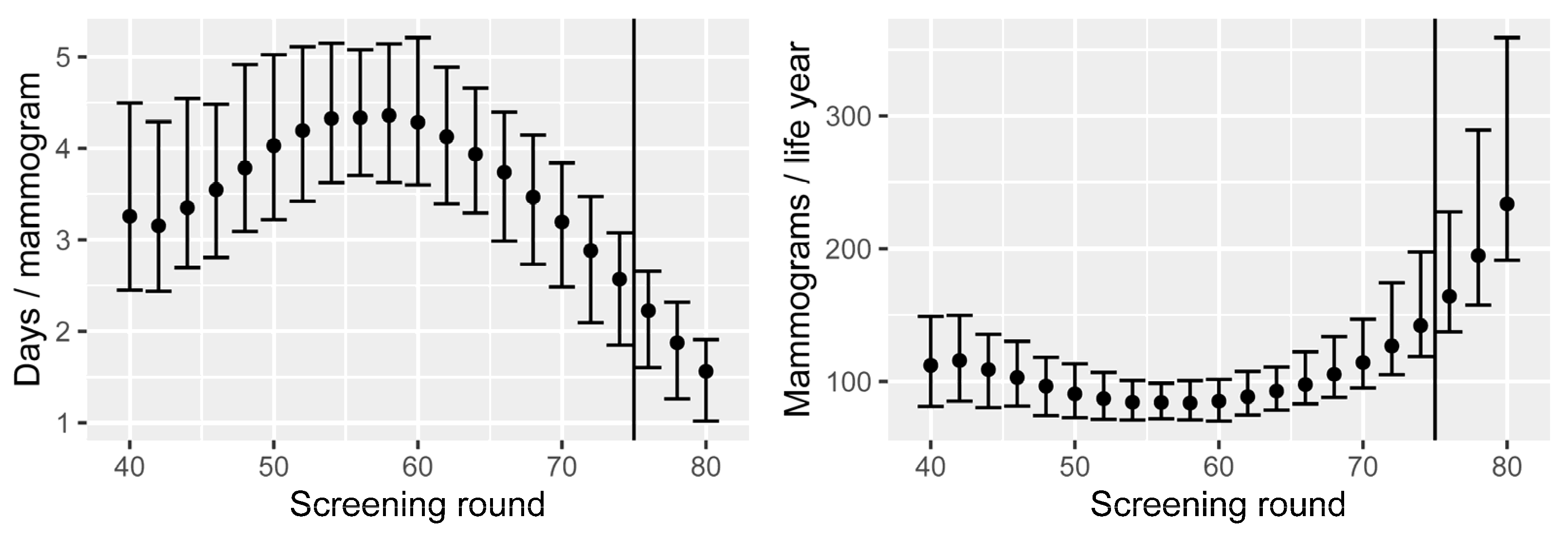
- Number of mammograms performed.
- Number of breast cancer cases.
- Number of cases detected through screening.
- Number of overdiagnosed cases.
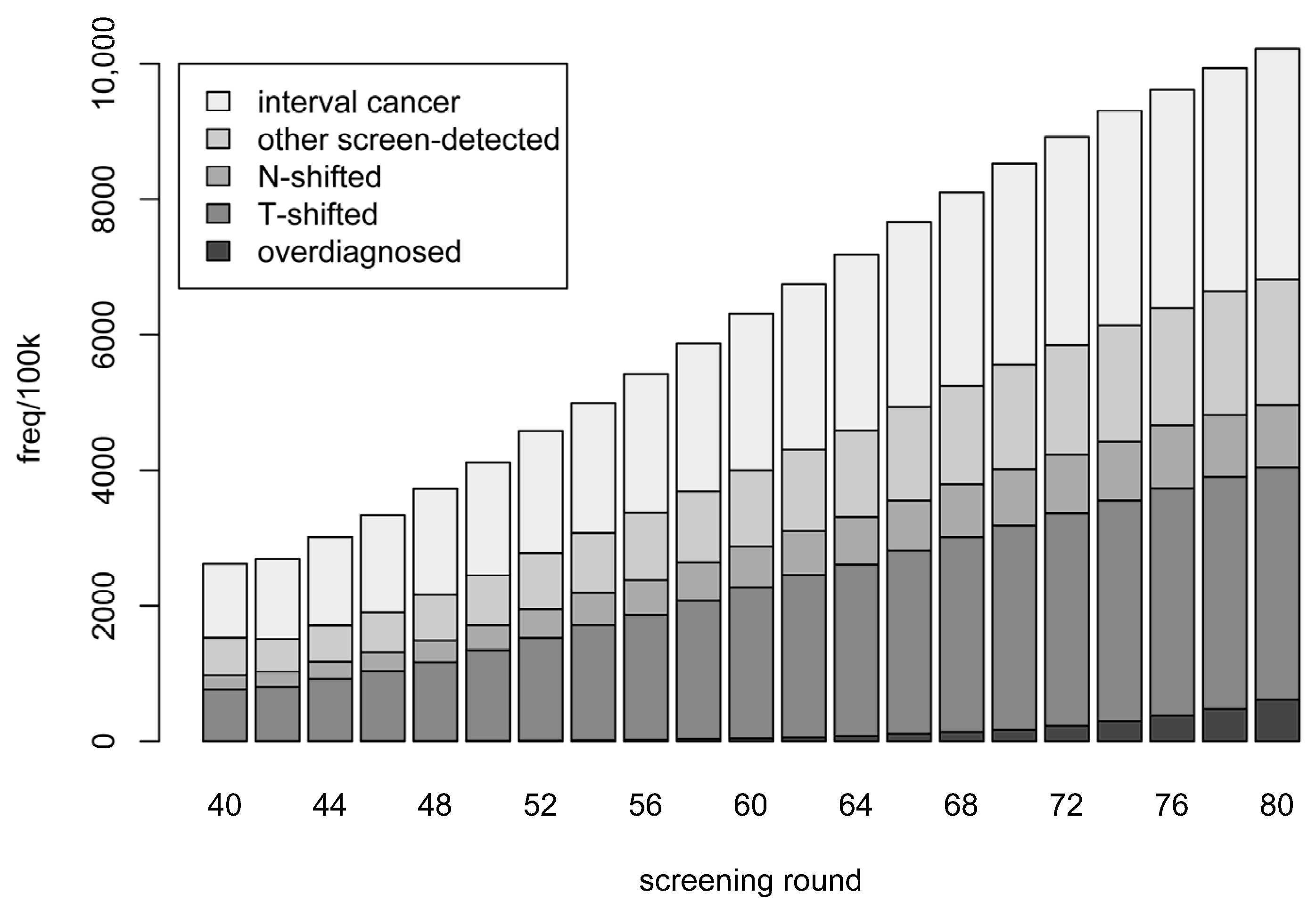
- Stage shift, which occurs when early detection causes the stages of either the primary tumour size or the number of affected lymph nodes to shift down one or more levels according to the categorisation as described in Table 1 (T: primary tumour or N: lymph node metastasis).
- Number of breast cancer deaths.
- Lead time, the time ”gained” between screen detection and would-be symptomatic detection.
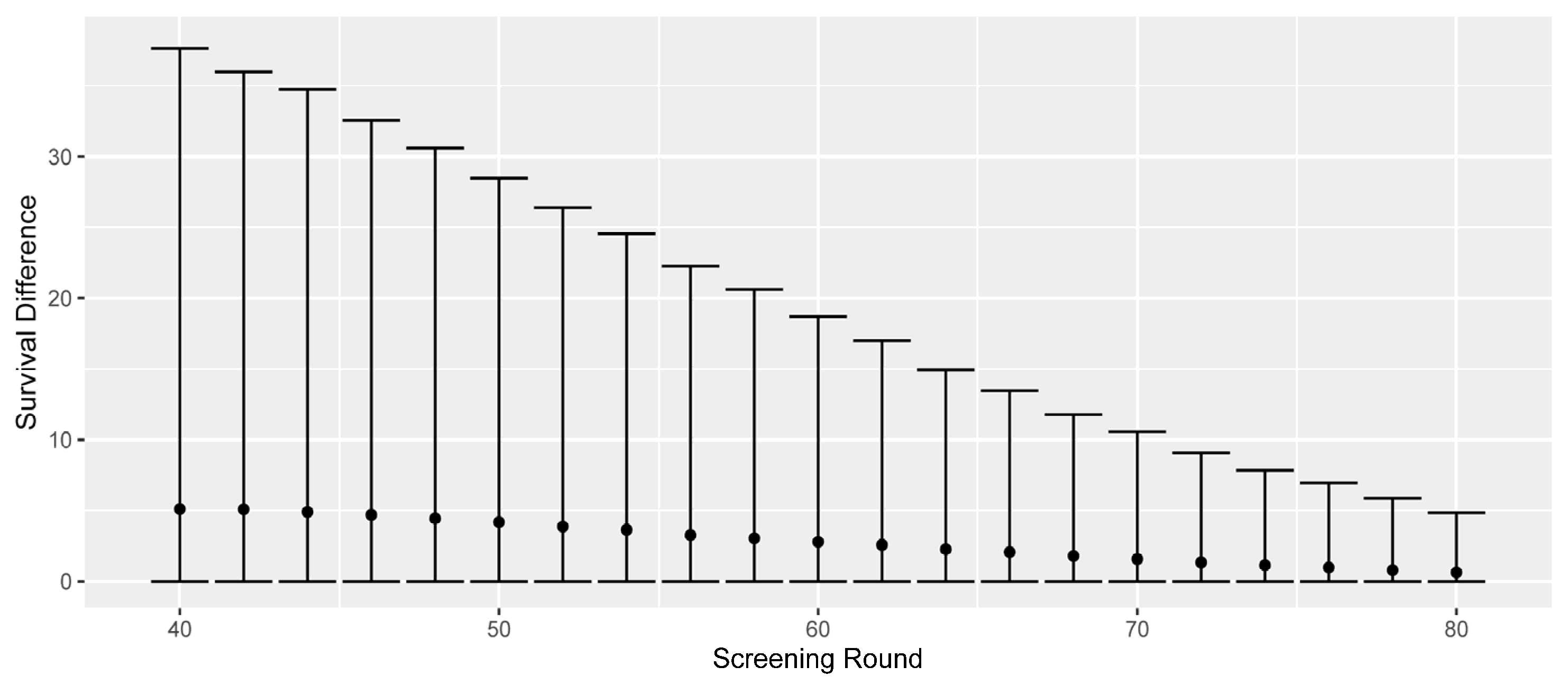
- Survival differences (all causes, difference between screen-detected and would-be symptomatic diagnosis).
3.3. Results
- Overdiagnosed—screen-detected cancer but has a non-cancer-related death before the time she would have been symptomatically detected.
- T-shifted—cases that were not overdiagnosed and where the T-stage was shifted due to early detection (but N-stage was not shifted).
- N-shifted—cases that were not overdiagnosed and where the N-stage was shifted. (This category also includes cases which where both T- and N-shifted.)
- Other screen-detected—the remainder of the screen-detected cases.
- Interval cancer—detected symptomatically between the current and next screening rounds.
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A. Submodels and Parameter Values

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Estimate | 2.5% | 97.5% |
|---|---|---|---|
| A | −0.0679 | −0.0329 | −0.1402 |
| B | 0.0017 | 0.0009 | 0.0033 |
| δ | 0.0990 | 0.0242 | 0.4055 |
| µ | 0.9706 | 0.7557 | 1.2467 |
| φ | 0.5266 | 0.4304 | 0.6443 |
| ln(η) | −8.8015 | −8.9698 | −8.6333 |
| β0 | −4.8344 | −5.2303 | −4.4386 |
| βs | 0.4948 | 0.4391 | 0.5504 |
| k | 1.9526 | 1.3950 | 2.7330 |
| ln(γ1) | −1.4237 | −1.5807 | −1.2667 |
| ln(γ2) | 5.7300 | 4.1528 | 7.3070 |
Appendix B. Survival Functions

| N-Stage | N0 | N1 | N2 | N3 |
|---|---|---|---|---|
| Hazard ratio | 1 | 1.35 | 2.19 | 3.48 |
References
- The Independent UK Panel on Breast Cancer Screening. The benefits and harms of breast cancer screening: An independent review. Lancet 2012, 380, 1778–1786. [Google Scholar] [CrossRef]
- Marmot, M.G.; Altman, D.G.; Cameron, D.A.; Dewar, J.A.; Thompson, S.G.; Wilcox, M. The benefits and harms of breast cancer screening: An independent review. Br. J. Cancer 2013, 108, 2205–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhry, Z.; Hédelin, G.; Colonna, M.; Asselain, B.; Arveux, P.; Rogel, A.; Exbrayat, C.; Guldenfels, C.; Courtial, I.; Soler-Michel, P.; et al. Multi-state Markov models in cancer screening evaluation: A brief review and case study. Stat. Methods Med. Res. 2010, 19, 463–486. [Google Scholar] [CrossRef]
- Duffy, S.W.; Chen, H.-H.; Tabar, L.; Day, N.E. Estimation of mean sojourn time in breast cancer screening using a Markov chain model of both entry to and exit from the preclinical detectable phase. Stat. Med. 1995, 14, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Alagoz, O.; Berry, D.A.; de Koning, H.J.; Feuer, E.J.; Lee, S.J.; Plevritis, S.K.; Schechter, C.B.; Stout, N.K.; Trentham-Dietz, A.; Mandelblatt, J.S. Introduction to the Cancer Intervention and Surveillance Modeling Network (CISNET) Breast Cancer Models. Med. Decis. Mak. 2018, 38, 3S–8S. [Google Scholar] [CrossRef]
- Berry, D.A.; Cronin, K.A.; Plevritis, S.K.; Fryback, D.G.; Clarke, L.; Zelen, M.; Mandelblatt, J.S.; Yakovlev, A.Y.; Habbema, J.D.F.; Feuer, E.J. Effect of Screening and Adjuvant Therapy on Mortality from Breast Cancer. N. Engl. J. Med. 2005, 353, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Plevritis, S.K.; Munoz, D.; Kurian, A.W.; Stout, N.K.; Alagoz, O.; Near, A.M.; Lee, S.J.; Broek, J.J.V.D.; Huang, X.; Schechter, C.B.; et al. Association of Screening and Treatment With Breast Cancer Mortality by Molecular Subtype in US Women, 2000–2012. JAMA 2018, 319, 154–164. [Google Scholar] [CrossRef]
- Cristiansen, S.R.; Autier, P.; Stovring, H. Change in effectiveness of mammography screening with decreasing breast cancer mortality: A population-based study. Eur. J. Public Health 2022, 32, 630–635. [Google Scholar] [CrossRef]
- Trentham-Dietz, A.; Alagoz, O.; Chapman, C.; Huang, X.; Jayasekera, J.; van Ravesteyn, N.T.; Lee, S.J.; Schechter, C.B.; Yeh, J.M.; Plevritis, S.K.; et al. Reflecting on 20 years of breast cancer modeling in CISNET: Recommendations for future cancer systems modeling efforts. PLoS Comput. Biol. 2021, 17, e1009020. [Google Scholar] [CrossRef]
- Brewer, N.T.; Salz, T.; Lillie, S.E. Systematic review: The long-term effects of false-positive mammograms. Ann. Intern. Med. 2007, 146, 502–510. [Google Scholar] [CrossRef]
- Román, R.; Sala, M.; De la Vega, M.; Natal, C.; Galceran, J.; Gonzalez-Roman, I.; Baroja, A.; Zubizarreta, R.; Ascunce, N.; Salas, D.; et al. Effect of false- positives and women’s char-acteristics on long-term adherence to breast cancer screening. Breast Cancer Res. Treat. 2011, 130, 543–552. [Google Scholar] [CrossRef]
- Gunsoy, N.B.; Garciaclosas, M.; Moss, S.M. Estimating breast cancer mortality reduction and overdiagnosis due to screening for different strategies in the United Kingdom. Br. J. Cancer 2014, 110, 2412–2419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryser, M.D.; Lange, J.; Inoue, L.Y.; O’Meara, E.S.; Gard, C.; Miglioretti, D.L.; Bulliard, J.-L.; Brouwer, A.F.; Hwang, E.S.; Etzioni, R.B. Estimation of Breast Cancer Overdiagnosis in a U.S. Breast Screening Cohort. Ann. Intern. Med. 2022, 175, 471–478. [Google Scholar] [CrossRef]
- Clift, A.K.; Dodwell, D.; Lord, S.; Petrou, S.; Brady, S.M.; Collins, G.S.; Hippisley-Cox, J. The current status of risk-stratified breast screening. Br. J. Cancer 2021, 126, 533–550. [Google Scholar] [CrossRef]
- Waters, E.A.; Taber, J.M.; McQueen, A.; Housten, A.J.; Studts, J.L.; Scherer, L.D. Translating Cancer Risk Prediction Models into Personalized Cancer Risk Assessment Tools: Stumbling Blocks and Strategies for Success. Cancer Epidemiol. Biomark. Prev. 2020, 29, 2389–2394. [Google Scholar] [CrossRef]
- The Age Limit of Mammography is Being Investigated by the National Board of Health and Welfare. Available online: https://www.dn.se/sverige/aldersgrans-for-mammografi-utreds-av-socialstyrelsen/ (accessed on 29 October 2022).
- Weedon-Fekjær, H.; Tretli, S.; Aalen, O.O. Estimating screening test sensitivity and tumour progression using tumour size and time since previous screening. Stat. Meth. Med. Res. 2010, 19, 507–527. [Google Scholar] [CrossRef] [PubMed]
- Weedon-Fekjaer, H.; Lindqvist, B.H.; Vatten, L.J.; Aalen, O.O.; Tretli, S. Breast cancer tumor growth estimated through mammography screening data. Breast Cancer Res. 2008, 10, R41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plevritis, S.K.; Salzman, P.; Sigal, B.M.; Glynn, P.W. A natural history model of stage progression applied to breast cancer. Stat. Med. 2007, 26, 581–595. [Google Scholar] [CrossRef]
- Abrahamsson, L.; Humphreys, K. A statistical model of breast cancer tumour growth with estimation of screening sensitivity as a function of mammographic density. Stat. Meth. Med. Res. 2016, 25, 1620–1637. [Google Scholar] [CrossRef]
- Strandberg, J.R.; Humphreys, K. Statistical models of tumour onset and growth for modern breast cancer screening cohorts. Math. Biosci. 2019, 318, 108270. [Google Scholar] [CrossRef]
- Isheden, G.; Abrahamsson, L.; Andersson, T.M.-L.; Czene, K.; Humphreys, K. Joint models of tumour size and lymph node spread for incident breast cancer cases in the presence of screening. Stat. Methods Med. Res. 2019, 28, 3822–3842. [Google Scholar] [CrossRef] [Green Version]
- Bartoszyński, R.; Edler, L.; Hanin, L.; Kopp-Schneider, A.; Pavlova, L.; Tsodikov, A.; Zorin, A.; Yakovlev, A.Y. Modeling cancer detection: Tumor size as a source of information on unobservable stages of carcinogenesis. Math. Biosci. 2001, 171, 113–142. [Google Scholar] [CrossRef]
- Heidenreich, W.F.; Luebeck, E.G.; Moolgavkaar, S.H. Some properties of the hazard function of the two-mutational clonal expansion model. Risk Anal. 1997, 17, 391–399. [Google Scholar] [CrossRef]
- Isheden, G.; Humphreys, K. A unifying framework for continuous tumour growth modelling of breast cancer screening data. Math. Biosci. 2022, 353, 108897. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.H.X.; Simonella, L.; Wee, H.L.; Roellin, A.A.; Lim, Y.-W.; Lim, W.-Y.; Chia, K.S.; Hartman, M.; Cook, A.R. Quantifying the natural history of breast cancer. Br. J. Cancer 2013, 109, 2035–2043. [Google Scholar] [CrossRef] [Green Version]
- Prevost, T.C.; Launoy, G.; Duffy, S.W.; Chen, H.-H. Estimating Sensitivity and Sojourn Time in Screening for Colorectal Cancer: A Comparison of Statistical Approaches. Am. J. Epidemiol. 1998, 148, 609–619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isheden, G.; Humphreys, K. Modeling breast cancer tumour growth for a stable disease population. Stat. Meth. Med. Res. 2019, 28, 681–702. [Google Scholar] [CrossRef]
- Strandberg, R.; Czene, K.; Eriksson, M.; Hall, P.; Humphreys, K. Estimating Distributions of Breast Cancer Onset and Growth in a Swedish Mammography Screening Cohort. Cancer Epidemiol. Biomark. Prev. 2022, 31, 569–577. [Google Scholar] [CrossRef]
- Weiss, N.S.; Rossing, M.A. Healthy Screenee Bias in Epidemiologic Studies of Cancer Incidence. Epidemiology 1996, 7, 319–322. [Google Scholar] [PubMed]
- Cox, B.; Sneid, M.J. Bias in breast cancer research in the screening era. Breast 2013, 22, 1041–1045. [Google Scholar] [CrossRef]
- Abrahamsson, L.; Isheden, G.; Czene, K.; Humphreys, K. Continuous tumour growth models, lead time estimation and length bias in breast cancer screening studies. Stat. Methods Med. Res. 2019, 29, 374–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahamsson, L.; Czene, K.; Hall, P.; Humphreys, K. Breast cancer tumour growth modelling for studying the association of body size with tumour growth rate and symptomatic detection using case-control data. Breast Cancer Res. 2015, 17, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isheden, G.; Czene, K.; Humphreys, K. Random effects models of lymph node metastases in breast cancer: Quantifying the roles of covariates and screening using a continuous growth model. Biometrics 2021, 78, 376–387. [Google Scholar] [CrossRef]
- Olsson, S.; Andersson, I.; Karlberg, I.; Bjurstam, N.; Frodis, E.; Håkansson, S. Implementation of service screening with mammog-raphy in Sweden: From pilot study to nationwide programme. J. Med. Screen. 2000, 7, 14–18. [Google Scholar] [CrossRef]
- Association of the Nordic Cancer Registries. NORDCAN. Available online: https://nordcan.iarc.fr/ (accessed on 23 August 2022).
- The Statistical Database of Statistics Sweden. Available online: https://www.statistikdatabasen.scb.se/sq/133506 (accessed on 23 August 2022).
- Alam Khan, S.; Hernandez-Villafuerte, K.V.; Muchadeyi, M.T.; Schlander, M. Cost-effectiveness of risk-based breast cancer screening: A systematic review. Int. J. Cancer 2021, 149, 790–810. [Google Scholar] [CrossRef]
- Mandelblatt, J.S.; Near, A.M.; Miglioretti, D.L.; Munoz, D.; Sprague, B.L.; Trentham-Dietz, A.; Gangnon, R.; Kurian, A.W.; Weedon-Fekjaer, H.; Cronin, K.A.; et al. Common Model Inputs Used in CISNET Collaborative Breast Cancer Modeling. Med. Decis. Mak. 2018, 38, 9S–23S. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene ex-pression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Zhou, F.; Ren, J.; Li, X.; Jiang, Y.; Ma, S. A Selective Review of Multi-Level Omics Data Integration Using Variable Selection. Biotech 2019, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Chootipongchaivat, S.; van Ravesteyn, N.T.; Li, X.; Huang, H.; Weedon-Fekjær, H.; Ryser, M.D.; Weaver, D.L.; Burnside, E.S.; Heckman-Stoddard, B.M.; de Koning, H.J.; et al. Modeling the natural history of ductal carcinoma in situ based on pop-ulation data. Breast Cancer Res. 2020, 22, 53. [Google Scholar] [CrossRef]
- Zheng, Y.-Z.; Wang, L.; Hu, X.; Shao, Z.-M. Effect of tumor size on breast cancer-specific survival stratified by joint hormone receptor status in a SEER population-based study. Oncotarget 2015, 6, 22985–22995. [Google Scholar] [CrossRef] [Green Version]
- Saadatmand, S.; Bretveld, R.; Siesling, S.; Tilanus-Linthorst, M.M.A. Influence of tumour stage at breast cancer detection on survival in modern times: Population based study in 173,797 patients. BMJ 2015, 351, h4901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
| Stage | 0 | 1 | 2 | 3 |
|---|---|---|---|---|
| T-stage (mm) | - | 1–20 | 20–50 | >50 |
| N-stage | 0 | 1–3 | 4–9 | >9 |
| No Screening | Current | Change | Extended | Change | |
|---|---|---|---|---|---|
| Mammograms (k) | 0 | 16,430 | 18,553 | +12.9% | |
| Total cases | 151,376 | 152,446 | +0.7% | 153,464 | +0.7% |
| Symptomatic/Interval | 151,376 | 46,640 | 21,678 | −46.5% | |
| Overdiagnosed | 0 | 1070 | 2088 | +95.1% | |
| N-shifted | 0 | 8686 | 10,528 | +21.2% | |
| T-shifted | 0 | 38,442 | 47,362 | +23.2% | |
| Other screen-detected | 0 | 57,608 | 71,808 | +24.7% | |
| Breast cancer deaths | 41,182 | 33,550 | −18.5% | 32,249 | −3.9% |
| Avg. lead time (yrs) | 0 | 2.18 | 2.21 | +1.4% | |
| Avg. surv. diff. (yrs) | 0 | 2.79 | 2.37 | −15.1% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strandberg, R.; Abrahamsson, L.; Isheden, G.; Humphreys, K. Tumour Growth Models of Breast Cancer for Evaluating Early Detection—A Summary and a Simulation Study. Cancers 2023, 15, 912. https://doi.org/10.3390/cancers15030912
Strandberg R, Abrahamsson L, Isheden G, Humphreys K. Tumour Growth Models of Breast Cancer for Evaluating Early Detection—A Summary and a Simulation Study. Cancers. 2023; 15(3):912. https://doi.org/10.3390/cancers15030912
Chicago/Turabian StyleStrandberg, Rickard, Linda Abrahamsson, Gabriel Isheden, and Keith Humphreys. 2023. "Tumour Growth Models of Breast Cancer for Evaluating Early Detection—A Summary and a Simulation Study" Cancers 15, no. 3: 912. https://doi.org/10.3390/cancers15030912