

Ethanol Metabolism and Melanoma
 , , , , and
, , , , and 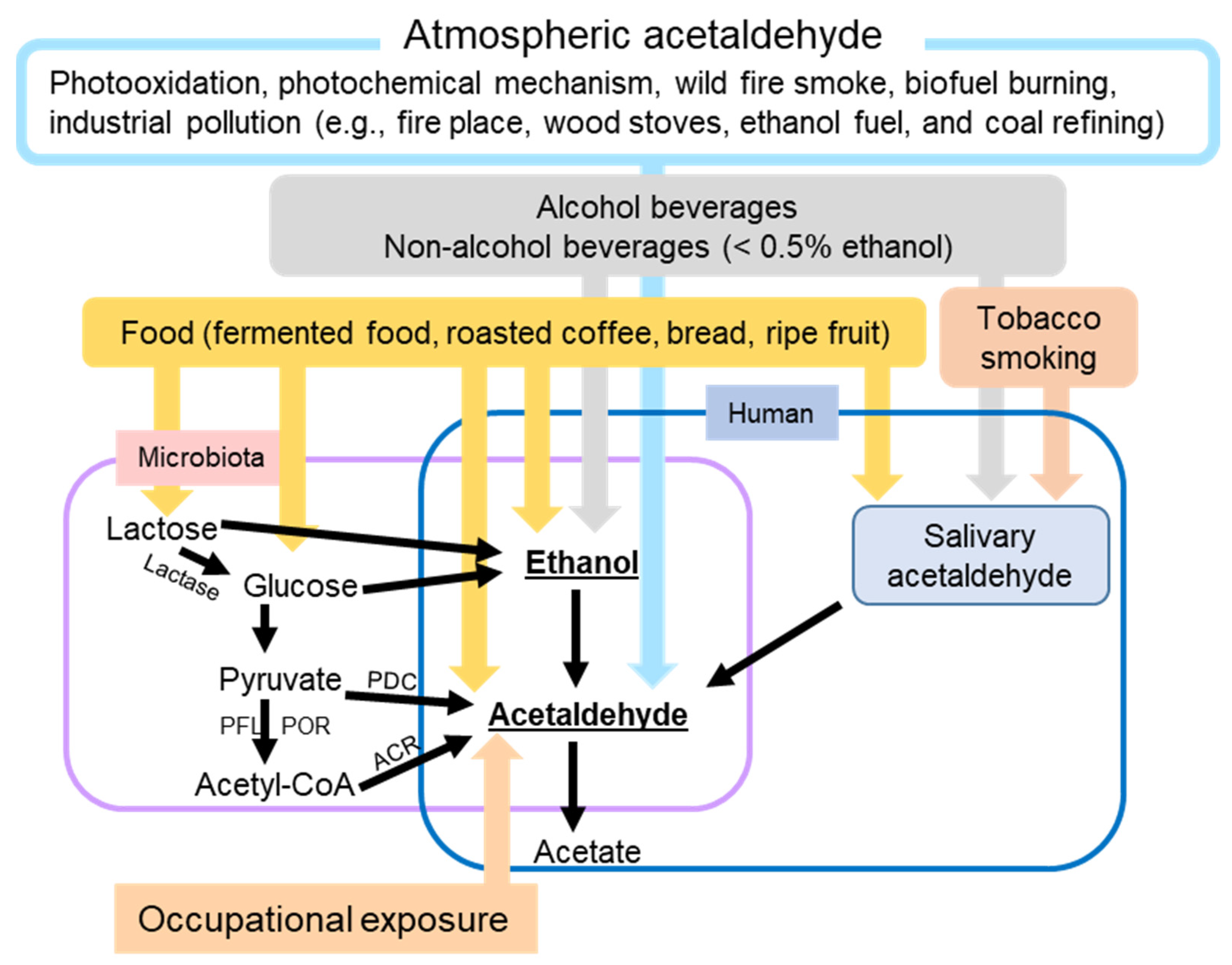{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Exposure to Ethanol and AcAH
2.1. Sources of Ethanol
2.2. Sources of AcAH
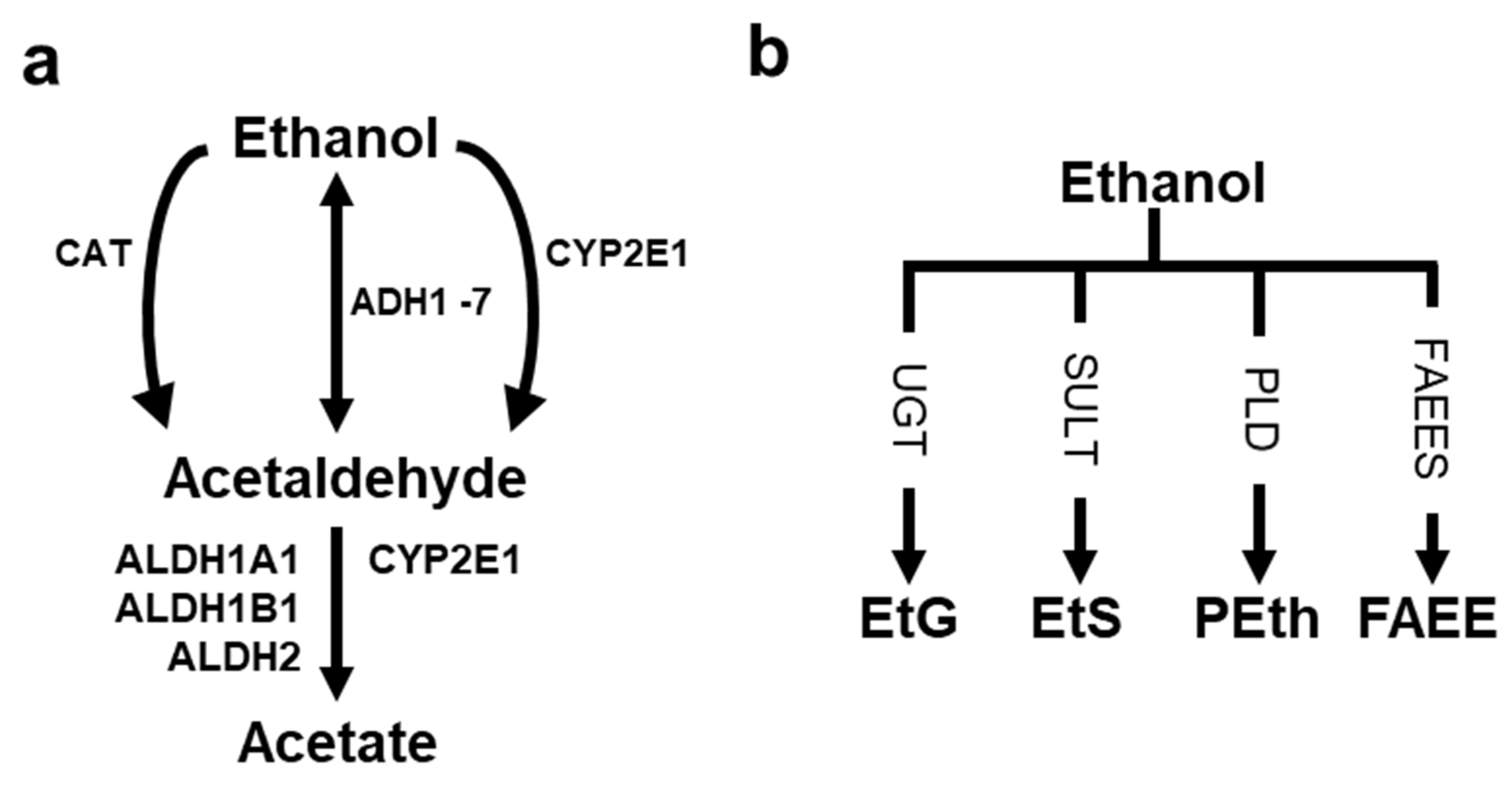
3. Ethanol Metabolism and Its Contribution to Human Diseases
3.1. Ethanol Metabolizing Enzymes and Their Impacts on Humans
3.2. AcAH Metabolizing Enzymes and Their Effects on Humans
4. Ethanol Metabolism and Its Contribution to Melanoma
4.1. Potential Roles of Dysfunctional Ethanol and AcAH Metabolism in Melanoma
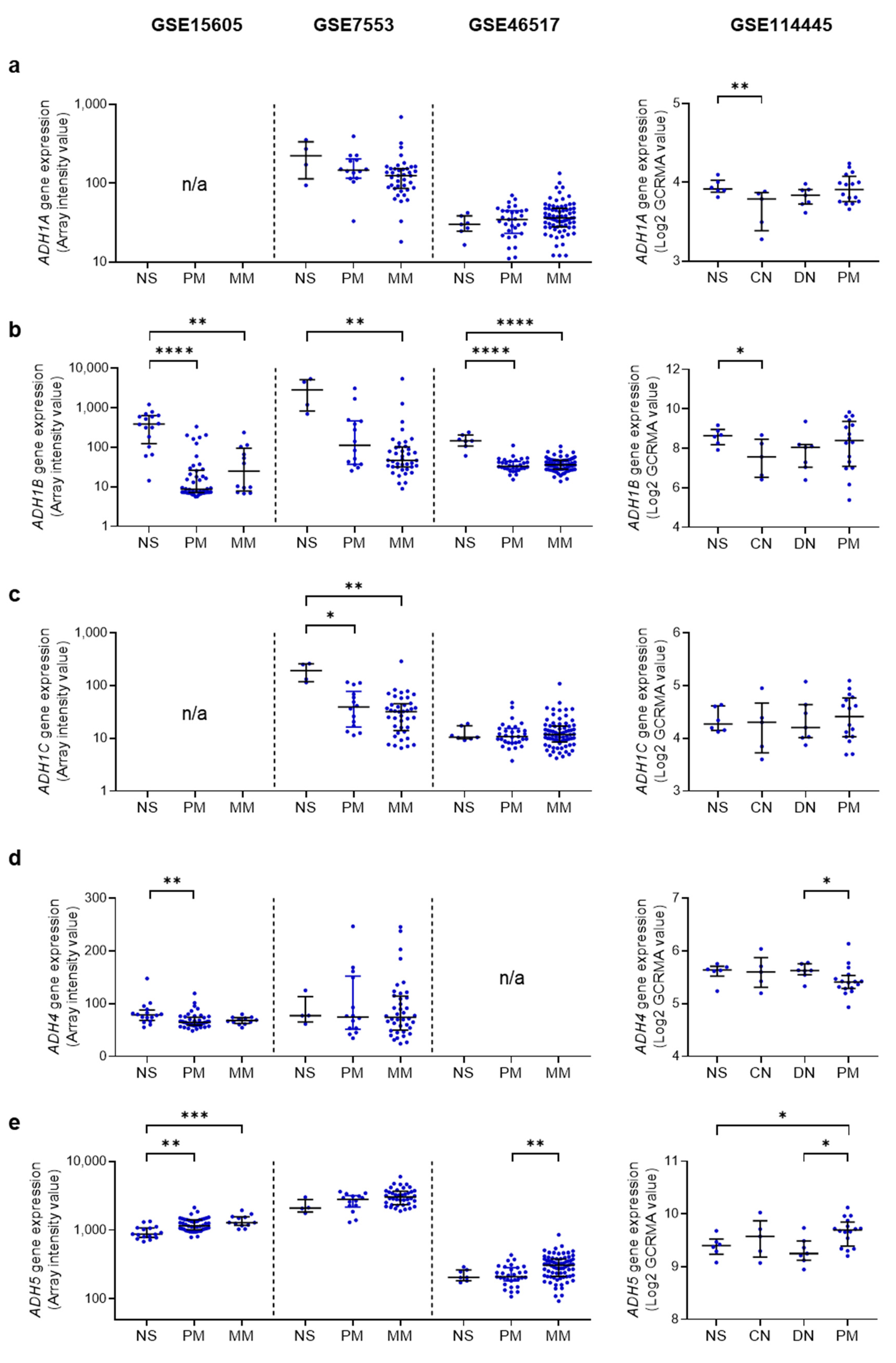
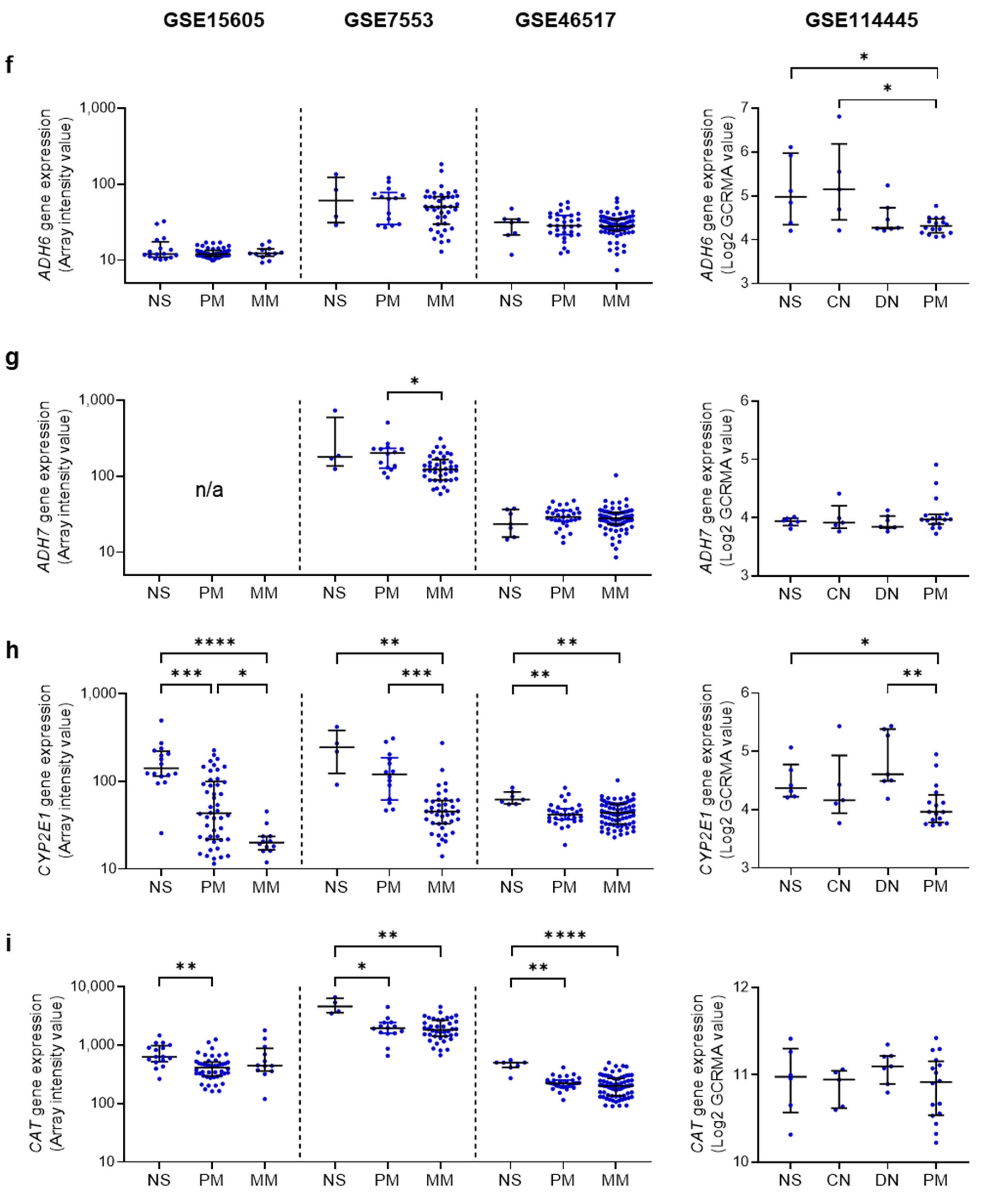
4.2. Ethanol Metabolizing Enzymes in Human Melanoma
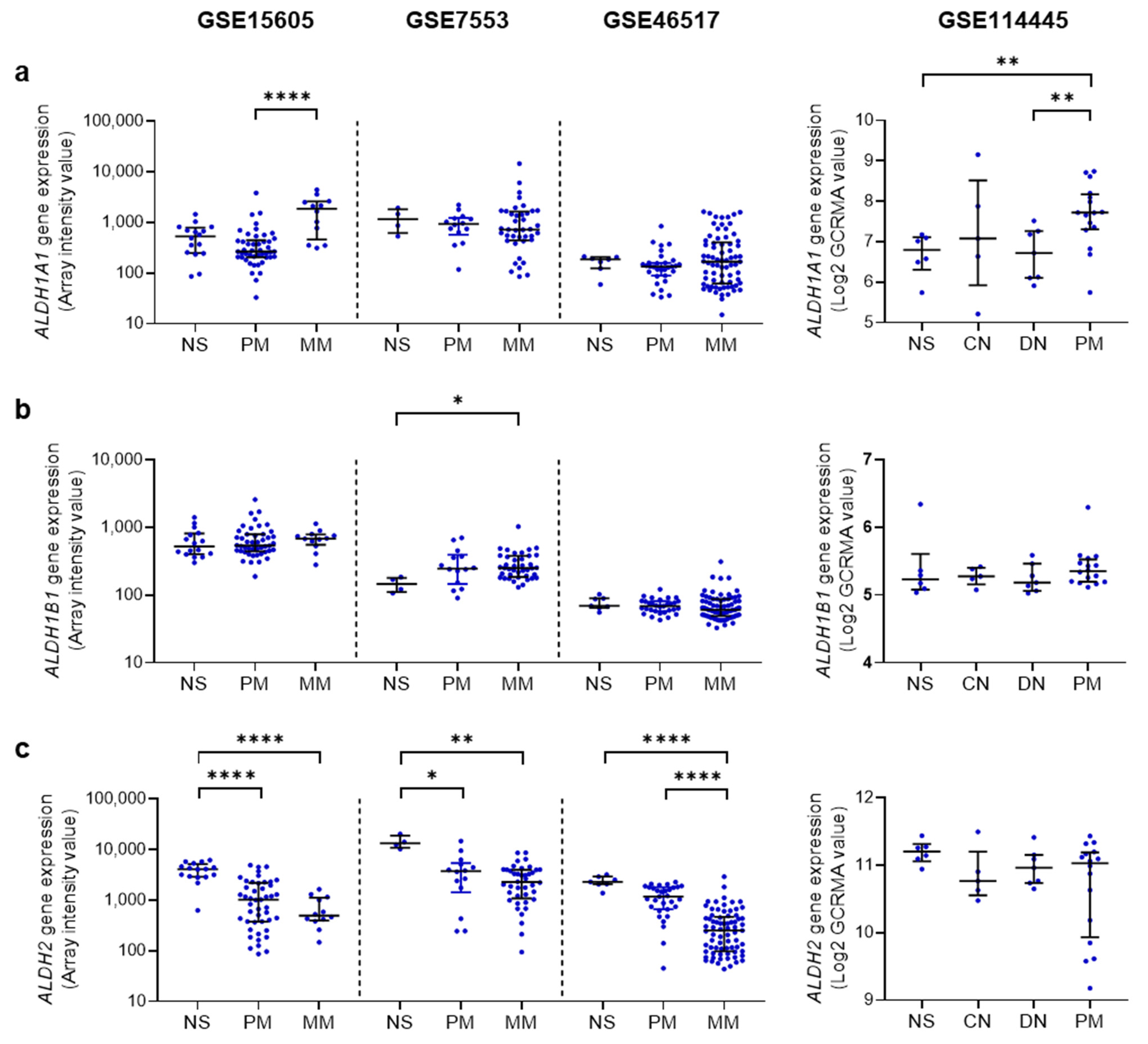
4.3. AcAH Metabolizing Enzymes in Human Melanoma
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Whiteman, D.C.; Green, A.C.; Olsen, C.M. The growing burden of invasive melanoma: Projections of incidence rates and numbers of new cases in six susceptible populations through 2031. J. Investig. Dermatol. 2016, 136, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Read, J.; Wadt, K.A.; Hayward, N.K. Melanoma genetics. J. Med. Genet. 2016, 53, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, J.E.; Truong, A.; Meyer, L.J. Genetic predisposition to melanoma. Semin. Oncol. 2016, 43, 591–597. [Google Scholar] [CrossRef]
- Sawada, Y.; Nakamura, M. Daily lifestyle and cutaneous malignancies. Int. J. Mol. Sci. 2021, 22, 5527. [Google Scholar] [CrossRef]
- Batta, N.; Shangraw, S.; Nicklawsky, A.; Yamauchi, T.; Zhai, Z.; Menon, D.R.; Gao, D.; Dellavalle, R.P.; Fujita, M. Global melanoma correlations with obesity, smoking, and alcohol consumption. JMIR Dermatol. 2021, 4, e31275. [Google Scholar] [CrossRef]
- Ribero, S.; Glass, D.; Bataille, V. Genetic epidemiology of melanoma. Eur. J. Dermatol. 2016, 26, 335–339. [Google Scholar] [CrossRef]
- Tagliabue, E.; Gandini, S.; Bellocco, R.; Maisonneuve, P.; Newton-Bishop, J.; Polsky, D.; Lazovich, D.; Kanetsky, P.A.; Ghiorzo, P.; Gruis, N.A.; et al. MC1R variants as melanoma risk factors independent of at-risk phenotypic characteristics: A pooled analysis from the M-SKIP project. Cancer Manag. Res. 2018, 10, 1143–1154. [Google Scholar] [CrossRef]
- Yamauchi, T.; Shangraw, S.; Zhai, Z.; Ravindran Menon, D.; Batta, N.; Dellavalle, R.P.; Fujita, M. Alcohol as a non-UV social-environmental risk factor for melanoma. Cancers 2022, 14, 5010. [Google Scholar] [CrossRef]
- Rota, M.; Pasquali, E.; Bellocco, R.; Bagnardi, V.; Scotti, L.; Islami, F.; Negri, E.; Boffetta, P.; Pelucchi, C.; Corrao, G.; et al. Alcohol drinking and cutaneous melanoma risk: A systematic review and dose-risk meta-analysis. Br. J. Dermatol. 2014, 170, 1021–1028. [Google Scholar] [CrossRef]
- Miura, K.; Zens, M.S.; Peart, T.; Holly, E.A.; Berwick, M.; Gallagher, R.P.; Mack, T.M.; Elwood, J.M.; Karagas, M.R.; Green, A.C. Alcohol consumption and risk of melanoma among women: Pooled analysis of eight case-control studies. Arch. Dermatol. Res. 2015, 307, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Bagnardi, V.; Rota, M.; Botteri, E.; Tramacere, I.; Islami, F.; Fedirko, V.; Scotti, L.; Jenab, M.; Turati, F.; Pasquali, E.; et al. Alcohol consumption and site-specific cancer risk: A comprehensive dose-response meta-analysis. Br. J. Cancer 2015, 112, 580–593. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.; Nan, H.; Li, T.; Qureshi, A.; Cho, E. Alcohol intake and risk of incident melanoma: A pooled analysis of three prospective studies in the United States. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Gandini, S.; Masala, G.; Palli, D.; Cavicchi, B.; Saieva, C.; Ermini, I.; Baldini, F.; Gnagnarella, P.; Caini, S. Alcohol, alcoholic beverages, and melanoma risk: A systematic literature review and dose-response meta-analysis. Eur. J. Nutr. 2018, 57, 2323–2332. [Google Scholar] [CrossRef]
- Rumgay, H.; Shield, K.; Charvat, H.; Ferrari, P.; Sornpaisarn, B.; Obot, I.; Islami, F.; Lemmens, V.; Rehm, J.; Soerjomataram, I. Global burden of cancer in 2020 attributable to alcohol consumption: A population-based study. Lancet Oncol. 2021, 22, 1071–1080. [Google Scholar] [CrossRef]
- Rumgay, H.; Murphy, N.; Ferrari, P.; Soerjomataram, I. Alcohol and Cancer: Epidemiology and biological mechanisms. Nutrients 2021, 13, 3173. [Google Scholar] [CrossRef]
- Heymann, H.M.; Gardner, A.M.; Gross, E.R. Aldehyde-induced DNA and protein adducts as biomarker tools for alcohol use disorder. Trends Mol. Med. 2018, 24, 144–155. [Google Scholar] [CrossRef]
- Cheung, C.; Smith, C.K.; Hoog, J.O.; Hotchkiss, S.A. Expression and localization of human alcohol and aldehyde dehydrogenase enzymes in skin. Biochem. Biophys. Res. Commun. 1999, 261, 100–107. [Google Scholar] [CrossRef]
- Pargoletti, E.; Rimoldi, L.; Meroni, D.; Cappelletti, G. Photocatalytic removal of gaseous ethanol, acetaldehyde and acetic acid: From a fundamental approach to real cases. Int. Mater. Rev. 2022, 67, 864–897. [Google Scholar] [CrossRef]
- Cheung, C.; Davies, N.G.; Hoog, J.O.; Hotchkiss, S.A.; Smith Pease, C.K. Species variations in cutaneous alcohol dehydrogenases and aldehyde dehydrogenases may impact on toxicological assessments of alcohols and aldehydes. Toxicology 2003, 184, 97–112. [Google Scholar] [CrossRef]
- Ahn, J.; Hayes, R.B. Environmental influences on the human microbiome and implications for noncommunicable disease. Annu. Rev. Public Health 2021, 42, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat. Rev. Cancer 2007, 7, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Goh, Y.I.; Verjee, Z.; Koren, G. Alcohol content in declared non-to low alcoholic beverages: Implications to pregnancy. Can. J. Clin. Pharmacol. 2010, 17, e47–e50. [Google Scholar] [PubMed]
- Kelber, O.; Steinhoff, B.; Nauert, C.; Biller, A.; Adler, M.; Abdel-Aziz, H.; Okpanyi, S.N.; Kraft, K.; Nieber, K. Ethanol in herbal medicinal products for children: Data from pediatric studies and pharmacovigilance programs. Wien. Med. Wochenschr. 2017, 167, 183–188. [Google Scholar] [CrossRef]
- Gorgus, E.; Hittinger, M.; Schrenk, D. Estimates of ethanol exposure in children from food not labeled as alcohol-containing. J. Anal. Toxicol. 2016, 40, 537–542. [Google Scholar] [CrossRef]
- Jones, A.W. Excretion of low-molecular weight volatile substances in human breath: Focus on endogenous ethanol. J. Anal. Toxicol. 1985, 9, 246–250. [Google Scholar] [CrossRef]
- Painter, K.; Cordell, B.J.; Sticco, K.L. Auto-brewery syndrome. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2022. [Google Scholar]
- Hafez, E.M.; Hamad, M.A.; Fouad, M.; Abdel-Lateff, A. Auto-brewery syndrome: Ethanol pseudo-toxicity in diabetic and hepatic patients. Hum. Exp. Toxicol. 2017, 36, 445–450. [Google Scholar] [CrossRef]
- Welch, B.T.; Coelho Prabhu, N.; Walkoff, L.; Trenkner, S.W. Auto-brewery syndrome in the setting of long-standing Crohn’s disease: A case report and review of the literature. J. Crohn’s Colitis 2016, 10, 1448–1450. [Google Scholar] [CrossRef]
- Malik, F.; Wickremesinghe, P.; Saverimuttu, J. Case report and literature review of auto-brewery syndrome: Probably an underdiagnosed medical condition. BMJ Open Gastroenterol. 2019, 6, e000325. [Google Scholar] [CrossRef]
- Vonghia, L.; Leggio, L.; Ferrulli, A.; Bertini, M.; Gasbarrini, G.; Addolorato, G.; Alcoholism Treatment Study, G. Acute alcohol intoxication. Eur. J. Intern. Med. 2008, 19, 561–567. [Google Scholar] [CrossRef]
- Bayoumy, A.B.; Mulder, C.J.J.; Mol, J.J.; Tushuizen, M.E. Gut fermentation syndrome: A systematic review of case reports. United Eur. Gastroenterol. J. 2021, 9, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Dinis-Oliveira, R.J. The auto-brewery syndrome: A perfect metabolic “storm” with clinical and forensic implications. J. Clin. Med. 2021, 10, 4637. [Google Scholar] [CrossRef] [PubMed]
- Pronk, J.T.; Yde Steensma, H.; Van Dijken, J.P. Pyruvate metabolism in Saccharomyces cerevisiae. Yeast 1996, 12, 1607–1633. [Google Scholar] [CrossRef]
- Homann, N.; Jousimies-Somer, H.; Jokelainen, K.; Heine, R.; Salaspuro, M. High acetaldehyde levels in saliva after ethanol consumption: Methodological aspects and pathogenetic implications. Carcinogenesis 1997, 18, 1739–1743. [Google Scholar] [CrossRef] [PubMed]
- Millet, D.B.; Guenther, A.; Siegel, D.A.; Nelson, N.B.; Singh, H.B.; de Gouw, J.A.; Warneke, C.; Williams, J.; Eerdekens, G.; Sinha, V.; et al. Global atmospheric budget of acetaldehyde: 3-D model analysis and constraints from in-situ and satellite observations. Atmos. Chem. Phys. 2010, 10, 3405–3425. [Google Scholar] [CrossRef]
- Singh, H.B.; Salas, L.J.; Chatfield, R.B.; Czech, E.; Fried, A.; Walega, J.; Evans, M.J.; Field, B.D.; Jacob, D.J.; Blake, D.; et al. Analysis of the atmospheric distribution, sources, and sinks of oxygenated volatile organic chemicals based on measurements over the Pacific during TRACE-P. J. Geophys. Res.-Atmos. 2004, 109, D15S07. [Google Scholar] [CrossRef]
- Custer, T.; Schade, G. Methanol and acetaldehyde fluxes over ryegrass. Tellus B 2007, 59, 673–684. [Google Scholar] [CrossRef]
- Cruz, L.P.S.; Luz, S.R.; Campos, V.P.; Santana, F.O.; Alves, R.S. Determination and risk assessment of formaldehyde and acetaldehyde in the ambient air of gas stations in Salvador, Bahia, Brazil. J. Brazil. Chem. Soc. 2020, 31, 1137–1148. [Google Scholar] [CrossRef]
- Hadei, M.; Shahsavani, A.; Hopke, P.K.; Kermani, M.; Yarahmadi, M.; Mahmoudi, B. Comparative health risk assessment of in-vehicle exposure to formaldehyde and acetaldehyde for taxi drivers and passengers: Effects of zone, fuel, refueling, vehicle’s age and model. Environ. Pollut. 2019, 254, 112943. [Google Scholar] [CrossRef]
- Naddafi, K.; Nabizadeh, R.; Rostami, R.; Ghaffari, H.R.; Fazlzadeh, M. Formaldehyde and acetaldehyde in the indoor air of waterpipe cafes: Measuring exposures and assessing health effects. Build. Environ. 2019, 165, 106392. [Google Scholar] [CrossRef]
- Chang, P.T.; Hung, P.C.; Tsai, S.W. Occupational exposures of flour dust and airborne chemicals at bakeries in Taiwan. J. Occup. Environ. Hyg. 2018, 15, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Hadei, M.; Hopke, P.K.; Shahsavani, A.; Moradi, M.; Yarahmadi, M.; Emam, B.; Rastkari, N. Indoor concentrations of VOCs in beauty salons; association with cosmetic practices and health risk assessment. J. Occup. Med. Toxicol. 2018, 13, 30. [Google Scholar] [CrossRef] [PubMed]
- Karaffa, L.S. The Merck Index: An Encyclopedia of Chemicals, Drugs, and Biologicals; RSC Publishing: London, UK, 2013. [Google Scholar]
- Miyake, T.; Shibamoto, T. Quantitative analysis of acetaldehyde in foods and beverages. J. Agric. Food Chem. 1993, 41, 1968–1970. [Google Scholar] [CrossRef]
- Uebelacker, M.; Lachenmeier, D.W. Quantitative determination of acetaldehyde in foods using automated digestion with simulated gastric fluid followed by headspace gas chromatography. J. Autom. Methods Manag. Chem. 2011, 2011, 907317. [Google Scholar] [CrossRef] [PubMed]
- Nieminen, M.T.; Salaspuro, M. Local acetaldehyde-An essential role in alcohol-related upper gastrointestinal tract carcinogenesis. Cancers 2018, 10, 11. [Google Scholar] [CrossRef]
- Talhout, R.; Opperhuizen, A.; van Amsterdam, J.G. Role of acetaldehyde in tobacco smoke addiction. Eur. Neuropsychopharmacol. 2007, 17, 627–636. [Google Scholar] [CrossRef]
- Rabinoff, M.; Caskey, N.; Rissling, A.; Park, C. Pharmacological and chemical effects of cigarette additives. Am. J. Public Health 2007, 97, 1981–1991. [Google Scholar] [CrossRef]
- Marttila, E.; Bowyer, P.; Sanglard, D.; Uittamo, J.; Kaihovaara, P.; Salaspuro, M.; Richardson, M.; Rautemaa, R. Fermentative 2-carbon metabolism produces carcinogenic levels of acetaldehyde in Candida albicans. Mol. Oral Microbiol. 2013, 28, 281–291. [Google Scholar] [CrossRef]
- Kumamoto, C.A. Inflammation and gastrointestinal Candida colonization. Curr. Opin. Microbiol. 2011, 14, 386–391. [Google Scholar] [CrossRef]
- Eram, M.S.; Ma, K. Decarboxylation of pyruvate to acetaldehyde for ethanol production by hyperthermophiles. Biomolecules 2013, 3, 578–596. [Google Scholar] [CrossRef]
- Barron, K.A.; Jeffries, K.A.; Krupenko, N.I. Sphingolipids and the link between alcohol and cancer. Chem. Biol. Interact. 2020, 322, 109058. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhang, T.; Kusumanchi, P.; Han, S.; Yang, Z.; Liangpunsakul, S. Alcohol metabolizing enzymes, microsomal ethanol oxidizing system, cytochrome P450 2E1, catalase, and aldehyde dehydrogenase in alcohol-associated liver disease. Biomedicines 2020, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Norberg, A.; Jones, A.W.; Hahn, R.G.; Gabrielsson, J.L. Role of variability in explaining ethanol pharmacokinetics: Research and forensic applications. Clin. Pharmacokinet. 2003, 42, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.W. Alcohol, its absorption, distribution, metabolism, and excretion in the body and pharmacokinetic calculations. WIREs Forensic Sci. 2019, 1, 26. [Google Scholar] [CrossRef]
- Le Dare, B.; Lagente, V.; Gicquel, T. Ethanol and its metabolites: Update on toxicity, benefits, and focus on immunomodulatory effects. Drug. Metab. Rev. 2019, 51, 545–561. [Google Scholar] [CrossRef]
- Estonius, M.; Svensson, S.; Hoog, J.O. Alcohol dehydrogenase in human tissues: Localisation of transcripts coding for five classes of the enzyme. FEBS Lett. 1996, 397, 338–342. [Google Scholar] [CrossRef]
- Hartman, J.H.; Miller, G.P.; Meyer, J.N. Toxicological implications of mitochondrial localization of CYP2E1. Toxicol. Res. 2017, 6, 273–289. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685. [Google Scholar] [CrossRef]
- Hurley, T.D.; Edenberg, H.J. Genes encoding enzymes involved in ethanol metabolism. Alcohol Res. 2012, 34, 339–344. [Google Scholar]
- Edenberg, H.J. The genetics of alcohol metabolism: Role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res. Health 2007, 30, 5–13. [Google Scholar]
- Guo, H.; Zhang, G.; Mai, R. Alcohol dehydrogenase-1B Arg47His polymorphism and upper aerodigestive tract cancer risk: A meta-analysis including 24,252 subjects. Alcohol. Clin. Exp. Res. 2012, 36, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Homann, N.; Stickel, F.; Konig, I.R.; Jacobs, A.; Junghanns, K.; Benesova, M.; Schuppan, D.; Himsel, S.; Zuber-Jerger, I.; Hellerbrand, C.; et al. Alcohol dehydrogenase 1C*1 allele is a genetic marker for alcohol-associated cancer in heavy drinkers. Int. J. Cancer 2006, 118, 1998–2002. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.C.; Lee, S.L.; Lee, Y.P.; Lai, C.L.; Yin, S.J. Modeling of human hepatic and gastrointestinal ethanol metabolism with kinetic-mechanism-based full-rate equations of the component alcohol dehydrogenase isozymes and allozymes. Chem. Res. Toxicol. 2018, 31, 556–569. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.L.; Wang, M.F.; Lee, A.I.; Yin, S.J. The metabolic role of human ADH3 functioning as ethanol dehydrogenase. FEBS Lett. 2003, 544, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Barnett, S.D.; Buxton, I.L.O. The role of S-nitrosoglutathione reductase (GSNOR) in human disease and therapy. Crit. Rev. Biochem. Mol. 2017, 52, 340–354. [Google Scholar] [CrossRef] [PubMed]
- Oka, Y.; Hamada, M.; Nakazawa, Y.; Muramatsu, H.; Okuno, Y.; Higasa, K.; Shimada, M.; Takeshima, H.; Hanada, K.; Hirano, T.; et al. Digenic mutations in ALDH2 and ADH5 impair formaldehyde clearance and cause a multisystem disorder, AMeD syndrome. Sci. Adv. 2020, 6, eabd7197. [Google Scholar] [CrossRef] [PubMed]
- Nadalutti, C.A.; Prasad, R.; Wilson, S.H. Perspectives on formaldehyde dysregulation: Mitochondrial DNA damage and repair in mammalian cells. DNA Repair 2021, 105, 103134. [Google Scholar] [CrossRef]
- Dingler, F.A.; Wang, M.; Mu, A.; Millington, C.L.; Oberbeck, N.; Watcham, S.; Pontel, L.B.; Kamimae-Lanning, A.N.; Langevin, F.; Nadler, C.; et al. Two aldehyde clearance systems are essential to prevent lethal formaldehyde accumulation in mice and humans. Mol. Cell 2020, 80, 996–1012.e9. [Google Scholar] [CrossRef]
- Hrycay, E.G.; Bandiera, S.M. Cytochrome P450 Enzymes. Preclinical Development Handbook: ADME and Biopharmaceutical Properties; Wiley: Hoboken, NY, USA, 2008; pp. 627–696. [Google Scholar]
- Katen, A.L.; Sipila, P.; Mitchell, L.A.; Stanger, S.J.; Nixon, B.; Roman, S.D. Epididymal CYP2E1 plays a critical role in acrylamide-induced DNA damage in spermatozoa and paternally mediated embryonic resorptionsdagger. Biol. Reprod. 2017, 96, 921–935. [Google Scholar] [CrossRef]
- Jin, M.; Ande, A.; Kumar, A.; Kumar, S. Regulation of cytochrome P450 2e1 expression by ethanol: Role of oxidative stress-mediated pkc/jnk/sp1 pathway. Cell Death. Dis. 2013, 4, e554. [Google Scholar] [CrossRef]
- Harjumaki, R.; Pridgeon, C.S.; Ingelman-Sundberg, M. CYP2E1 in alcoholic and non-alcoholic liver injury. Roles of ROS, reactive intermediates and lipid overload. Int. J. Mol. Sci. 2021, 22, 8221. [Google Scholar] [CrossRef] [PubMed]
- Koechling, U.M.; Amit, Z. Relationship between blood catalase activity and drinking history in a human population, a possible biological marker of the affinity to consume alcohol. Alcohol Alcohol. 1992, 27, 181–188. [Google Scholar] [PubMed]
- Plemenitas, A.; Kastelic, M.; Porcelli, S.; Serretti, A.; Rus Makovec, M.; Kores Plesnicar, B.; Dolzan, V. Genetic variability in CYP2E1 and catalase gene among currently and formerly alcohol-dependent male subjects. Alcohol Alcohol. 2015, 50, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.D.; Sun, Y.; Chen, N.; Huang, L.; Huang, J.W.; Zhu, M.; Wang, T.; Ji, Y.L. The role of catalase C262T gene polymorphism in the susceptibility and survival of cancers. Sci. Rep. 2016, 6, 26973. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Liu, X.; Wang, M.; Wang, X.; Kang, H.; Lin, S.; Yang, P.; Dai, C.; Xu, P.; Li, S.; et al. Two common functional catalase gene polymorphisms (rs1001179 and rs794316) and cancer susceptibility: Evidence from 14,942 cancer cases and 43,285 controls. Oncotarget 2016, 7, 62954–62965. [Google Scholar] [CrossRef]
- Shindo, Y.; Witt, E.; Han, D.; Epstein, W.; Packer, L. Enzymic and non-enzymic antioxidants in epidermis and dermis of human skin. J. Investig. Dermatol. 1994, 102, 122–124. [Google Scholar] [CrossRef]
- Wagener, F.A.; Carels, C.E.; Lundvig, D.M. Targeting the redox balance in inflammatory skin conditions. Int. J. Mol. Sci. 2013, 14, 9126–9167. [Google Scholar] [CrossRef]
- Rhie, G.E.; Seo, J.Y.; Chung, J.H. Modulation of catalase in human skin in vivo by acute and chronic UV radiation. Mol. Cells 2001, 11, 399–404. [Google Scholar]
- Maresca, V.; Flori, E.; Briganti, S.; Mastrofrancesco, A.; Fabbri, C.; Mileo, A.M.; Paggi, M.G.; Picardo, M. Correlation between melanogenic and catalase activity in in vitro human melanocytes: A synergic strategy against oxidative stress. Pigment Cell Melanoma Res. 2008, 21, 200–205. [Google Scholar] [CrossRef]
- Schallreuter, K.U.; Wood, J.M.; Berger, J. Low catalase levels in the epidermis of patients with vitiligo. J. Investig. Dermatol. 1991, 97, 1081–1085. [Google Scholar] [CrossRef]
- Schallreuter, K.U.; Moore, J.; Wood, J.M.; Beazley, W.D.; Peters, E.M.; Marles, L.K.; Behrens-Williams, S.C.; Dummer, R.; Blau, N.; Thony, B. Epidermal H(2)O(2) accumulation alters tetrahydrobiopterin (6BH4) recycling in vitiligo: Identification of a general mechanism in regulation of all 6BH4-dependent processes? J. Investig. Dermatol. 2001, 116, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Schallreuter, K.U.; Moore, J.; Wood, J.M.; Beazley, W.D.; Gaze, D.C.; Tobin, D.J.; Marshall, H.S.; Panske, A.; Panzig, E.; Hibberts, N.A. In vivo and in vitro evidence for hydrogen peroxide (H2O2) accumulation in the epidermis of patients with vitiligo and its successful removal by a UVB-activated pseudocatalase. J. Investig. Dermatol. Symp. Proc. 1999, 4, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, H.R.; Ged, C.; Bouadjar, B.; de Verneuil, H.; Taieb, A. Catalase overexpression reduces UVB-induced apoptosis in a human xeroderma pigmentosum reconstructed epidermis. Cancer Gene Ther. 2008, 15, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Dinis-Oliveira, R.J. Oxidative and non-oxidative metabolomics of ethanol. Curr. Drug Metab. 2016, 17, 327–335. [Google Scholar] [CrossRef]
- Heier, C.; Xie, H.; Zimmermann, R. Nonoxidative ethanol metabolism in humans-from biomarkers to bioactive lipids. IUBMB Life 2016, 68, 916–923. [Google Scholar] [CrossRef]
- Appenzeller, B.M.; Agirman, R.; Neuberg, P.; Yegles, M.; Wennig, R. Segmental determination of ethyl glucuronide in hair: A pilot study. Forensic Sci. Int. 2007, 173, 87–92. [Google Scholar] [CrossRef]
- D’Alessandro, A.; Fu, X.; Reisz, J.A.; Stone, M.; Kleinman, S.; Zimring, J.C.; Busch, M.; Recipient Epidemiology and Donor Evaluation Study-III (REDS III). Ethyl glucuronide, a marker of alcohol consumption, correlates with metabolic markers of oxidant stress but not with hemolysis in stored red blood cells from healthy blood donors. Transfusion 2020, 60, 1183–1196. [Google Scholar] [CrossRef]
- Paprocki, S.; Qassem, M.; Kyriacou, P.A. Review of ethanol intoxication sensing technologies and techniques. Sensors 2022, 22, 6819. [Google Scholar] [CrossRef]
- Lewis, S.S.; Hutchinson, M.R.; Zhang, Y.; Hund, D.K.; Maier, S.F.; Rice, K.C.; Watkins, L.R. Glucuronic acid and the ethanol metabolite ethyl-glucuronide cause toll-like receptor 4 activation and enhanced pain. Brain Behav. Immun. 2013, 30, 24–32. [Google Scholar] [CrossRef]
- Seishima, M.; Aoyama, Y.; Mori, S.; Nozawa, Y. Involvement of phospholipase D in ganglioside GQ1b-induced biphasic diacylglycerol production in human keratinocytes. J. Investig. Dermatol. 1995, 104, 835–838. [Google Scholar] [CrossRef]
- Hill, R.A.; Xu, W.; Yoshimura, M. Role of an adenylyl cyclase isoform in ethanol’s effect on cAMP regulated gene expression in NIH 3T3 cells. Biochem. Biophys. Rep. 2016, 8, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, M.; Tabakoff, B. Ethanol’s actions on cAMP-mediated signaling in cells transfected with type VII adenylyl cyclase. Alcohol. Clin. Exp. Res. 1999, 23, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Torres-Quesada, O.; Mayrhofer, J.E.; Stefan, E. The many faces of compartmentalized PKA signalosomes. Cell Signal. 2017, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Stickel, F. Acetaldehyde as an underestimated risk factor for cancer development: Role of genetics in ethanol metabolism. Genes Nutr. 2010, 5, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.J.; Malek, K.; Crabb, D.W. Distribution of messenger RNAs for aldehyde dehydrogenase 1, aldehyde dehydrogenase 2, and aldehyde dehydrogenase 5 in human tissues. J. Investig. Med. 1996, 44, 42–46. [Google Scholar] [PubMed]
- Chen, C.H.; Ferreira, J.C.; Gross, E.R.; Mochly-Rosen, D. Targeting aldehyde dehydrogenase 2: New therapeutic opportunities. Physiol. Rev. 2014, 94, 1–34. [Google Scholar] [CrossRef]
- Singh, S.; Arcaroli, J.; Thompson, D.C.; Messersmith, W.; Vasiliou, V. Acetaldehyde and retinaldehyde-metabolizing enzymes in colon and pancreatic cancers. Adv. Exp. Med. Biol. 2015, 815, 281–294. [Google Scholar]
- Lind, P.A.; Eriksson, C.J.; Wilhelmsen, K.C. The role of aldehyde dehydrogenase-1 (ALDH1A1) polymorphisms in harmful alcohol consumption in a Finnish population. Hum. Genomics 2008, 3, 24–35. [Google Scholar] [CrossRef]
- Linneberg, A.; Gonzalez-Quintela, A.; Vidal, C.; Jorgensen, T.; Fenger, M.; Hansen, T.; Pedersen, O.; Husemoen, L.L. Genetic determinants of both ethanol and acetaldehyde metabolism influence alcohol hypersensitivity and drinking behaviour among Scandinavians. Clin. Exp. Allergy 2010, 40, 123–130. [Google Scholar] [CrossRef]
- Zhang, Y.; Ren, J. ALDH2 in alcoholic heart diseases: Molecular mechanism and clinical implications. Pharmacol. Ther. 2011, 132, 86–95. [Google Scholar] [CrossRef]
- Song, B.J.; Abdelmegeed, M.A.; Yoo, S.H.; Kim, B.J.; Jo, S.A.; Jo, I.; Moon, K.H. Post-translational modifications of mitochondrial aldehyde dehydrogenase and biomedical implications. J. Proteom. 2011, 74, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A. The bidirectional effect of defective ALDH2 polymorphism and disease prevention. Adv. Exp. Med. Biol. 2019, 1193, 69–87. [Google Scholar] [PubMed]
- Chang, J.S.; Hsiao, J.R.; Chen, C.H. ALDH2 polymorphism and alcohol-related cancers in Asians: A public health perspective. J. Biomed. Sci. 2017, 24, 19. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Yabushita, H.; Kanaly, R.A.; Shibutani, S.; Yokoyama, A. Increased DNA damage in ALDH2-deficient alcoholics. Chem. Res. Toxicol. 2006, 19, 1374–1378. [Google Scholar] [CrossRef] [PubMed]
- Haschemi Nassab, M.; Rhein, M.; Hagemeier, L.; Kaeser, M.; Muschler, M.; Glahn, A.; Pich, A.; Heberlein, A.; Kornhuber, J.; Bleich, S.; et al. Impaired regulation of ALDH2 protein expression revealing a yet unknown epigenetic impact of rs886205 on specific methylation of a negative regulatory promoter region in alcohol-dependent patients. Eur. Addict. Res. 2016, 22, 59–69. [Google Scholar] [CrossRef]
- Pathak, H.; Frieling, H.; Bleich, S.; Glahn, A.; Heberlein, A.; Haschemi Nassab, M.; Hillemacher, T.; Burkert, A.; Rhein, M. Promoter polymorphism rs886205 genotype interacts with DNA methylation of the ALDH2 regulatory region in alcohol dependence. Alcohol Alcohol. 2017, 52, 269–276. [Google Scholar] [CrossRef]
- Xue, L.; Xu, F.; Meng, L.; Wei, S.; Wang, J.; Hao, P.; Bian, Y.; Zhang, Y.; Chen, Y. Acetylation-dependent regulation of mitochondrial ALDH2 activation by SIRT3 mediates acute ethanol-induced eNOS activation. FEBS Lett. 2012, 586, 137–142. [Google Scholar] [CrossRef]
- Shankarappa, B.; Mahadevan, J.; Murthy, P.; Purushottam, M.; Viswanath, B.; Jain, S.; Devarbhavi, H.; Mysore, A.V. Genetics and epigenetics of aldehyde dehydrogenase (ALDH2) in alcohol related liver disease. medRxiv 2021. [Google Scholar] [CrossRef]
- Stewart, M.J.; Dipple, K.M.; Stewart, T.R.; Crabb, D.W. The role of nuclear factor NF-Y/CP1 in the transcriptional regulation of the human aldehyde dehydrogenase 2-encoding gene. Gene 1996, 173, 155–161. [Google Scholar] [CrossRef]
- Stewart, M.J.; Dipple, K.M.; Estonius, M.; Nakshatri, H.; Everett, L.M.; Crabb, D.W. Binding and activation of the human aldehyde dehydrogenase 2 promoter by hepatocyte nuclear factor 4. Biochim. Biophys. Acta 1998, 1399, 181–186. [Google Scholar] [CrossRef]
- Pinaire, J.; Hasanadka, R.; Fang, M.; Chou, W.Y.; Stewart, M.J.; Kruijer, W.; Crabb, D. The retinoid X receptor response element in the human aldehyde dehydrogenase 2 promoter is antagonized by the chicken ovalbumin upstream promoter family of orphan receptors. Arch. Biochem. Biophys. 2000, 380, 192–200. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Fischer, M.; Cho, W.K.; Crabb, D. Transcriptional control of the human aldehyde dehydrogenase 2 promoter by hepatocyte nuclear factor 4: Inhibition by cyclic AMP and COUP transcription factors. Arch. Biochem. Biophys. 2002, 398, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Zuo, M.L.; Hu, G.H.; Duan, X.M.; Yang, Z.B. mir-193 targets ALDH2 and contributes to toxic aldehyde accumulation and tyrosine hydroxylase dysfunction in cerebral ischemia/reperfusion injury. Oncotarget 2017, 8, 99681–99692. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, Q.; Zhang, Y.; Geng, M.; Wei, Y.; Liu, Y.; Liu, S.; Petersen, R.B.; Yue, J.; Huang, K.; et al. Multigenerational maternal obesity increases the incidence of HCC in offspring via miR-27a-3p. J. Hepatol. 2020, 73, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Song, B.J.; Akbar, M.; Abdelmegeed, M.A.; Byun, K.; Lee, B.; Yoon, S.K.; Hardwick, J.P. Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox Biol. 2014, 3, 109–123. [Google Scholar] [CrossRef]
- Zhong, S.; Li, L.; Zhang, Y.L.; Zhang, L.; Lu, J.; Guo, S.; Liang, N.; Ge, J.; Zhu, M.; Tao, Y.; et al. Acetaldehyde dehydrogenase 2 interactions with LDLR and AMPK regulate foam cell formation. J. Clin. Investig. 2019, 129, 252–267. [Google Scholar] [CrossRef]
- Zhang, H.; Fu, L. The role of ALDH2 in tumorigenesis and tumor progression: Targeting ALDH2 as a potential cancer treatment. Acta Pharm. Sin. B 2021, 11, 1400–1411. [Google Scholar] [CrossRef]
- Zuo, W.; Zhan, Z.; Ma, L.; Bai, W.; Zeng, S. Effect of ALDH2 polymorphism on cancer risk in Asians: A meta-analysis. Medicine 2019, 98, e14855. [Google Scholar] [CrossRef] [PubMed]
- Masaoka, H.; Ito, H.; Soga, N.; Hosono, S.; Oze, I.; Watanabe, M.; Tanaka, H.; Yokomizo, A.; Hayashi, N.; Eto, M.; et al. Aldehyde dehydrogenase 2 (ALDH2) and alcohol dehydrogenase 1B (ADH1B) polymorphisms exacerbate bladder cancer risk associated with alcohol drinking: Gene-environment interaction. Carcinogenesis 2016, 37, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.T.; Wong, T.Y.; Ou, C.Y.; Fang, S.Y.; Chen, K.C.; Hsiao, J.R.; Huang, C.C.; Lee, W.T.; Lo, H.I.; Huang, J.S.; et al. The interplay between alcohol consumption, oral hygiene, ALDH2 and ADH1B in the risk of head and neck cancer. Int. J. Cancer 2014, 135, 2424–2436. [Google Scholar] [CrossRef] [PubMed]
- Peng, G.S.; Yin, S.J. Effect of the allelic variants of aldehyde dehydrogenase ALDH2*2 and alcohol dehydrogenase ADH1B*2 on blood acetaldehyde concentrations. Hum. Genomics 2009, 3, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, Y.N.; Suzuki, E.; Imoto, I.; Kasugai, Y.; Oze, I.; Ugai, T.; Iwase, M.; Usui, Y.; Kawakatsu, Y.; Sawabe, M.; et al. Across-site differences in the mechanism of alcohol-induced digestive tract carcinogenesis: An evaluation by mediation analysis. Cancer. Res. 2020, 80, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Kunitoh, S.; Imaoka, S.; Hiroi, T.; Yabusaki, Y.; Monna, T.; Funae, Y. Acetaldehyde as well as ethanol is metabolized by human CYP2E1. J. Pharmacol. Exp. Ther. 1997, 280, 527–532. [Google Scholar] [PubMed]
- Ye, X.; Wang, X.; Shang, L.; Zhu, G.; Su, H.; Han, C.; Qin, W.; Li, G.; Peng, T. Genetic variants of ALDH2-rs671 and CYP2E1-rs2031920 contributed to risk of hepatocellular carcinoma susceptibility in a Chinese population. Cancer Manag. Res. 2018, 10, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Subramaniyan, V.; Chakravarthi, S.; Jegasothy, R.; Seng, W.Y.; Fuloria, N.K.; Fuloria, S.; Hazarika, I.; Das, A. Alcohol-associated liver disease: A review on its pathophysiology, diagnosis and drug therapy. Toxicol. Rep. 2021, 8, 376–385. [Google Scholar] [CrossRef]
- Bode, C.; Bode, J.C. Alcohol’s role in gastrointestinal tract disorders. Alcohol Health Res. World 1997, 21, 76–83. [Google Scholar]
- Azimian Zavareh, P.; Silva, P.; Gimhani, N.; Atukorallaya, D. Effect of embryonic alcohol exposure on craniofacial and skin melanocyte development: Insights from zebrafish (Danio rerio). Toxics 2022, 10, 544. [Google Scholar] [CrossRef]
- Bang, J.; Zippin, J.H. Cyclic adenosine monophosphate (cAMP) signaling in melanocyte pigmentation and melanomagenesis. Pigment Cell Melanoma Res. 2021, 34, 28–43. [Google Scholar] [CrossRef]
- Larribere, L.; Utikal, J. NF1-dependent transcriptome regulation in the melanocyte lineage and in melanoma. J. Clin. Med. 2021, 10, 3350. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef]
- Rodriguez-Zavala, J.S.; Calleja, L.F.; Moreno-Sanchez, R.; Yoval-Sanchez, B. Role of aldehyde dehydrogenases in physiopathological processes. Chem. Res. Toxicol. 2019, 32, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Denat, L.; Kadekaro, A.L.; Marrot, L.; Leachman, S.A.; Abdel-Malek, Z.A. Melanocytes as instigators and victims of oxidative stress. J. Investig. Dermatol. 2014, 134, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, K.; Kazimierczak, U.; Kolenda, T. Oxidative stress in melanogenesis and melanoma development. Contemp. Oncol. 2022, 26, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.D.; Halevy, S. Alcohol intake, immune response, and the skin. Clin. Dermatol. 1999, 17, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Brand, R.M.; Stottlemyer, J.M.; Paglia, M.C.; Carey, C.D.; Falo, L.D., Jr. Ethanol consumption synergistically increases ultraviolet radiation induced skin damage and immune dysfunction. J. Dermatol. Sci. 2021, 101, 40–48. [Google Scholar] [CrossRef]
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; de Lichy, M.; Bille, K.; Dessen, P.; d’Hayer, B.; Mohamdi, H.; Remenieras, A.; et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 2011, 480, 94–98. [Google Scholar] [CrossRef]
- Meadows, G.G.; Zhang, H. Effects of alcohol on tumor growth, metastasis, immune response, and host survival. Alcohol Res. 2015, 37, 311–322. [Google Scholar]
- Zhang, H.; Zhu, Z.; Meadows, G.G. Chronic alcohol consumption impairs distribution and compromises circulation of B cells in B16BL6 melanoma-bearing mice. J. Immunol. 2012, 189, 1340–1348. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, Z.; Meadows, G.G. Chronic alcohol consumption decreases the percentage and number of NK cells in the peripheral lymph nodes and exacerbates B16BL6 melanoma metastasis into the draining lymph nodes. Cell. Immunol. 2011, 266, 172–179. [Google Scholar] [CrossRef]
- Hoyt, L.R.; Randall, M.J.; Ather, J.L.; DePuccio, D.P.; Landry, C.C.; Qian, X.; Janssen-Heininger, Y.M.; van der Vliet, A.; Dixon, A.E.; Amiel, E.; et al. Mitochondrial ROS induced by chronic ethanol exposure promote hyper-activation of the NLRP3 inflammasome. Redox Biol. 2017, 12, 883–896. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Goda, N.; Kanai, M.; Niwa, D.; Osanai, K.; Yamamoto, Y.; Senoo-Matsuda, N.; Johnson, R.S.; Miura, S.; Kabe, Y.; et al. HIF-1alpha induction suppresses excessive lipid accumulation in alcoholic fatty liver in mice. J. Hepatol. 2012, 56, 441–447. [Google Scholar] [CrossRef]
- D’Aguanno, S.; Mallone, F.; Marenco, M.; Del Bufalo, D.; Moramarco, A. Hypoxia-dependent drivers of melanoma progression. J. Exp. Clin. Cancer Res. 2021, 40, 159. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, M.; Ke, Z.J.; Luo, J. Cellular and molecular mechanisms underlying alcohol-induced aggressiveness of breast cancer. Pharmacol. Res. 2017, 115, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sun, J.; Han, D.; Cui, S.; Yan, X. Identification of potential biomarkers and small molecule drugs for cutaneous melanoma using integrated bioinformatic analysis. Front. Cell Dev. Biol. 2022, 10, 858633. [Google Scholar] [CrossRef]
- Sumantran, V.N.; Mishra, P.; Bera, R.; Sudhakar, N. Microarray analysis of differentially-expressed genes encoding CYP450 and phase II drug metabolizing enzymes in psoriasis and melanoma. Pharmaceutics 2016, 8, 4. [Google Scholar] [CrossRef]
- Raskin, L.; Fullen, D.R.; Giordano, T.J.; Thomas, D.G.; Frohm, M.L.; Cha, K.B.; Ahn, J.; Mukherjee, B.; Johnson, T.M.; Gruber, S.B. Transcriptome profiling identifies HMGA2 as a biomarker of melanoma progression and prognosis. J. Investig. Dermatol. 2013, 133, 2585–2592. [Google Scholar] [CrossRef]
- Riker, A.I.; Enkemann, S.A.; Fodstad, O.; Liu, S.; Ren, S.; Morris, C.; Xi, Y.; Howell, P.; Metge, B.; Samant, R.S.; et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med. Genomics. 2008, 1, 13. [Google Scholar] [CrossRef]
- Kabbarah, O.; Nogueira, C.; Feng, B.; Nazarian, R.M.; Bosenberg, M.; Wu, M.; Scott, K.L.; Kwong, L.N.; Xiao, Y.; Cordon-Cardo, C.; et al. Integrative genome comparison of primary and metastatic melanomas. PLoS ONE 2010, 5, e10770. [Google Scholar] [CrossRef]
- Yan, B.Y.; Garcet, S.; Gulati, N.; Kiecker, F.; Fuentes-Duculan, J.; Gilleaudeau, P.; Sullivan-Whalen, M.; Shemer, A.; Mitsui, H.; Krueger, J.G. Novel immune signatures associated with dysplastic naevi and primary cutaneous melanoma in human skin. Exp. Dermatol. 2019, 28, 35–44. [Google Scholar] [CrossRef]
- Cichorek, M.; Wachulska, M.; Stasiewicz, A.; Tyminska, A. Skin melanocytes: Biology and development. Postep. Dermatol. Alergol. 2013, 30, 30–41. [Google Scholar] [CrossRef]
- Reemann, P.; Reimann, E.; Ilmjarv, S.; Porosaar, O.; Silm, H.; Jaks, V.; Vasar, E.; Kingo, K.; Koks, S. Melanocytes in the skin--comparative whole transcriptome analysis of main skin cell types. PLoS ONE 2014, 9, e115717. [Google Scholar] [CrossRef]
- Luo, Y.; Dallaglio, K.; Chen, Y.; Robinson, W.A.; Robinson, S.E.; McCarter, M.D.; Wang, J.; Gonzalez, R.; Thompson, D.C.; Norris, D.A.; et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells 2012, 30, 2100–2113. [Google Scholar] [CrossRef]
- Mameishvili, E.; Serafimidis, I.; Iwaszkiewicz, S.; Lesche, M.; Reinhardt, S.; Bolicke, N.; Buttner, M.; Stellas, D.; Papadimitropoulou, A.; Szabolcs, M.; et al. Aldh1b1 expression defines progenitor cells in the adult pancreas and is required for Kras-induced pancreatic cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 20679–20688. [Google Scholar] [CrossRef]
- Feng, Z.; Hom, M.E.; Bearrood, T.E.; Rosenthal, Z.C.; Fernandez, D.; Ondrus, A.E.; Gu, Y.; McCormick, A.K.; Tomaske, M.G.; Marshall, C.R.; et al. Targeting colorectal cancer with small-molecule inhibitors of ALDH1B1. Nat. Chem. Biol. 2022, 18, 1065–1075. [Google Scholar] [CrossRef]
- Chang, P.M.; Chen, C.H.; Yeh, C.C.; Lu, H.J.; Liu, T.T.; Chen, M.H.; Liu, C.Y.; Wu, A.T.H.; Yang, M.H.; Tai, S.K.; et al. Transcriptome analysis and prognosis of ALDH isoforms in human cancer. Sci. Rep. 2018, 8, 2713. [Google Scholar] [CrossRef]
- Ma, B.; Liu, Z.; Xu, H.; Liu, L.; Huang, T.; Meng, L.; Wang, L.; Zhang, Y.; Li, L.; Han, X. Molecular characterization and clinical relevance of ALDH2 in human cancers. Front. Med. 2021, 8, 832605. [Google Scholar] [CrossRef]
- Wu, L.; Dong, B.; Zhang, F.; Li, Y.; Liu, L. Prediction of the engendering mechanism and specific genes of primary melanoma by bioinformatics analysis. Dermatol. Sin. 2016, 34, 6. [Google Scholar] [CrossRef]
- Dimitriou, F.; Krattinger, R.; Ramelyte, E.; Barysch, M.J.; Micaletto, S.; Dummer, R.; Goldinger, S.M. The world of melanoma: Epidemiologic, genetic, and anatomic differences of melanoma across the globe. Curr. Oncol. Rep. 2018, 20, 87. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhai, Z.; Yamauchi, T.; Shangraw, S.; Hou, V.; Matsumoto, A.; Fujita, M. Ethanol Metabolism and Melanoma. Cancers 2023, 15, 1258. https://doi.org/10.3390/cancers15041258
Zhai Z, Yamauchi T, Shangraw S, Hou V, Matsumoto A, Fujita M. Ethanol Metabolism and Melanoma. Cancers. 2023; 15(4):1258. https://doi.org/10.3390/cancers15041258
Chicago/Turabian StyleZhai, Zili, Takeshi Yamauchi, Sarah Shangraw, Vincent Hou, Akiko Matsumoto, and Mayumi Fujita. 2023. "Ethanol Metabolism and Melanoma" Cancers 15, no. 4: 1258. https://doi.org/10.3390/cancers15041258
APA StyleZhai, Z., Yamauchi, T., Shangraw, S., Hou, V., Matsumoto, A., & Fujita, M. (2023). Ethanol Metabolism and Melanoma. Cancers, 15(4), 1258. https://doi.org/10.3390/cancers15041258